
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
3d single-ion magnets
Gavin A.
Craig
and
Mark
Murrie
 *
*
WestCHEM, School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: mark.murrie@glasgow.ac.uk
First published on 26th February 2015
Abstract
One of the determining factors in whether single-molecule magnets (SMMs) may be used as the smallest component of data storage, is the size of the barrier to reversal of the magnetisation, Ueff. This physical quantity depends on the magnitude of the magnetic anisotropy of a complex and the size of its spin ground state. In recent years, there has been a growing focus on maximising the anisotropy generated for a single 3d transition metal (TM) ion, by an appropriate ligand field, as a means of achieving higher barriers. Because the magnetic properties of these compounds arise from a single ion in a ligand field, they are often referred to as single-ion magnets (SIMs). Here, the synthetic chemist has a significant role to play, both in the design of ligands to enforce propitious splitting of the 3d orbitals and in the judicious choice of TM ion. Since the publication of the first 3d-based SIM, which was based on Fe(II), many other contributions have been made to this field, using different first row TM ions, and exploring varied coordination environments for the paramagnetic ions.
 Gavin A. Craig | Gavin A. Craig graduated with an MSci (Hons.) in Chemistry from the University of Glasgow in 2008. Under the supervision of Guillem Aromí at the Universitat de Barcelona, he obtained an MSc (2010) and then a PhD (2013) working in the field of spin crossover. During his doctoral studies he undertook several placements in the group of Azzedine Bousseksou at the LCC in Toulouse. Since November 2013 he is a Post-Doctoral Research Associate in the group of Mark Murrie in the School of Chemistry at the University of Glasgow, studying the effect of pressure on molecular magnetic materials. |
 Mark Murrie | Mark Murrie received his PhD from the University of Manchester in 1997 under the supervision of Professor Dave Garner FRS and Professor David Collison. Following postdoctoral positions at the University of Edinburgh, with Professor Richard Winpenny and at the University of Bern in Switzerland, with Professor Hans Güdel, he joined the School of Chemistry at the University of Glasgow in 2003, where he is now Reader. His current research interests are in the synthesis & characterisation of molecular magnets and magnetic nanoparticles and the application of high pressure science to magnetic systems. |
Key learning points(1) Slow relaxation of the magnetisation arises from a single 3d ion under an appropriate ligand field.(2) Slow relaxation in single-ion magnets (SIMs) can be observed, and energy barriers measured, using alternating current (ac) susceptibility measurements. (3) Quantum tunnelling of the magnetisation (QTM) may lead to no out-of-phase component of the dynamic magnetic susceptibility being observed. It may be possible to hinder the QTM by applying an additional external dc field. (4) Design principles based on the 3d ion used, coordination number, and ligand field generated to target and attain SIM behaviour. |
1. Introduction
If a single molecule could be used to encode binary information, then vast increases in data storage density could be achieved with respect to traditional media. The exploration of this possibility for the compound [Mn12O12(OAc)16(H2O)4]·2AcOH·4H2O (Mn12ac) led to the establishment of a new class of materials called single-molecule magnets (SMMs).1 Mn12ac has a preferential direction for the resultant magnetisation that arises from the precession of the spin in a magnetic field, caused by the anisotropy associated with the metal ions in the complex. At low temperature, by flipping the orientation of the field, this preferential direction can be reversed; that is, switched from lying along the z-axis, to lying along the −z-axis. Crucially, magnetisation in either direction is retained when the field is removed. Therefore, it can be imagined that “1” in binary coding could be assigned to the magnetisation along the +z direction, and “0” to the magnetisation along the −z direction.Since then this field, which has more generally studied molecular nanomagnets, has undergone several developments. Often, this has involved looking at the potential of molecular nanomagnets to fulfil applications in areas such as quantum computing or magnetic refrigeration, as well as how to deposit these molecules on surfaces.2 On a synthetic level, it has also led to the study of the one-dimensional analogues of SMMs, known as single-chain magnets (SCMs).3,4 Another synthetic strategy has been the use of a single ion to develop monometallic SMMs, an approach which first used lanthanide ions.5
In this Tutorial Review, we will focus on the relatively recent approach to obtain SMMs whose magnetic properties arise from a single first row transition metal (TM) ion in a suitable ligand field that creates magnetic anisotropy. In the literature, these are often referred to as either single-ion magnets (SIMs) or mononuclear SMMs, neither of which are perfect descriptors. Monometallic SMM is probably better but herein, we have chosen to use the SIM acronym rather than e.g. MSMM, which is more awkward. In Section 2, we briefly describe how slow relaxation of the magnetisation may arise, how it is observed, and why SIMs have become a focus of attention. Section 3 highlights some of the different strategies employed to induce a large magnetic anisotropy using a single 3d ion.
2. Single-molecule magnets: towards monometallic complexes
For a single-molecule magnet to function effectively as a means of data storage, there must be a barrier to the re-orientation of the molecule's magnetisation, to prevent a loss of information. The origin of the barrier lies in magnetic anisotropy. When the spin ground state of a molecule is S > 1/2, then zero-field splitting (ZFS) may arise if the symmetry is lower than cubic. The symmetry lowering may lead to the separation of excited states, which can then mix through spin–orbit coupling. The Hamiltonian associated with ZFS can be expressed as:| Ĥ = D[Ŝ2z − S(S + 1)/3] + E(Ŝ2x − Ŝ2y) | (1) |
| Ueff = |D|·S2 | (2a) |
| |D|·(S2 − ¼) | (2b) |
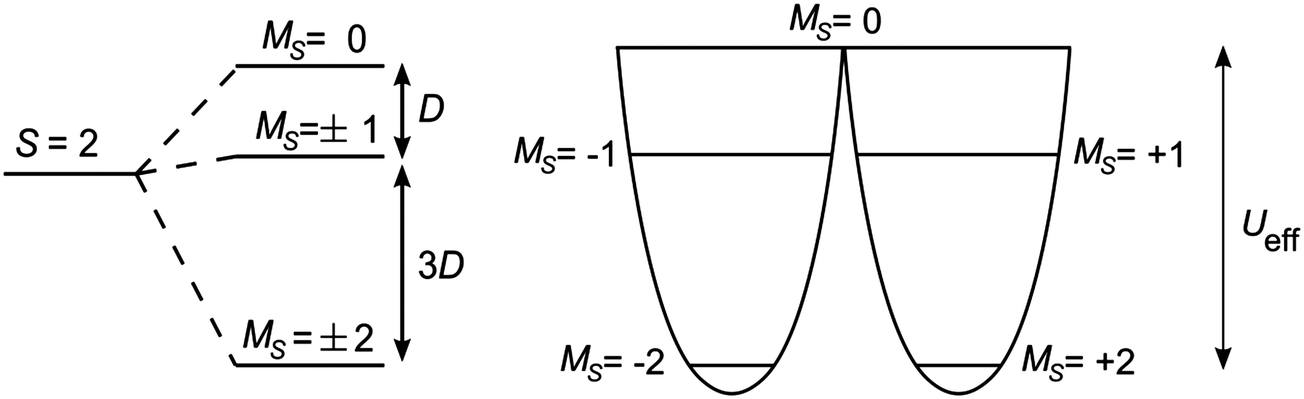 | ||
| Fig. 1 (left) Splitting of an S = 2 state into its constituent MS levels, induced by negative axial ZFS. (right) A view of the double-well thus generated, with the barrier to relaxation shown as Ueff. | ||
2.1 Measurement of the barrier to relaxation
The barrier to relaxation in SMMs is usually determined by alternating current (ac) susceptibility measurements. Under these conditions, the magnetic susceptibility of a compound consists of two components, corresponding to a real (in-phase) contribution, χM′, and an imaginary (out-of-phase) contribution, χM′′, which depend on the angular frequency with which the magnetic field oscillates.1 The inability of the magnetisation to follow the progressively faster switching field causes a decrease of the in-phase component and an increase of the out-of-phase component. The out-of-phase component will reach a maximum in χM′′ before decreasing again with the highest frequencies of switching fields. At this maximum, the angular frequency (ω) can be related to the relaxation time, τ, through:| ωτ = 2πντ = 1 | (3) |
By scanning the frequency to measure the dynamic susceptibility at several different temperatures, the relationship between τ and T may be determined through an Arrhenius plot of ln(τ) vs. 1/T. At higher temperatures, a linear fit will normally be possible, corresponding to eqn (4)
ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) τ = lnτ0 + Ueff/kBT τ = lnτ0 + Ueff/kBT | (4) |
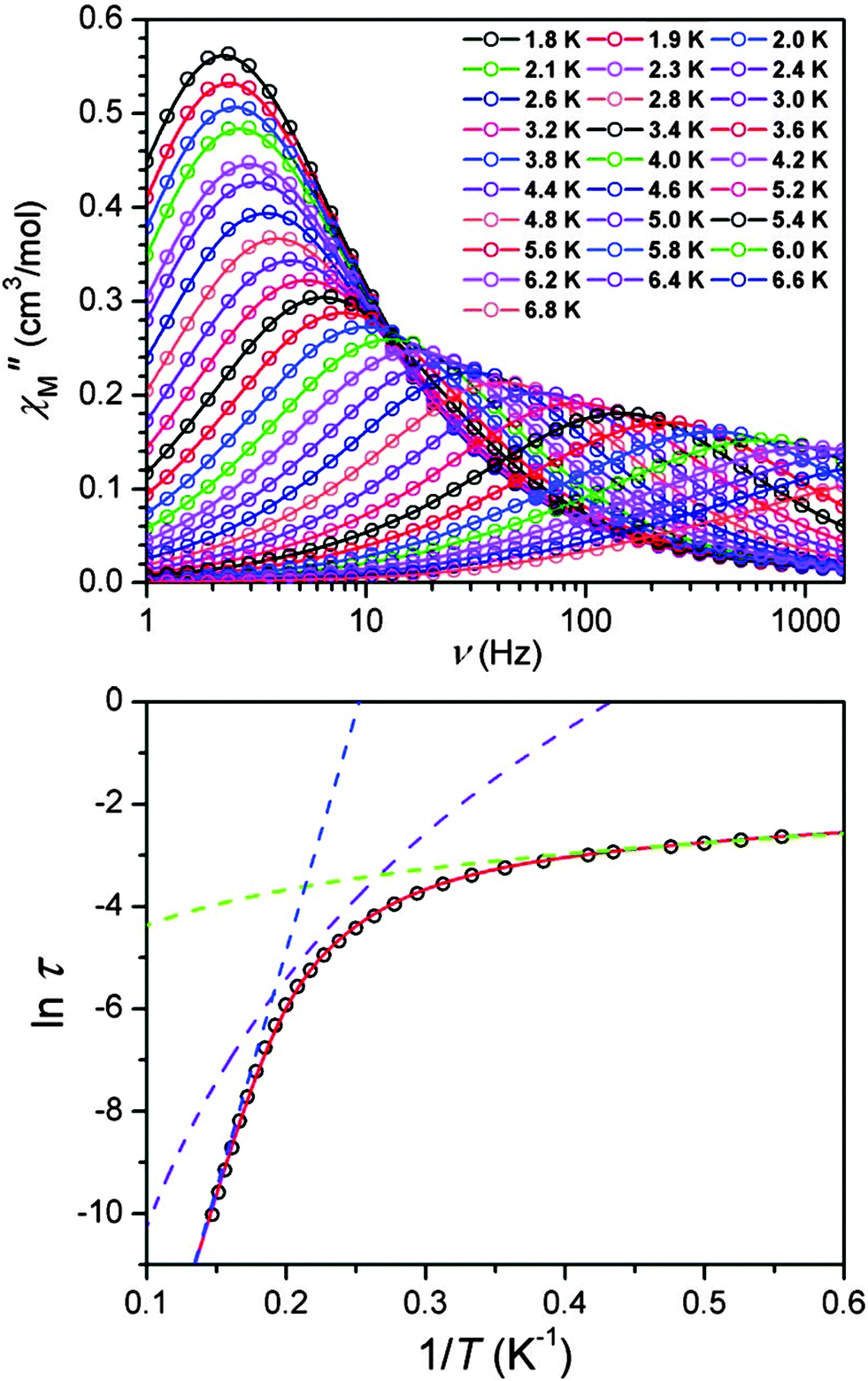 | ||
| Fig. 2 Top: variable-frequency out-of-phase ac susceptibility data for Na[(tpat-Bu)Fe]·THF, under a 1500 Oe dc field at various temperatures. Bottom: Arrhenius plot constructed from data. Dashed lines represent data fits to an Orbach (blue), Raman (purple), and direct (green) process. The solid red line represents a fit to the three processes simultaneously. Reprinted with permission from ref. 11. Copyright (2010) American Chemical Society. | ||
2.2 Increasing Ueff
The relationship between the barrier to relaxation of the magnetisation and the ground spin state S led to a vast effort towards building the compounds with the highest possible nuclearity, in an attempt to maximise Ueff.2 However, the stumbling block encountered was that D was found to be inversely proportional to S2.12 Therefore, incorporating large numbers of paramagnetic transition metal ions in a compound may be antagonistic to generating a large magnetic anisotropy. Often, this is because the anisotropy axis for a given ion within a polymetallic complex is not aligned with that of another ion within the same complex, leading to a diminished overall anisotropy.13 By employing a single ion, this scenario would be avoided, but leads to a clear limiting factor: MS can only ever be as large as the maximum spin of the single ion. The challenge thus presented is how to achieve the greatest possible anisotropy, given by D, and so attain larger barriers for the relaxation of the magnetisation.The discussion in Section 2 described situations where MS states are split by ZFS. In the majority of these cases, first order orbital angular momentum is quenched. The axial anisotropy that arises is due to second order spin–orbit coupling, which admixes relevant excited states into the ground state. The large first order orbital angular momentum in the lanthanides has already led to many studies of monometallic 4f single-molecule magnets.2 Therefore, many of the examples described below seek coordination environments in which first order orbital angular momentum is largely unquenched, potentially leading to much higher barriers.
3. 3d single-ion magnets
The first example of a monometallic 3d SMM was the high spin Fe(II) compound K[(tpaMes)Fe] (1, H3tpaMes = Tris((5-mesityl-1H-pyrrol-2-yl)methyl)amine).14 The Fe(II) ion lies in a trigonal pyramidal geometry, with an N4 coordination sphere (Fig. 3). The bulky ligand promotes the unusual geometry around the metal centre by impeding access to the second axial site. The orbital splitting thus induced generates a large magnetic anisotropy due to the unequal occupation of the 1e orbitals shown in Fig. 3, which leads to unquenched orbital angular momentum. The non-superposition of the variable field magnetisation measurements confirms that a large axial zero-field splitting is attained, with fits of the data yielding D = −39.6 cm−1, together with a small rhombic contribution, E = −0.4 cm−1. This rhombic contribution arises from a small structural distortion around the Fe(II) ion, which lowers the three-fold symmetry.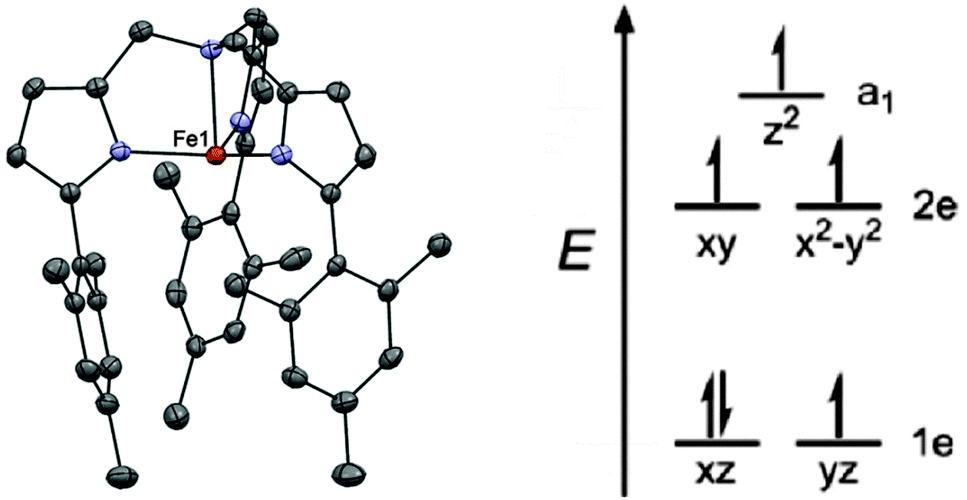 | ||
| Fig. 3 (left) A view of the anion [(tpaMes)Fe]−; Fe, red; N, blue; and C, grey. (right) A simplified view of the orbital splitting. Reprinted with permission from ref. 14. Copyright (2010) American Chemical Society. | ||
No out-of-phase signal for the magnetic susceptibility could be detected in the absence of an applied dc field, which was attributed to efficient QTM. To lower the rate of tunnelling, a dc field of 1500 Oe was applied during the alternating current (ac) susceptibility measurements leading to maxima in χ′′. While the value of D extracted from the fits of the static magnetic properties may have suggested a very large barrier (U = S2·|D| = 4 × 39.6 = 158 cm−1), the effective barrier, Ueff = 42 cm−1, derived from the Arrhenius plot was much lower due to tunnelling processes.
Subsequently, 1 was used as a platform to develop compounds in which other sterically demanding derivatives of tpa enforced similar geometries upon Fe(II) (see Table 1, compounds 2 and 3).11 Compound 2 is the only analogue to have crystallographically imposed three-fold symmetry and it shows the highest barrier. Since the study of this family of high spin Fe(II) compounds, comparable strategies have been used for other first row transition metals. As will be shown, many of these complexes share some features with 1–3, such as low coordination numbers and unusual geometries, and efficient QTM in the absence of an applied dc field.
| Compound | U eff/cm−1 (applied field/Oe) | τ 0/s | Ref. |
|---|---|---|---|
| Mn(III) | |||
| Ph4P[Mn(opbaCl2)(py)2] (4) | 12.6 (1000) | 1.2 × 10−7 | 15 |
| [Mn(5-TMAM(R)-salmen)(H2O)Co(CN)6]·7H2O·MeCN (5) | 11.5 (4500) | 2.9 × 10−7 | 16 |
| [Mn((OPPh2)2N)3] (6) | 8.3 (2250) | 0.5 × 10−7 | 17 |
| Fe(I) | |||
| [K(crypt-222)][Fe(C(SiMe3)3)2] (7) | 226 (0) | 1.3 × 10−9 | 19, 20 |
| [(cAAC)2Fe][B(C6F5)4] (9) | <20 (3000) | — | 21 |
| (cAAC)2FeCl (10) | 22.4 (500) | 7.0 × 10−8 | 21 |
| Fe(II) | |||
| K[(tpaMes)Fe] (1) | 42 (1500) | 2.0 × 10−9 | 14 |
| Na[(tpat-Bu)Fe] (2) | 65 (1500) | 6.7 × 10−11 | 11 |
| Na[(tpaPh)Fe] (3) | 25 (1500) | — | 11 |
| [Fe(C(SiMe3)3)2] (8) | 146 (500) | 4 × 10−9 | 18, 20 |
| Fe[N(SiMe3)(Dipp)]2 (11) | 181 (500) | 1 × 10−11 | 18 |
| Fe[N(H)Ar′]2 (12) | 109 (1800) | 5 × 10−9 | 18 |
| Fe[N(H)Ar*]2 (13) | 104 (875) | 4 × 10−8 | 18 |
| Fe(OAr′)2 (14) | 43 (2500) | 3 × 10−7 | 18 |
| Fe[N(H)Ar#]2 (15) | — | — | 18 |
| [Fe(N(SiMe3)2)2(PCy3)] (16) | 29.2/16 (600) | 6.0 × 10−7/1.6 × 10−6 | 22, 23 |
| [5CpFe(C6H3iPr3-2,6)] (20) | 28.0 (750)/99.7 (2500) | 6.0 × 10−6/7.8 × 10−9 | 24 |
| [Fe(1-ptz)6](BF4)2 (21) | 15 (2000) | 4.2 × 10−8 | 25 |
PhB(MesIm)3Fe-N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3 (22) PPh3 (22) |
15 (1000) | 8.7 × 10−7 | 27 |
| Fe(III) | |||
| [(PNP)FeCl2] (23) | 32 (0) | 2 × 10−8 | 28 |
| Co(II) | |||
| [Li(15-crown-5)][Co(N(SiMe3)2)3] (17) | 16.1 (800) | 3.5 × 10−7 | 22 |
| [Co(N(SiMe3)2)2(THF)] (18) | 18.1 (600) | 9.3 × 10−8 | 22 |
| [Co(N(SiMe3)2)2(PCy3)] (19) | 19.1 (750) | 3.0 × 10−7 | 22 |
| [(ArNCMe)2(NPh)]Co(NCS)2 (24) |
11.1 (2000) | 3.6 × 10−6 | 30 |
| [(ArNCPh)2(NPh)]Co(NCS)2 (25) |
16.7/17.4 (2000) | 5.1 × 10−7/1.6 × 10−6 | 30 |
| [Co(terpy)Cl2] (26) | 19.5 (600)/2.8 (5600) | 1.1 × 10−6/7.4 × 10−2 | 31 |
| [Co(terpy)(NCS)2] (27) | 11.8 (600)/2.1 (5600) | 5.9 × 10−6/0.11 | 31 |
| [Co(P(S)([N(CH3)NCHC3N2H3]3))](NO3)2 (28) |
23 (2000) | 4 × 10−6 | 32 |
| K(Co(N[CH2C(O)NC(CH3)3]3)) (29) | 8.7 (1500) | 8 × 10−6 | 32 |
| cis-[Co(dmphen)2(NCS)2]·0.25EtOH (30) | 16.2 (1000) | 4 × 10−7 | 33 |
| [(L)4CoIII2CoII(H2O)2](NO3)4·6H2O (31) | 5.6 (1000) | 1.0 × 10−5 | 34 |
| [(3G)CoCl](CF3SO3) (32) | 24 (1500) | 1.9 × 10−9 | 35 |
| [dmphCoBr2] (33) | 22.9 (1000) | 3.7 × 10−10 | 37 |
| [Co(L)(OAc)Y(NO3)2] (34) | 15.7 (1000) | 8.9 × 10−7 | 38 |
| [Co(acac)2(H2O)2] (35) | ∼16 (various) | — | 39 |
| [Co(abpt)2(tcm)2] (36) | 59.9 (3000) | 1.4 × 10−9 | 40 |
| [Co(12C4)2](I3)2(12C4) (37) | 17 (500) | 1.5 × 10−6 | 41 |
| [Co(L1)2] (38, L1 = 2-(4,5-diphenyl-1H-imidazol-2-yl)phenol) | 34.1 (400)/61.9 (1000) | 7.5 × 10−8/1.0 × 10−10 | 45 |
| [Co(L3)2] (39, L3 = 2-(4,5-diphenyl-1H-imidazol-2-yl)-6-methoxyphenol) | 29.2 (400)/43.8 (1000) | 1.4 × 10−7/2.6 × 10−9 | 45 |
| Co(hpbdti)2 (40) | 39.4/29.7/10.6 (2000) | 1.3 × 10−8/5.4 × 10−7/1.3 × 10−5 | 46 |
| [Co(PPh3)2Br2] (41) | 27.8 (1000) | 5.9 × 10−11 | 42 |
| [Co(PPh3)2Cl2] (42) | 25.8 (1000) | 1.2 × 10−9 | 44 |
| [Co(DPEphos)Cl2] (43) | 24.3 (1000) | 2.1 × 10−10 | 44 |
| [Co(Xantphos)Cl2] (44) | 20.8 (1000) | 6.0 × 10−9 | 44 |
| (Ph4P)2[Co(OPh)4]·CH3CN (45) | 21.1 (1400) | 7 × 10−10 | 43 |
| K(Ph4P)[Co(OPh)4] (46a) | Required dilution, see 46b | — | 43 |
| K(Ph4P)[Co0.06Zn0.94(OPh)4] (46b) | 34.0 (0) | 1.0 × 10−9 | 43 |
| (Ph4P)2[Co(SPh)4] (47) | 21.1 (0) | 1.0 × 10−6 | 43, 47 |
| (Ph4P)2[Co(SePh)4] (48) | 19.1 (0) | 3 × 10−6 | 43 |
| (Ph4P)2[Co(C3S5)2] (49) | 33.9 (0) | 4.5 × 10−6 | 48 |
| (HNEt3)(CoIICoIII3L6) (50) | 75.8 (0) | 1 × 10−7 | 49 |
| [CoIIICoII(LH2)2(Cl)(H2O)](H2O)4 (51) | 7.9 (1000) | 6.1 × 10−6 | 50 |
| [CoIIICoII(LH2)2(Br)(H2O)](H2O)4 (52) | 14.5 (1000) | 1.0 × 10−6 | 50 |
| Ni(I) | |||
| [Ni(6-Mes)2]Br (53) | 11.8 (600) | 4.6 × 10−6 | 51 |
3.1 Mn(III)
Mn(III) has been widely used in the field of molecular nanomagnets. For the d4 Mn(III) ion, the Jahn–Teller effect leads to a tetragonal distortion away from Oh symmetry towards D4h, most commonly an elongation along the z-axis. This splits the 5E ground state and mixing of the 5B1 state with excited states, through second order spin–orbit coupling, gives rise to the zero-field splitting where D is almost always negative in a tetragonally elongated environment.The most comprehensive study of a monometallic Mn(III) SMM so far was performed by Vallejo and co-workers on the complex Ph4P[Mn(opbaCl2)(py)2] (4, where H4opbaCl2 = N,N′-3,4-dichloro-o-phenylenebis(oxamic acid)).15 This compound contains an axially elongated Mn(III) ion, with a mixed N2O2 donor set in the equatorial positions, provided by the ligand, and two axial N atoms provided by pyridine molecules (Fig. 4). The coordination of the ligand forms three chelate rings that impose a distortion around the metal centre.
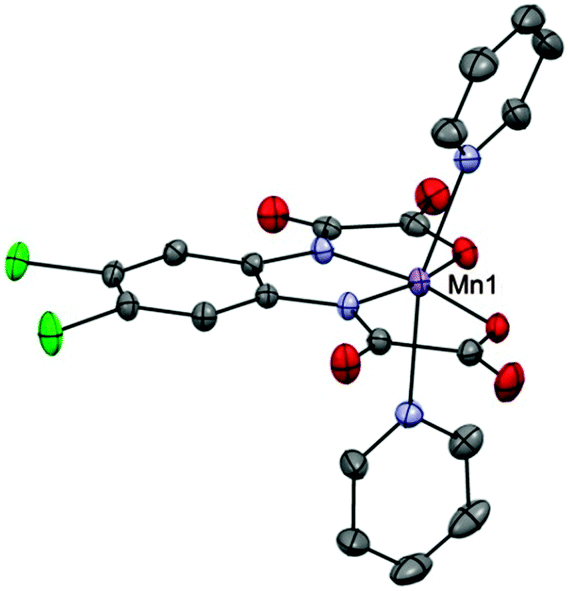 | ||
| Fig. 4 A view of the complex [Mn(opbaCl2)(py)2]−. Mn, lilac; N, blue; O, red; C, grey; Cl, green. | ||
A high-field EPR (HFEPR) powder study was performed yielding a value for the ZFS of D = −3.421(2) cm−1 and a transverse component E of −0.152(2) cm−1. The axial magnetic anisotropy was shown by complete active space (CAS) calculations to be mainly due to second order spin–orbit coupling, with DSOC = −2.97 cm−1 and a smaller spin–spin contribution. Maxima in the out-of-phase ac susceptibility were seen on the application of a dc field of 1000 Oe, and the Arrhenius plot gave Ueff = 12.6 cm−1 (Fig. 5(a)). Measurement of a single crystal using a micro-SQUID (Fig. 5(b)) reveals closed hysteresis loops at zero field, due to the fast QTM, consistent with the absence of χ′′ signals in zero dc field.
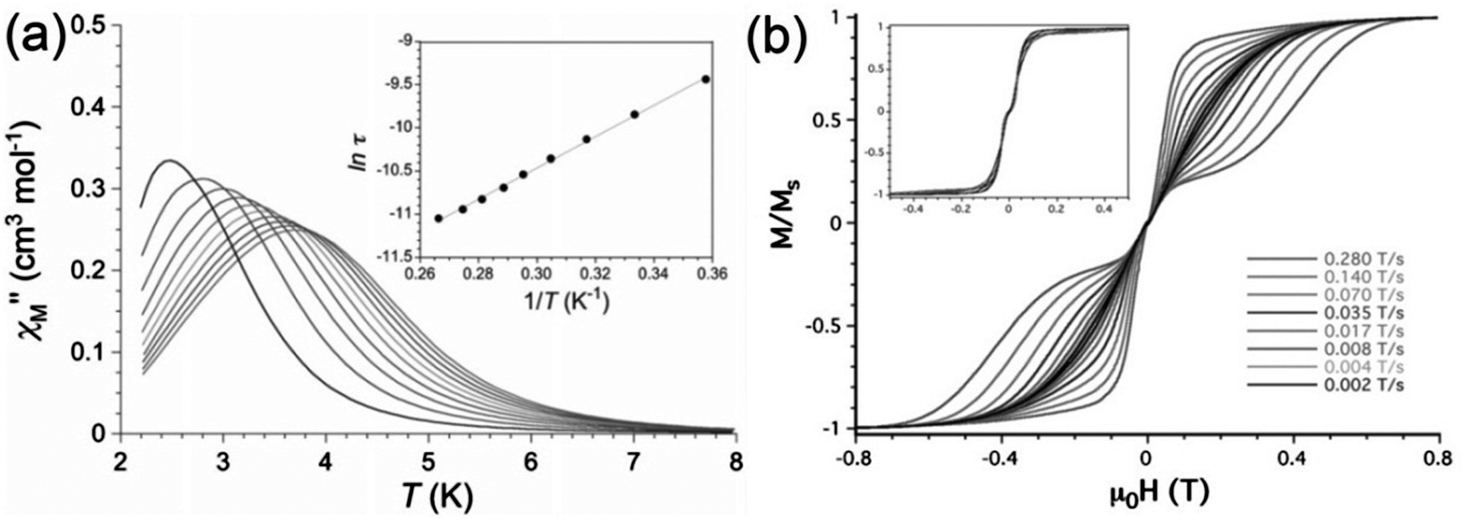 | ||
| Fig. 5 (a) Temperature dependence of χ′′ under a dc field of 1000 Oe, and (inset) the derived Arrhenius plot for compound Ph4P[Mn(opbaCl2)(py)2]. (b) Sweep rate dependence of the normalised magnetisation, as measured at 0.5 K and (inset) at 0.03 K. Reprinted with permission from ref. 15. Copyright (2013) Wiley-VCH. | ||
The barrier to relaxation observed for 4 is of the same order of magnitude as that found for the other examples of Mn(III)-based SIMs in the literature. The dimetallic complex [Mn(5-TMAM(R)-salmen)(H2O)Co(CN)6]·7H2O·MeCN 5, 5-TMAM(R)-salmen = (R)-N,N′-(1-methylethylene)bis(5-trimethylammoniomethylsalicylideneiminate), which contains a diamagnetic Co(III) ion, was shown to have an axial ZFS of D = −3.3 cm−1, by fitting the isofield magnetisation curves.16 Despite displaying a small frequency dependence for χ′ and χ′′ in zero applied dc field, maxima in the out-of-phase component were not observed, even on application of a dc field of 4500 Oe. Fits of the dynamic susceptibility data yielded an effective barrier Ueff = 9.3 or 11.5 cm−1 (for zero field and 4500 Oe, respectively). A slightly lower barrier (Ueff = 8 cm−1, under a field of 2250 Oe) was determined for the monometallic complex [Mn(OPPh2)2N3] (6).17
3.2 Fe(I)
The rather low values of D found for the six-coordinate Mn(III) containing compounds are due to the large energy gap between the ground state and excited states. The magnitude of D is inversely proportional to the gap between ground and excited states, and also depends upon which d orbitals are involved (the interested reader can find this point illustrated in Fig. 2 of ref. 32). The relatively strong crystal field arising from coordinative saturation can be reduced by lowering the coordination number. This allows better mixing with excited states and often an almost unquenched orbital angular momentum than can produce large increases in the magnetic anisotropy. The two-coordinate linear Fe(I) complex [K(crypt-222)][Fe(C(SiMe3)3)2] (7), can be synthesised by single electron reduction of the neutral Fe(II)-based SIM18 [Fe(C(SiMe3)3)2] (8) with KC8 (Fig. 6).19 The Fe(I) oxidation state was unequivocally established using Mössbauer spectroscopy. Fe(I) has a half-integer spin of S = 3/2. According to Kramers′ theorem, QTM should be minimised in such half-integer systems, which should show slow relaxation of the magnetisation, even in the absence of an applied dc field. Ab initio calculations confirmed the success of the strategy in reducing the ligand field around the metal centre, as well as indicating large energy splittings of the MJ sublevels, which should lead to a significant energy barrier.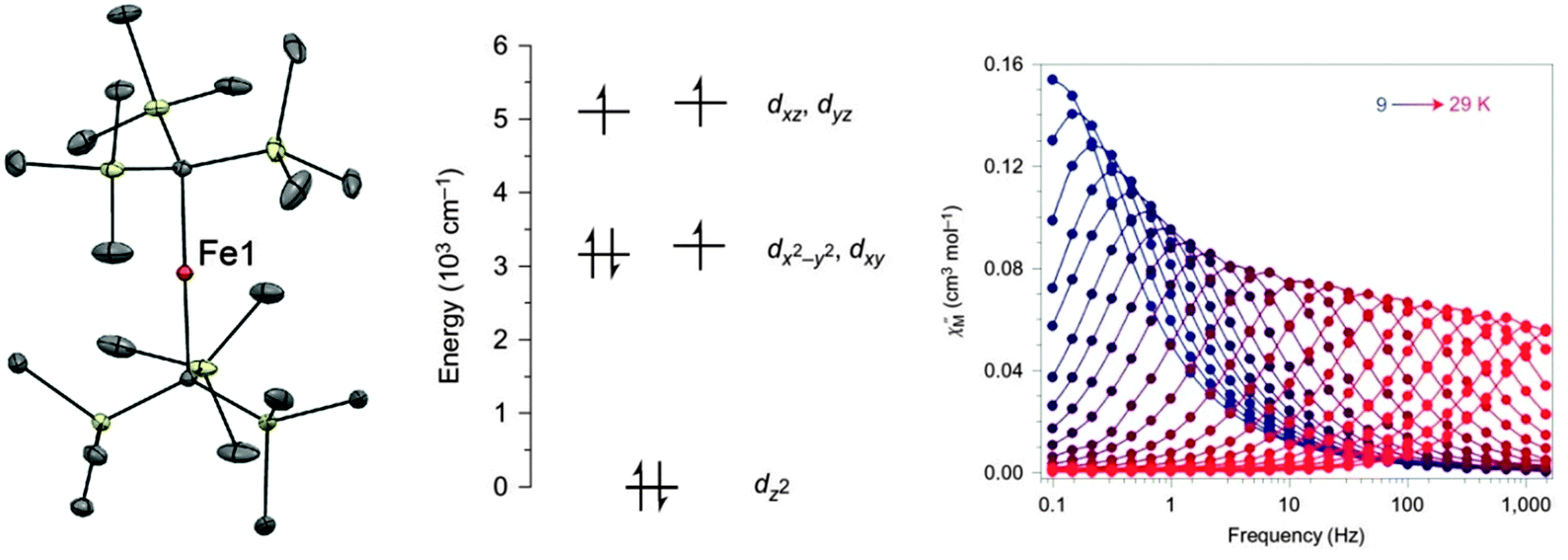 | ||
| Fig. 6 (left) A view of the anion [Fe(C(SiMe3)3)2]−; Fe, orange; C, grey; Si, pale yellow. (middle) A view of the energies of the 3d orbitals in 7. (right) Dynamic magnetic susceptibility data for 7, measured under zero applied field. Reprinted with permission from ref. 19. Copyright (2013) Nature Chemistry, Macmillan Publishers Ltd. | ||
Frequency-dependence of the out-of-phase susceptibility was observed without the need for an applied field (Fig. 6). In fact, the barrier to relaxation in 7 is Ueff = 226 cm−1, which is the highest value recorded to date for monometallic 3d SMMs. This barrier was derived from the linear region of the Arrhenius plot between 29 and 20 K. Below 20 K, the Arrhenius plot curves, as the relaxation deviates from an Orbach mechanism. Intermolecular dipolar interactions may aid QTM in these types of complexes, so dynamic susceptibility measurements were performed on a frozen solution to reduce the interactions. Dilution had previously been shown as an effective means of mitigating tunneling in the case of the compound (Ph4P)2[Co(SPh)4] (47, vide infra). The resulting plot deviated to a lesser extent from Arrhenius behaviour, but still displayed curvature, which could arise from lower than axial symmetry around the Fe(I) ion, permitting mixing of the MJ states. In a subsequent work Mössbauer spectroscopy was used to extend the high temperature range over which the relaxation could be measured.20 Interestingly, above 50 K on the timescale of the experiment, 7 appears to follow an Arrhenius law associated with a larger effective barrier (420 cm−1) than in the range 9 to 50 K. This barrier is close to the energy difference between the MJ = ±7/2 and MJ = ±3/2 levels, derived from theoretical calculations.
However, the strategy of using Fe(I) in linear environments is not a guarantee of large barriers to relaxation. In the compound [(cAAC)2Fe][B(C6F5)4] (9, where cAAC = a cyclic alkyl(amino) carbene), the barrier was found to be Ueff < 20 cm−1 under a field of 3000 Oe.21 Compared to 7, compound 9 has a different electronic structure, with additional π-bonding interactions. Theoretical studies suggest that the carbene πz orbitals reduce the axial nature of the ground state doublet. Compound 9 can be synthesised from the trigonal Fe(I) complex (cAAC)2FeCl (10), which has an easy-plane anisotropy (vide infra) and a barrier of 22.4 cm−1 in an applied field of 500 Oe.
3.3 Other examples containing Fe(II)
As mentioned above, compound 7 is derived from the neutral Fe(II) SIM complex [Fe(C(SiMe3)3)2] (8). This system was described as part of a family of two-coordinate Fe(II) complexes which present either a strictly linear geometry, as in the cases of 8, Fe[N(SiMe3)(Dipp)]2 (11, Dipp = C6H3-2,6-Pri2), Fe[N(H)Ar′]2 (12, Ar′ = C6H3-2,6-(C6H3-2,6-Pri2)2), Fe[N(H)Ar*]2 (13, Ar* = C6H3-2,6-(C6H2-2,4,6-Pri3)2), and Fe(OAr′)2 (14), or a bent geometry, as found for Fe[N(H)Ar#]2 (15, L–Fe–L = 140.9°, Ar# = C6H3-2,6-(C6H2-2,4,6-Me3)2).18 With the exception of compound 15, for which only the onset of slow magnetic relaxation was observed under an applied dc field, maxima in the out-of-phase component under different field strengths allowed Arrhenius plots to be derived for compounds 8 and 11–14. These plots were found to be significantly curved, which is attributed to the applied field inhibiting QTM while at the same time promoting direct relaxation processes.This methodology of unsaturated coordination environments is the most common method of achieving high anisotropy for Fe(II). To this end, three trigonal planar Fe(II) compounds were prepared as part of a comparative study with their Co(II) analogues. However only one of the ferrous compounds, [Fe(N(SiMe3)2)2(PCy3)] (16, where Cy = cyclohexyl) displayed slow relaxation of the magnetisation, while all three Co(II) compounds did (17–19, Table 1) and all four compounds were studied under applied dc fields.22 Complex 16 had been previously reported,23 and in both cases its dynamic magnetic properties were studied. Somewhat unusually, the barriers reported for the same compound differ in the two papers, as recognised by the authors of the later work. In the 2011 paper, the barrier was calculated as 29.2 cm−1, while the more recent paper found a value of 16.0 cm−1, both determined in an applied dc field of 600 Oe. The reason is unclear, but this does highlight the importance of reporting exactly how the barrier is determined.
Two different barriers were found for the same compound [5CpFe(C6H3iPr3-2,6)] (20) under different fields.24 Compound 20 can be prepared from the reaction of the diamagnetic complex [5CpFeBr(dme)] (5Cp = pentaisopropylcyclopentadienide, dme = 1,2-dimethoxyethane) with 2,6-diisopropylphenylmagnesium bromide in THF. Fits of the reduced magnetisation data for 20 give D = −51.4 cm−1 and E = −0.3 cm−1. Under an applied field, the ac susceptibility measurements revealed two types of relaxation process. The first, observed under a field of 750 Oe, process I was assigned to direct relaxation between the MS = ±2 states of the S = 2 ground state. The second, slower process II (under a field of 2500 Oe) was ascribed to a phonon-induced excitation to the MS = ±1 levels prior to relaxation. Accordingly, process I had a lower barrier (28.0 cm−1) than process II (99.6 cm−1).
In some of the cases already described, the importance of the symmetry around the central metal ion is apparent and this is also crucial for the compound [Fe(1-ptz)6](BF4)2 (21), where 1-ptz = 1-propyltetrazole (Fig. 7).25 Complex 21 is a classic spin crossover (SCO) compound which was the first system to be reversibly photo-switched from the diamagnetic low spin (LS) to the paramagnetic high spin (HS) form in the solid state.26 Extensive crystallographic studies have shown that the symmetry of 21 is intimately related to how the compound is thermally treated. Slow cooling of the compound causes it to undergo a phase transition with a loss of symmetry, while flash cooling causes 21 to retain its high temperature space group (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ) with the now LS Fe(II) lying in a D3d local symmetry. Irradiation of this LS high symmetry form of 21 gives access to a HS high symmetry phase, where the local symmetry of the Fe(II) ions gives rise to an axial magnetic anisotropy: HFEPR data gives D = −14.8 cm−1, together with a transverse component of E = −0.95 cm−1. The barrier to relaxation of the magnetisation under an applied dc field of 2000 Oe, is 15 cm−1. The ability to turn the SIM behaviour on and off by photo-switching between the HS and LS states, together with the possible states within the HS form render 21 a tristable system (|0〉, |−2〉, |+2〉) (Fig. 7).
) with the now LS Fe(II) lying in a D3d local symmetry. Irradiation of this LS high symmetry form of 21 gives access to a HS high symmetry phase, where the local symmetry of the Fe(II) ions gives rise to an axial magnetic anisotropy: HFEPR data gives D = −14.8 cm−1, together with a transverse component of E = −0.95 cm−1. The barrier to relaxation of the magnetisation under an applied dc field of 2000 Oe, is 15 cm−1. The ability to turn the SIM behaviour on and off by photo-switching between the HS and LS states, together with the possible states within the HS form render 21 a tristable system (|0〉, |−2〉, |+2〉) (Fig. 7).
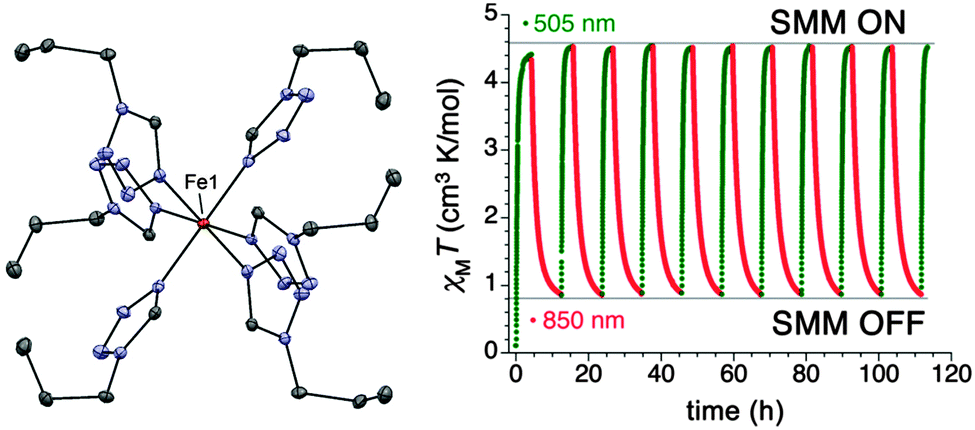 | ||
| Fig. 7 (left) A view of the cation [Fe(1-ptz)6]2+, Fe, orange; N, blue; C, grey. (right) Photo-excitation cycles of the high symmetry form of 21, represented as the variation in χT with time. Reprinted with permission from ref. 25. Copyright (2013) American Chemical Society. | ||
Another example of an SCO compound that shows slow relaxation of the magnetisation is PhB(MesIm)3Fe-NPPh3 (22) (MesIm = mesitylimidazole).27 Unlike 21, here the Fe(II) centre is four-coordinate and displays a pseudo three-fold symmetry around the metal ion. Photo-excitation of the LS state at low temperatures allows the meta-stable HS state to be generated. Under an applied dc field of 1000 Oe, a frequency dependence in χ′′ could be observed, and the fit of the Arrhenius plot yielded a barrier to relaxation of 15 cm−1. While the relaxation barriers for compounds 21 and 22 are significantly lower than those found for Co(II) complexes (vide infra), both systems are fascinating for illustrating the overlap between molecular nanomagnets and SCO compounds.
3.4 Fe(III)
Partial SCO was observed in the only existing SIM containing Fe(III), [(PNP)FeCl2] (23, PNP = N[2-P(CHMe2)2-4-methylphenyl]2−), where the Fe(III) is in a distorted 5-coordinate environment.28 At temperatures above 80 K, 23 is found in the HS S = 5/2 state, as evidenced by the dc magnetic measurements, and also by an extensive variable temperature crystallographic study. Below 80 K, 23 is found in an intermediate S = 3/2 spin state. Ac susceptibility measurements reveal that 23 is the first (and to date, only) example of a monometallic Fe(III) complex to display slow relaxation of the magnetisation, even in the absence of dc field, with a barrier of 36 cm−1.3.5 Co(II)
The promise of Co(II) for use in polymetallic SMMs has been previously highlighted, with a particular emphasis placed on the effects of the molecular shape, size and symmetry.29 One of the first examples of a monometallic Co(II) SIM takes advantage of the first order spin–orbit coupling displayed by the d7 ion when it lies just above the basal plane of a square-based pyramid. In the compounds [(ArNCMe)2(NPh)]Co(NCS)2 (24, (ArNCMe)2(NPh) = 2,6-bis(1-[(2,6-diisopropylphenyl)imino]ethyl)pyridine) and [(ArNCPh)2(NPh)]Co(NCS)2 (25, (ArNCPh)2(NPh) = 2,6-bis(1-[(2,6-diisopropylphenyl)imino]benzyl)pyridine), the neutral bis(imino)pyridine ligands coordinate to the metal centre in three of its equatorial positions and the two monodentate thiocyanate ligands fill an axial and equatorial position.30 Due to the steric constraints of the pincer ligands, the transition metal sits above the basal plane of the pyramid. In the regular geometry, in which the metal ion lies in the basal plane of the pyramid, the degenerate dxz and dyz orbitals are fully occupied, while in the distorted environment they are unequally occupied, giving rise to an appreciable magnetic anisotropy (Fig. 8).
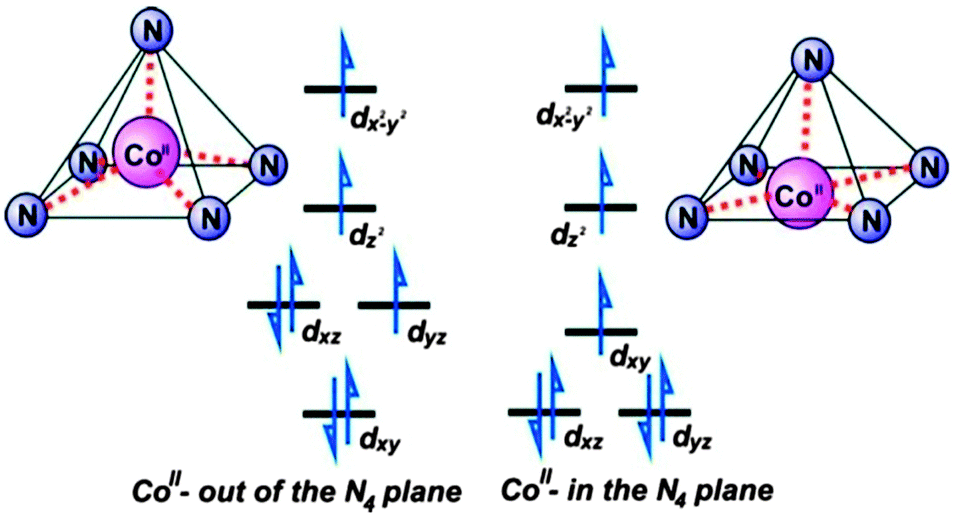 | ||
| Fig. 8 A simplified scheme illustrating the relative energies of the d-orbitals in a distorted square-based pyramid (left) and ideal square-based pyramid (right). Reprinted with permission from ref. 30. Copyright (2011) American Chemical Society. | ||
The static magnetic properties reveal intermolecular ferromagnetic interactions between molecules of 25 at low temperature. To disrupt these contacts, 25 was dissolved in THF and the magnetic measurements repeated. As expected, χMT was seen to decrease at low temperature, as was the case for compound 24. The barrier to relaxation recorded for the solid state and frozen solution dynamic magnetic experiments performed on 25 was similar (16.7 and 17.4 cm−1, respectively, under a dc field of 2000 Oe), while 24 displayed a smaller barrier of 11.1 cm−1 (also at 2000 Oe). In the compound [Co(terpy)Cl2] (26), the basal plane of the pyramid is formed by one Cl− ion and the three donor N-atoms of the terpyridine ligand, with the metal ion also sitting just above this plane.31 The coordination sphere is therefore different compared to those of 24 and 25, leading to a different d-orbital splitting. In [Co(terpy)(NCS)2] (27), the thiocyanate ligands both point out of the plane defined by the coordination of terpy to the metal ion. Through ac susceptibility measurements under two different fields, two different relaxation processes – fast and slow – could be observed for compounds 26 and 27, with barriers (fast/slow) of 19.5/2.8 cm−1 and 11.8/2.1 cm−1, respectively.
A wider theoretical study of how variations in coordination geometry can lead to magnetic anisotropy in first row transition metals has been carried out.32 Using CASSCF calculations, it can be shown how D may be expected to vary with the number of d-electrons and the symmetry around the metal ion. Using this predictive strategy, two systems from the literature were identified that ought to show high magnetic anisotropy, [Co(P(S)([N(CH3)NCHC3N2H3]3))](NO3)2 (28) and K(Co(N[CH2C(O)NC(CH3)3]3)) (29). Compound 28 (Fig. 9) was predicted to have a large negative value of D, which was found to be −72 cm−1 based on the temperature and field-dependence of the magnetisation. Under a field of 2000 Oe, one clearly resolved temperature dependent maximum in χ′′ could be observed, with a barrier of Ueff = 23 cm−1. For 29, the barrier at 1500 Oe was observed to be 8.7 cm−1, and the anisotropy derived from the magnetisation measurements was consistent with theoretical calculations, with D = +16 cm−1.
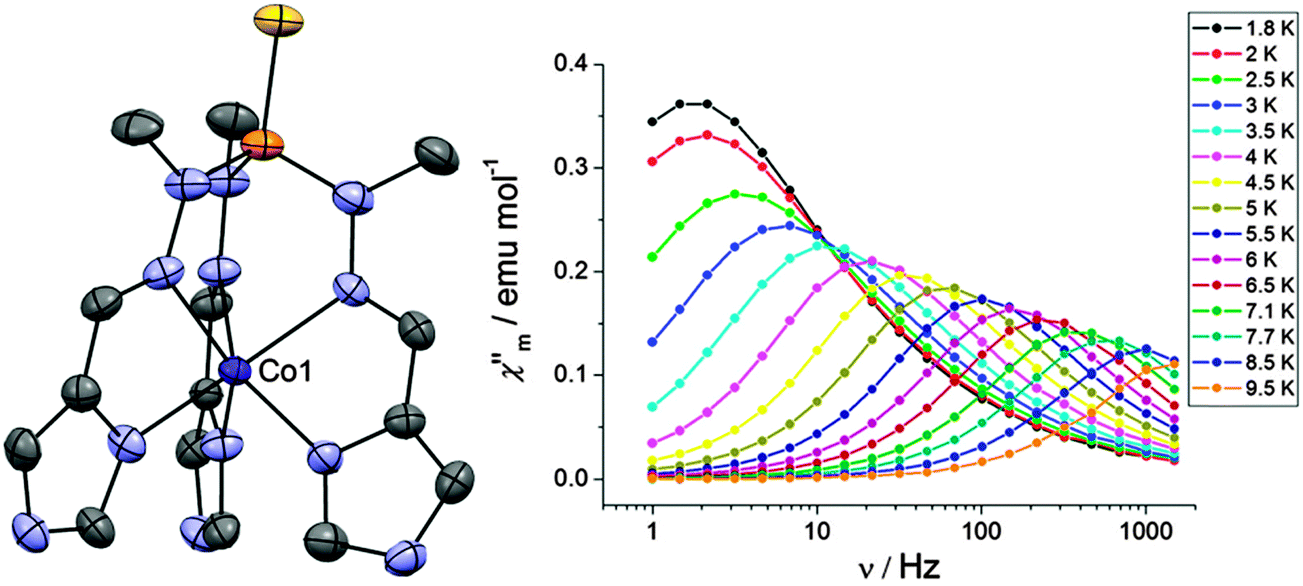 | ||
| Fig. 9 (left) A view of the cation [Co(P(S)([N(CH3)NCHC3N2H3]3))]2+; Co, purple; N, blue; P, orange; S, yellow; C, grey. (right) Frequency dependence of χ′′ under a dc field of 2000 Oe. Reprinted with permission from ref. 32. Copyright (2013) American Chemical Society. | ||
Positive D values leading to SMM-like behaviour in monometallic Co(II) complexes is not uncommon, despite the perception that this might prevent slow relaxation processes.33 The first example of a hexa-coordinate Co(II) SIM, the compound cis-[Co(dmphen)2(NCS)2]·0.25EtOH (30, dmphen = 2,9-dimethyl-1,10-phenanthroline) displays slow relaxation of the magnetisation with Ueff = 17.0 cm−1 under a dc field of 1000 Oe, with D = +98 cm−1. The slow relaxation is a result of a transverse anisotropy (xy plane). Under these conditions, the magnetisation would have a preferred orientation in the xy plane, either along the x- or the y-axis, rather than along the z-axis. The barrier to reorientation in the opposite direction along the same axis would then be governed by the parameter E. Through the relation Ueff ∼ 2E, a theoretical barrier of 16.8 cm−1 was determined, based on the value of E = +8.4 cm−1 obtained from low temperature magnetisation data. This was also the explanation put forward for the compound [(L)4CoIII2CoII(H2O)2](NO3)4·6H2O (31) (where L is a carbohydrazide derivative).34
An alternative explanation of how positive axial anisotropy can result in slow relaxation of the magnetisation has been proposed for the pseudotetrahedral Co(II) complex [(3G)CoCl](CF3SO3) (32, where 3G = 1,1,1-tris-[2N-(1,1,3,3-tetramethylguanidino)methyl]ethane).35 For 32, slow magnetic relaxation was observed with a barrier of 24 cm−1 under a dc field of 1500 Oe, and EPR determined D = +12.7 cm−1 and E = 1.2 cm−1. Here it is proposed that relaxation from the MS = +1/2 to the MS = −1/2 level is slowed by a phonon bottleneck,36 such that relaxation involves excitation to the higher lying MS = ±3/2 levels. From the EPR and magnetisation data, an energy gap of the order of 24 cm−1 was found to separate the MS = ±1/2 levels from the higher lying MS = ±3/2 levels, consistent with the barrier determined from the dynamic susceptibility measurements. A similar situation was found for the compound [dmphCoBr2] (33, dmph = 2,9-dimethyl-1,10-phenanthroline).37 The easy-plane anisotropy in this compound, with D = +11.68 cm−1 and E = −2.60 cm−1, yielded Ueff = 22.9 cm−1 (under a field of 1000 Oe).
A phonon bottleneck has been ruled out for the compound [Co(L)(OAc)Y(NO3)2] (34, where LH2 = N,N′,N′′-trimethyl-N,N′′-bis(2-hydroxy-3-methoxy-5-methylbenzyl)-diethylenetriamine) (Fig. 10).38 Here, the magnetic anisotropy is of an easy-plane nature, with D = +47 cm−1 and E = 1.6 cm−1. Inelastic neutron scattering (INS) revealed an excitation with an associated energy of 95.2 cm−1, corresponding to the energy gap (= 2D) between the MS = ±3/2 and MS = ±1/2 levels. Dilution experiments with the purely diamagnetic species [Zn(L)(OAc)Y(NO3)2] did not cause any increase in the relaxation, suggesting that a phonon bottleneck is not at the root of the observed barrier. The determined barrier of Ueff = 15.7 cm−1, was far lower than the energy gap between the MS = ±3/2 and MS = ±1/2 doublets. In fact, fits of the Arrhenius plot to a T−n law indicate that an optical acoustic Raman process is a viable proposal for the relaxation mechanism, mixed with direct processes.
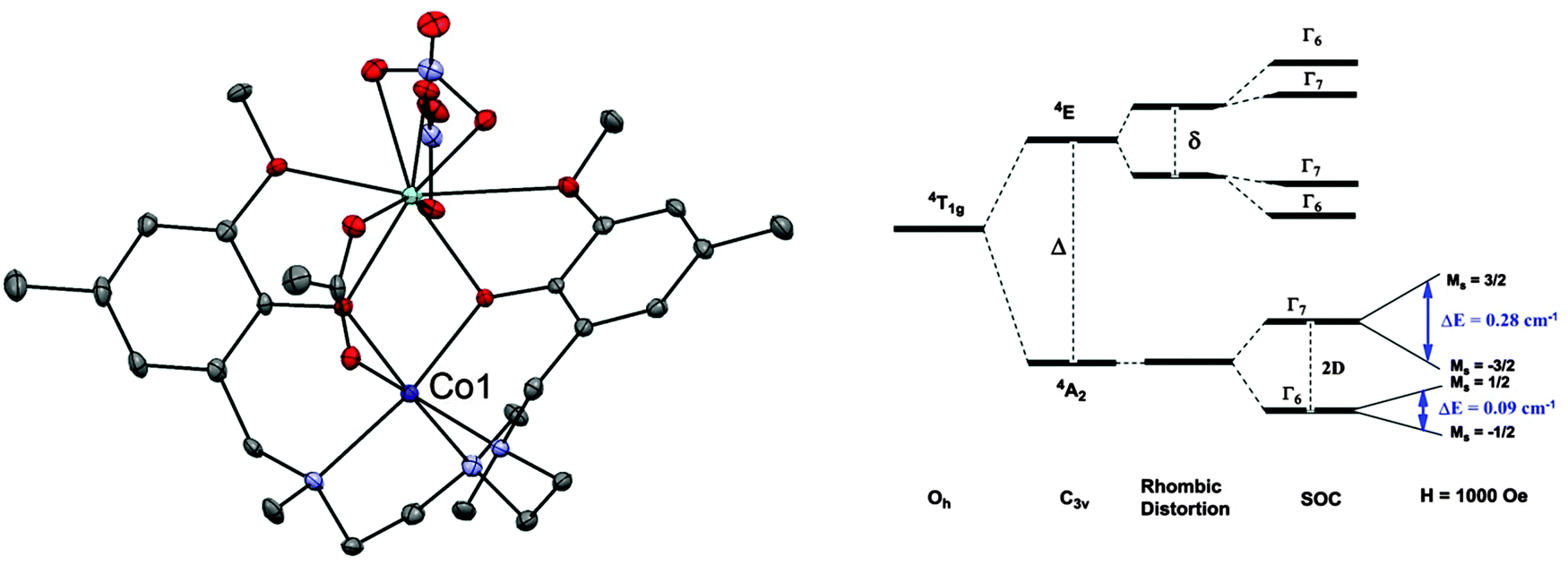 | ||
| Fig. 10 (left) A view of [Co(L)(OAc)Y(NO3)2] (34); Co, purple; Y, pale blue; C, grey; O, red; N, blue. (right) A simplified representation of the energy levels in 34. Reprinted with permission from ref. 38. Copyright (2013) Wiley-VCH. | ||
The most concerted effort to clarify how and why slow magnetic relaxation occurs in complexes with an easy plane anisotropy has been carried out on the hexa-coordinate Co(II) complex [Co(acac)2(H2O)2] (35, acac = acetylacetonate).39 The dynamic ac susceptibility can be accounted for with a linear fit to an Arrhenius plot, suggesting a thermally-activated Orbach process at higher temperatures. At lower temperatures, Raman and direct processes are responsible for the relaxation. Because Orbach processes proceed through phonon-induced transitions to higher energy states, their associated energy barrier should reflect this and be of the order of 2D, which in this case would imply a barrier of around 130 cm−1. However, the barrier to thermal relaxation was found to be ∼16 cm−1.
Formally, direct phonon-induced transitions between the two states that describe the low temperature electronic states of Co(II), shown in Fig. 11 as |Φ〉 and |Ψ〉, are forbidden. In theory, this leaves two-phonon Orbach and Raman relaxation processes available, with the proviso that ZFS is often far larger than the available thermal energies at low temperatures, hindering Orbach-driven relaxation. However, this clear-cut two level description is not entirely accurate (Fig. 11(a)). Hyperfine interactions with the nuclear spin of the Co(II) ion (I = 7/2) broaden these two levels into a manifold in which some phonon-induced transitions are permitted (Fig. 11(b)). The application of an external magnetic field exacerbates this splitting, and confers upon each state a measureable magnetic moment (Fig. 11(c)). Finally, an interaction between the nuclear spin and molecular vibrations in the lattice allow phonon-induced transitions between different nuclear spin states (Fig. 11(d)). The combination of these perturbations allows slow relaxation in Co(II) ions with easy plane anisotropy to be observed at low magnetic fields. Hence, it is proposed that hyperfine interactions should be minimised to limit the relaxation pathways available to a SIM. In fact, the low coordinate Fe(I) compound 7, which has the highest barrier found for a transition metal SIM fits with this strategy. The highest barrier for a Co(II) SIM with easy plane anisotropy was reported for the octahedral Co(II) complex [Co(abpt)2(tcm)2] (36), where abpt = 4-amino-3,5-bis(2-pyridyl)-1,2,4-triazole and tcm = tricyanomethanide, which displays a barrier of Ueff = 59.9 cm−1 for an applied field of 3000 Oe, and D = +55 cm−1 and E/D = 0.27.40
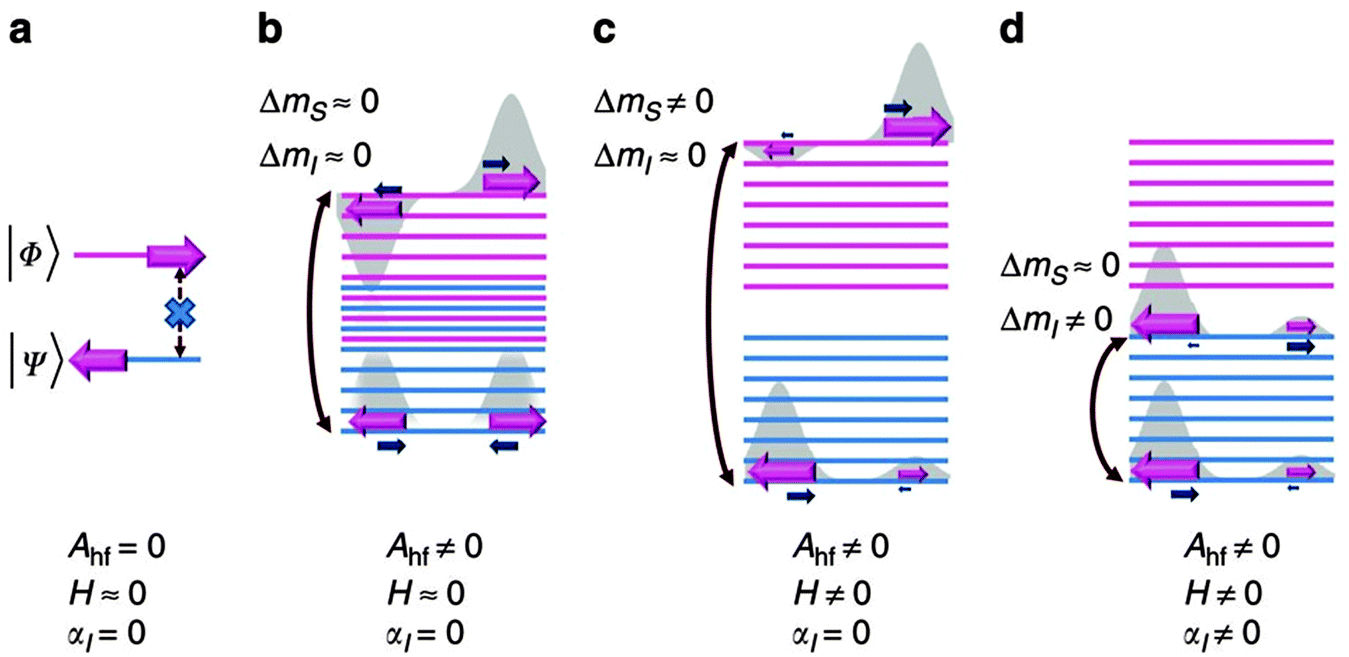 | ||
| Fig. 11 (top) A schematic view of the energy levels in the Kramers ion Co(II) with positive axial anisotropy. Ahf = hyperfine coupling; H = an applied magnetic field; αl = lattice-phonon interactions. See text for details. Reprinted with permission from ref. 39. Copyright (2014) Nature Communications, Macmillan Publishers Ltd. | ||
However, the majority of Co(II)-based SIMs display negative D values. In the cobalt(II)-12-crown-4 (12C4) complex [Co(12C4)2](I3)2(12C4) (37) (Fig. 12) the Co(II) ion is coordinated by four oxygen atoms from the two crown molecules, leading to a distorted square anti-prismatic geometry.41 Density functional theory (DFT) calculations show that the partially filled dxz and dyz orbitals are almost degenerate. This gives rise to a low-lying excited state and coupling with the ground state can give rise to a large axial anisotropy.
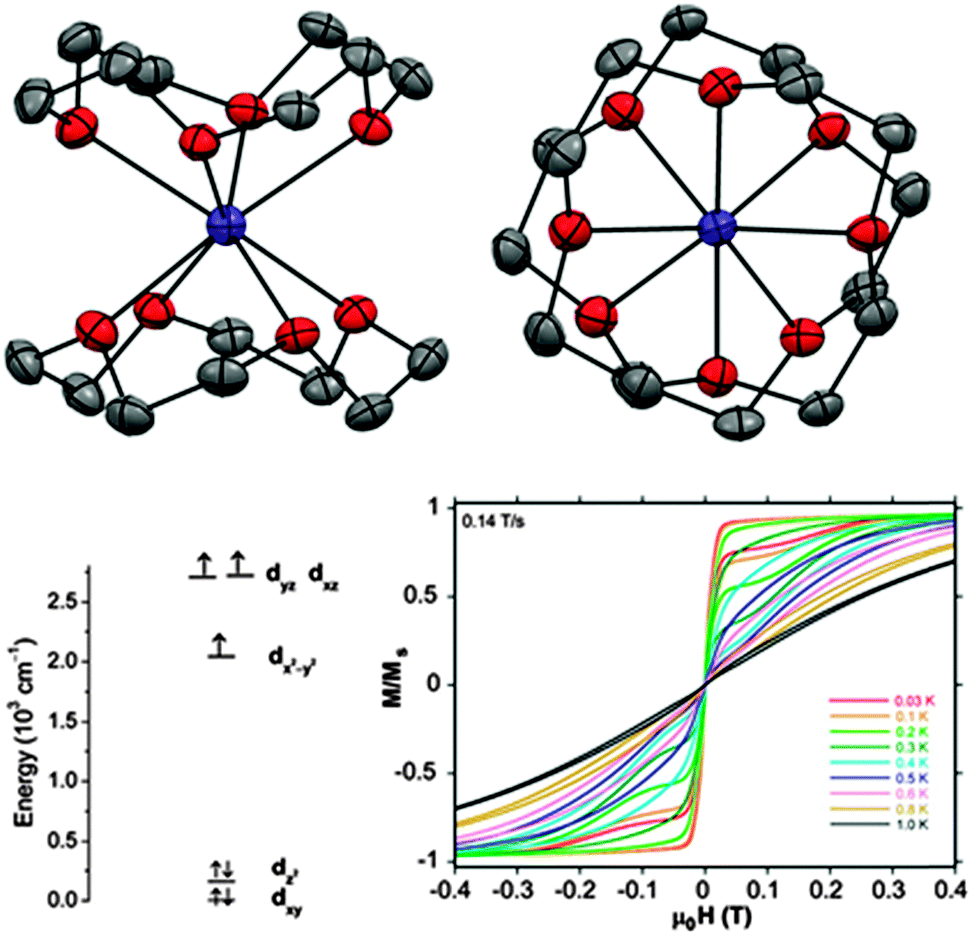 | ||
| Fig. 12 (top) A view of the cation [Co(12C4)2]2+; Co, purple; O, red; C, grey. (bottom, left) d-orbital energy diagram derived from DFT calculations. (bottom, right) Field dependence of the normalised magnetisation of 37. Reprinted with permission from ref. 41. Copyright (2014) American Chemical Society. | ||
Through fits of the dc susceptibility measurements, values of D = −37.6 cm−1 and E = 0.1 cm−1 were obtained. HFEPR studies showed the compound to be nearly “silent”. This was attributed to a combination of only the ±3/2 doublet being populated at low temperatures together with a large, negative D causing a large energy gap to the ±1/2 doublet. Simulations put a lower limit on this axial anisotropy of |D| > 20 cm−1 and further theoretical calculations also confirm the large, negative D value. Suppression of QTM using an applied dc field of 500 Oe allows the observation of an out-of-phase signal in the ac susceptibility measurements, with an associated barrier to relaxation of 17.0 cm−1.
Of the variety of coordination environments that have been employed to try to induce slow magnetic relaxation in Co(II) complexes, the most successful is tetrahedral or pseudo-tetrahedral, as in the case of 32, and 38–48 (see Table 1).42–47 The value of D can be increased by increasing the softness of the donor atom in a family of compounds based on the complex anion [Co(EPh)4]2− (E = O, S, Se): (Ph4P)2[Co(OPh)4]·CH3CN (45), K(Ph4P)[Co(OPh)4] (46), (Ph4P)2[Co(SPh)4] (47), (Ph4P)2[Co(SePh)4] (48). However, it does not necessarily follow that the relaxation barrier increases in line with increasing D, and the reported barriers all lie in the range 19–34 cm−1.43,47 Similarly, the tetragonally-elongated pseudotetrahedral Co(II) compound (Ph4P)2[Co(C3S5)2] (49) (C3S52− = 4,5-dimercapto-1,3-dithiole-2-thione) displays a large magnetic anisotropy, leading to a barrier of Ueff = 33.9 cm−1 in the absence of an applied dc field.48
Although compounds 46–49 display slow relaxation of the magnetisation without an applied dc field, this phenomenon is still rather rare for 3d SIMs. (HNEt3)(CoIICoIII3L6) (50, where H2L = R-4-bromo-2-((2-hydroxy-1-phenylethylimino)methyl)phenol) displays zero-field slow magnetic relaxation.49 The coordination environment around the Co(II) ion is twisted away from octahedral towards D3 symmetry. The effect of the coordination geometry on the energies of the d-orbitals is shown in Fig. 13, consistent with the presence of significant unquenched orbital angular momentum. Fits of the magnetisation data give D = −115 cm−1 and E = 2.8 cm−1. The strong axial anisotropy leads to slow relaxation of the magnetisation, with a barrier of Ueff = 75.8 cm−1. It is suggested that having the central Co(II) ion surrounded by three diamagnetic Co(III) ions helps to isolate the d7 centre, enhancing the SIM behaviour. A similar point was made for [CoIIICoII(LH2)2(X)(H2O)](H2O)4 (51, X = Cl; 52, X = Br, LH4 = 2-[((2-hydroxy-3-methoxyphenyl)methylene)amino]-2-(hydroxymethyl)-1,3-propanediol) where the presence of the diamagnetic Co(III) ion aids the observation of SIM behaviour, by reducing Co(II)⋯Co(II) intermolecular interactions.50
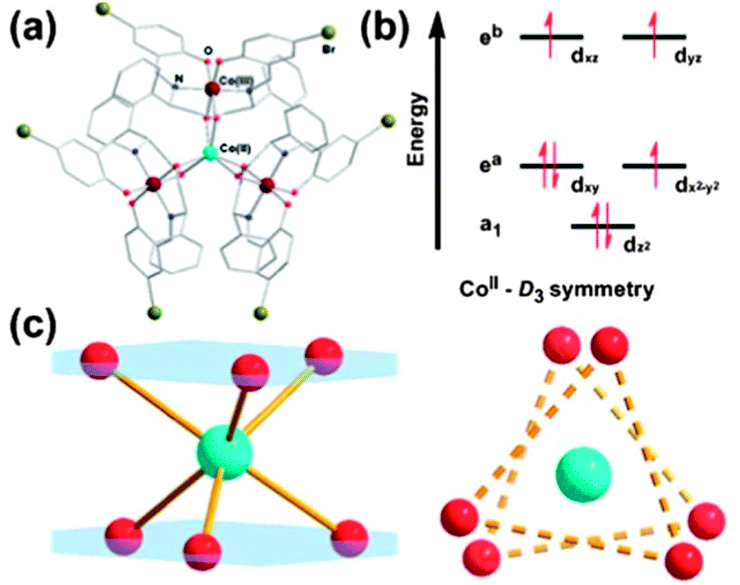 | ||
| Fig. 13 (a) A view of the cation in 50. (b) Approximate energy splittings of the d orbitals in D3 symmetry. (c) A view of the coordination sphere and geometry around the Co(II) ion. Reprinted with permission from ref. 49. Copyright (2013) Royal Society of Chemistry. | ||
3.6 Ni(I)
The only example of SIM behaviour described to date involving Ni(I) is for the linear, two-coordinate geometry, similar to that of 7. In [Ni(6-Mes)2]Br (53) (6-Mes = 1,3-bis(2,4,6-trimethylphenyl)-3,4,5,6-tetrahydropyrimidin-2-ylidene), the mesityl groups of the bulky aromatic ligand are sufficient to block the equatorial sites on the Ni(I) ion, leading to the low coordination number.51 The geometry is found to be nearly linear, with a C–Ni–C angle of 179.27(13)°. The dc magnetic measurements show that the room temperature value of χT (1.12 cm3 mol−1 K) is well above that expected for an S = 1/2 ion (0.375 cm3 mol−1 K), indicative of the presence of unquenched orbital momentum. An applied field is required to observe slow magnetic relaxation, the barrier for which was determined as 11.8 cm−1, based on the linear region of the Arrhenius plot.3.7 Ni(II)
Despite promising studies that have shown very large magnetic anisotropy in several Ni(II) complexes, as of yet there has been no experimental demonstration of slow relaxation of the magnetisation in monometallic Ni(II) compounds. For example, in the trigonal bipyramidal compound [Ni(Me6tren)Cl](ClO4) (54), D is estimated to be between −120 and −180 cm−1 and E = 1.6 cm−1 from HFEPR measurements.524. Conclusions and outlook
In the relatively short time in which they have been studied, 3d SIMs or monometallic SMMs have already yielded some fascinating results and possibilities. Amongst these, the linear Fe(I) compound 7 stands out, both for the huge barrier to relaxation of the magnetisation observed and also for the synthetic challenge such a compound represents. The ability to switch on slow relaxation of the magnetisation in the SCO compounds 21 and 22 also raises the question of how many “dormant” SIMs could be found in photo-switchable SCO complexes.Attempts to understand the slow magnetic relaxation in Kramers ions with positive axial anisotropy have outlined the importance of physical considerations beyond the symmetry around a 3d ion, such as the importance of hyperfine interactions. Control of the relaxation pathways available to a single-ion magnet should allow for improvement in performance as a potential data carrier. The physics of these compounds and how this relates to other possible applications for nanomagnets, such as quantum computing, still has wide scope for exploration. This point was addressed for compound 49, which was shown to have a large axial anisotropy that enabled slow relaxation of the magnetisation to be observed. On the other hand, this anisotropy was so large that 49 was found to be EPR-silent, rendering it ineffective for use in quantum computing, for which EPR would need to be used to observe and address spin transitions.
The chemical synthesis of these compounds presents a huge opportunity. If factors such as nuclear spin, symmetry around the metal ion, and modification of ligands are taken into account, then there are clearly a large number of experimental parameters to be tuned and explored. The d8 ion Ni(II), for example, remains an extremely promising candidate, given the huge magnetic anisotropy already displayed in several compounds.
Acknowledgements
The UK Engineering and Physical Sciences Research Council are thanked for financial support (grant ref. EP/K033662/1).References
-
D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, OUP, 2006 Search PubMed
.
- All of these advances are touched upon in: Molecular Nanomagnets and Related Phenomena, ed. S. Gao, Springer-Verlag, Berlin, 2014 Search PubMed
- H. Miyasaka, M. Julve, M. Yamashita and R. Clérac, Inorg. Chem., 2009, 48, 3420 CrossRef CAS PubMed
- L. Bogani, A. Vindigni, R. Sessoli and D. Gatteschi, J. Mater. Chem., 2008, 18, 4750 RSC
- H. L. C. Feltham and S. Brooker, Coord. Chem. Rev., 2014, 276, 1 CrossRef CAS PubMed
-
J. Ribas Gispert, Coordination Chemistry, Wiley-VCH, Weinheim, 2008 Search PubMed
-
R. L. Carlin, Magnetochemistry, Springer, Berlin, 1986 Search PubMed
- D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268 CrossRef CAS PubMed
- K. S. Pedersen, J. Bendix and R. Clerac, Chem. Commun., 2014, 50, 4396 RSC
-
Molecular Magnets: Physics and Applications, ed. J. Bartolomé, F. Luis and J. F. Fernández, Springer-Verlag, Berlin, 2014 Search PubMed
- W. H. Harman, T. D. Harris, D. E. Freedman, H. Fong, A. Chang, J. D. Rinehart, A. Ozarowski, M. T. Sougrati, F. Grandjean, G. J. Long, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 18115 CrossRef CAS PubMed
- O. Waldmann, Inorg. Chem., 2007, 46, 10035 CrossRef CAS PubMed
- P. Parois, S. A. Moggach, J. Sánchez-Benítez, K. V. Kamenev, A. R. Lennie, J. E. Warren, E. K. Brechin, S. Parsons and M. Murrie, Chem. Commun., 2010, 46, 1881 RSC
- D. E. Freedman, W. H. Harman, T. D. Harris, G. J. Long, C. J. Chang and J. R. Long, J. Am. Chem. Soc., 2010, 132, 1224 CrossRef CAS PubMed
- J. Vallejo, A. Pascual-Álvarez, J. Cano, I. Castro, M. Julve, F. Lloret, J. Krzystek, G. De Munno, D. Armentano, W. Wernsdorfer, R. Ruiz-García and E. Pardo, Angew. Chem., Int. Ed., 2013, 52, 14075 CrossRef CAS PubMed
- R. Ishikawa, R. Miyamoto, H. Nojiri, B. K. Breedlove and M. Yamashita, Inorg. Chem., 2013, 52, 8300 CrossRef CAS PubMed
- A. Grigoropoulos, M. Pissas, P. Papatolis, V. Psycharis, P. Kyritsis and Y. Sanakis, Inorg. Chem., 2013, 52, 12869 CrossRef CAS PubMed
- J. M. Zadrozny, M. Atanasov, A. M. Bryan, C.-Y. Lin, B. D. Rekken, P. P. Power, F. Neese and J. R. Long, Chem. Sci., 2013, 4, 125 RSC
- J. M. Zadrozny, D. J. Xiao, M. Atanasov, G. J. Long, F. Grandjean, F. Neese and J. R. Long, Nat. Chem., 2013, 5, 577 CrossRef CAS PubMed
- J. M. Zadrozny, D. J. Xiao, J. R. Long, M. Atanasov, F. Neese, F. Grandjean and G. J. Long, Inorg. Chem., 2013, 52, 13123 CrossRef CAS PubMed
- P. P. Samuel, K. C. Mondal, N. Amin Sk, H. W. Roesky, E. Carl, R. Neufeld, D. Stalke, S. Demeshko, F. Meyer, L. Ungur, L. F. Chibotaru, J. Christian, V. Ramachandran, J. van Tol and N. S. Dalal, J. Am. Chem. Soc., 2014, 136, 11964 CrossRef CAS PubMed
- A. Eichhöfer, Y. Lan, V. Mereacre, T. Bodenstein and F. Weigend, Inorg. Chem., 2014, 53, 1962 CrossRef PubMed
- P.-H. Lin, N. C. Smythe, S. I. Gorelsky, S. Maguire, N. J. Henson, I. Korobkov, B. L. Scott, J. C. Gordon, R. T. Baker and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 15806 CrossRef CAS PubMed
- D. Weismann, Y. Sun, Y. Lan, G. Wolmershäuser, A. K. Powell and H. Sitzmann, Chem. – Eur. J., 2011, 17, 4700 CrossRef CAS PubMed
- X. Feng, C. Mathonière, I.-R. Jeon, M. Rouzières, A. Ozarowski, M. L. Aubrey, M. I. Gonzalez, R. Clérac and J. R. Long, J. Am. Chem. Soc., 2013, 135, 15880 CrossRef CAS PubMed
- A. Hauser, Chem. Phys. Lett., 1986, 124, 543 CrossRef CAS
- C. Mathonière, H.-J. Lin, D. Siretanu, R. Clérac and J. M. Smith, J. Am. Chem. Soc., 2013, 135, 19083 CrossRef PubMed
- S. Mossin, B. L. Tran, D. Adhikari, M. Pink, F. W. Heinemann, J. Sutter, R. K. Szilagyi, K. Meyer and D. J. Mindiola, J. Am. Chem. Soc., 2012, 134, 13651 CrossRef CAS PubMed
- M. Murrie, Chem. Soc. Rev., 2010, 39, 1986 RSC
- T. Jurca, A. Farghal, P.-H. Lin, I. Korobkov, M. Murugesu and D. S. Richeson, J. Am. Chem. Soc., 2011, 133, 15814 CrossRef CAS PubMed
- F. Habib, O. R. Luca, V. Vieru, M. Shiddiq, I. Korobkov, S. I. Gorelsky, M. K. Takase, L. F. Chibotaru, S. Hill, R. H. Crabtree and M. Murugesu, Angew. Chem., Int. Ed., 2013, 52, 11290 CrossRef CAS PubMed
- S. Gómez-Coca, E. Cremades, N. Aliaga-Alcalde and E. Ruiz, J. Am. Chem. Soc., 2013, 135, 7010 CrossRef PubMed
- J. Vallejo, I. Castro, R. Ruiz-García, J. Cano, M. Julve, F. Lloret, G. De Munno, W. Wernsdorfer and E. Pardo, J. Am. Chem. Soc., 2012, 134, 15704 CrossRef CAS PubMed
- D. Wu, X. Zhang, P. Huang, W. Huang, M. Ruan and Z. W. Ouyang, Inorg. Chem., 2013, 52, 10976 CrossRef CAS PubMed
- J. M. Zadrozny, J. Liu, N. A. Piro, C. J. Chang, S. Hill and J. R. Long, Chem. Commun., 2012, 48, 3927 RSC
- A more detailed discussion and description of phonon bottlenecks and their effects can be found in: R. Schenker, M. N. Leuenberger, G. Chaboussant, D. Loss and H. U. Güdel, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 184403 CrossRef
- W. Huang, T. Liu, D. Wu, J. Cheng, Z. W. Ouyang and C. Duan, Dalton Trans., 2013, 42, 15326 RSC
- E. Colacio, J. Ruiz, E. Ruiz, E. Cremades, J. Krzystek, S. Carretta, J. Cano, T. Guidi, W. Wernsdorfer and E. K. Brechin, Angew. Chem., Int. Ed., 2013, 52, 9130 CrossRef CAS PubMed
- S. Gómez-Coca, A. Urtizberea, E. Cremades, P. J. Alonso, A. Camón, E. Ruiz and F. Luis, Nat. Commun., 2014, 5, 4300 Search PubMed
- R. Herchel, L. Váhovská, I. Potočňák and Z. Trávníček, Inorg. Chem., 2014, 53, 5896 CrossRef CAS PubMed
- L. Chen, J. Wang, J.-M. Wei, W. Wernsdorfer, X.-T. Chen, Y.-Q. Zhang, Y. Song and Z.-L. Xue, J. Am. Chem. Soc., 2014, 136, 12213 CrossRef CAS PubMed
- R. Boča, J. Miklovič and J. Titiš, Inorg. Chem., 2014, 53, 2367 CrossRef PubMed
- J. M. Zadrozny, J. Telser and J. R. Long, Polyhedron, 2013, 64, 209 CrossRef CAS PubMed
- F. Yang, Q. Zhou, Y. Zhang, G. Zeng, G. Li, Z. Shi, B. Wang and S. Feng, Chem. Commun., 2013, 49, 5289 RSC
- A. Buchholz, A. O. Eseola and W. Plass, C. R. Chim., 2012, 15, 929 CrossRef CAS PubMed
- D.-K. Cao, J.-Q. Feng, M. Ren, Y.-W. Gu, Y. Song and M. D. Ward, Chem. Commun., 2013, 49, 8863 RSC
- J. M. Zadrozny and J. R. Long, J. Am. Chem. Soc., 2011, 133, 20732 CrossRef CAS PubMed
- M. S. Fataftah, J. M. Zadrozny, D. M. Rogers and D. E. Freedman, Inorg. Chem., 2014, 53, 10716 CrossRef CAS PubMed
- Y. Y. Zhu, C. Cui, Y. Q. Zhang, J. H. Jia, X. Guo, C. Gao, K. Qian, S. D. Jiang, B. W. Wang, Z. M. Wang and S. Gao, Chem. Sci., 2013, 4, 1802 RSC
- V. Chandrasekhar, A. Dey, A. J. Mota and E. Colacio, Inorg. Chem., 2013, 52, 4554 CrossRef CAS PubMed
- R. C. Poulten, M. J. Page, A. G. Algarra, J. J. Le Roy, I. López, E. Carter, A. Llobet, S. A. Macgregor, M. F. Mahon, D. M. Murphy, M. Murugesu and M. K. Whittlesey, J. Am. Chem. Soc., 2013, 135, 13640 CrossRef CAS PubMed
- R. Ruamps, R. Maurice, L. Batchelor, M. Boggio-Pasqua, R. Guillot, A. L. Barra, J. Liu, E.-E. Bendeif, S. Pillet, S. Hill, T. Mallah and N. Guihéry, J. Am. Chem. Soc., 2013, 135, 3017 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2015 |