
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling the thermoelectric properties of polymers: application to PEDOT and polypyrrole
Mario
Culebras
,
Belén
Uriol
,
Clara M.
Gómez
and
Andrés
Cantarero
 *
*
Materials Science Institute, University of Valencia, PO Box 22085, 46071 Valencia, Spain. E-mail: andres.cantarero@uv.es; Fax: +34 963543633; Tel: +34 963544713
First published on 28th April 2015
Abstract
Poly(3,4-ethylenedioxythiophene) (PEDOT) and polypyrrole (PPy) films have been prepared by an electrochemical method in a three electrode cell. The films have been obtained at different oxidation levels regarded as bipolaron, polaron and neutral states by varying the voltage, as is usually done in conjugated heterocyclic polymers. The voltage (−0.2 < V < 1.0 V) has been applied versus a Ag/AgCl reference electrode, producing a variation of one order of magnitude in the electrical conductivity and the Seebeck coefficient of the films. In the voltage range explored, the electrical conductivity increases from 80 to 766 S cm−1 in PEDOT and from 15 to 160 S cm−1 in PPy, while the Seebeck coefficient decreases from 37.0 to 9.6 μV K−1 for PEDOT and from 51.0 to 6.7 μV K−1 for PPy. The thermal conductivity remains unchanged with the oxidation state of the film, κ ≈ 0.35 ± 0.02 W m−1 K−1 for PEDOT and 0.17 ± 0.02 W m−1 K−1 for PPy. A maximum thermoelectric efficiency of 1.4 × 10−2 for PEDOT and 6.8 × 10−3 for PPy has been achieved. These changes are related to the doping level of the polymer films and they can be accurately controlled by the applied voltage. In this work, we provide a very simple method to control and optimize the power factor or the figure of merit of conducting polymers.
1 Introduction
The lack of energy resources is a global problem. On one side, the oil reservoirs are limited, while on the other the energy demand is increasing over the years not only in developed countries, but also due to new demands which came into the market from countries like China and India with a growth rate of 5–8%. Water will also become a problem in the future, thus probably hydraulic plants will provide energy for a short period of time difficult to evaluate. With this panorama in mind, new energy resources must be in operation soon at least to compensate the growth of the energy market. The most plausible alternative at the moment is nuclear plants; being pragmatic they must be used at least for a few decades until enough energy can be supplied by photovoltaics, mill farms, biomass, and other clean/green energy production processes compatible with the sustainability of our planet. In the last few years, thermoelectricity has arisen as a promising alternative to transform waste heat into electricity, via the Seebeck effect, or to complement photovoltaic cells, to keep them working at maximum efficiency.1 Unfortunately, the efficiency of thermoelectric devices has not been improved much in the last few decades. But in a few years from now, the scientific community will start to work hard on thermoelectricity and we can expect substantial growth of the efficiency in the next few years. How good a thermoelectric material is can be quantitatively written in terms of the dimensionless figure of merit ZT, which is defined as: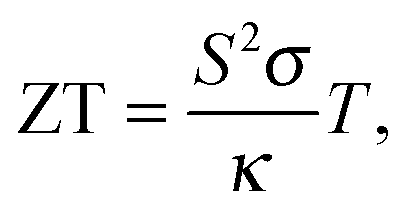 | (1) |
Nowadays, the best thermoelectric materials in terms of ZT are semiconductors. For instance, Bi2Sb3 is a good thermoelectric material used in low temperature applications,2 while Bi2Te3 and PbTe can be used for room temperature applications3–7 and Si1−xGex alloys8 and half-Heusler compounds9 in high temperature applications, and skutterudites in medium-temperature thermoelectric modules.10 The figure of merit was around one for many years, until the beginning of the 90's of the last century. After the seminal work of Hicks and Dresselhaus11 it was clear that by reducing the dimensionality (for instance by building a superlattice) of the semiconductor, the figure of merit would increase, whenever the Fermi energy can be located in a maximum of the density of electronic states. Unfortunately, in more than 20 years of research, ZT has increased only by a factor of 3.
In the last 10–15 years, organic materials have become important in optoelectronics,12,13 both in solar cells and light emitting diodes, and more recently in thermoelectricity,14,15 especially in thin films, where the polymer chains remain basically in two dimensions.16,17 Several intrinsically conducting polymers (ICPs) have been successfully used, increasing the figure of merit by several orders of magnitude,15,18 until the values were very close to those of inorganic materials. Polymers present, in addition, many advantages over inorganic materials: nonscarcity of raw materials, lack of toxicity, lower cost of production and many others. In the case of inorganic compounds, one of the barriers which does not allow the improvement of ZT is their high thermal conductivity, which is mainly produced by the heat transported by phonons. Polymers have a thermal conductivity at least one order of magnitude smaller than any semiconductor. In the next few years, we need to work on the improvement of the Seebeck coefficient and the electrical conductivity of ICPs.
The efficiency of thermoelectric polymers is largely dependent on the doping level of the material since it modifies both S and σ. These parameters are actually not completely independent. Thus, by increasing the doping level the electrical conductivity increases but the Seebeck coefficient decreases making it necessary to find a balance between them in order to obtain the optimum value of ZT, which is actually given by the power factor PF.19–23 One of the methods more commonly used to control the doping level of polymers is chemical de-doping.19–21 This method changes the doping level by using reducing agents such as hydrazine or tetrakis(dimethylamino)ethylene (TDAE).19–21 Another procedure to control the doping level is electrochemical doping/de-doping,22,23 which offers certain advantages. The first one is that the oxidation state (doping level) can be easily controlled by using an electrochemical cell. The second advantage is the absence of purification steps, required in other methods, which again simplifies the process. Finally, it produces a free standing film, which is moreover a novelty, indicating that it does not need an electrode in order to work.
In this work, we have synthesized free-standing films by electrochemical deposition of two conducting polymers, poly(3,4-ethylenedioxythiophene) (PEDOT) and polypyrrole (PPy). The electrochemical cell has been employed to change the doping level of the polymers in order to control the thermoelectric properties. We describe a new methodology able to control the thermoelectric properties of conducting polymers, such as PEDOT and PPy, and able to supply the required properties for a given application. This is of great importance in future applications as for instance the use of polymer matrices in thermoelectric nanocomposites or as sensitive elements in temperature sensors.
2 Experimental
2.1 Materials
The reactants used in this work, pyrrole, 3,4-ethylenedioxythiophene (EDOT), lithium perchlorate (LiClO4), ethanol and acetonitrile, were purchased from Sigma-Aldrich.2.2 Synthesis of PEDOT
The electrochemical synthesis of PEDOT was carried out at room temperature in a three electrode cell following the procedure described in a previous work.19 The working electrode was a gold coated PET surface for SEM and transport measurements, while an ITO coated glass was used for cyclic voltammetry and absorbance measurements. A platinum grid acted as the counter electrode and the Ag/AgCl electrode acted as the reference electrode. PEDOT was polymerized from a 0.01 M solution of EDOT and LiClO4 0.1 M in acetonitrile. Electrochemical polymerization was performed at a deposition intensity of 3 mA for 1.5 min over the working electrode surface. Films of 120–130 nm thickness were obtained. The gold layer of the corresponding working electrode was removed with an acid solution (HNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HCl ratio 1:3) after film deposition. Finally, the deposited PEDOT films were rinsed several times with water and ethanol to remove the untreated monomer and then dried in air at room temperature.
:HCl ratio 1:3) after film deposition. Finally, the deposited PEDOT films were rinsed several times with water and ethanol to remove the untreated monomer and then dried in air at room temperature.
2.3 Synthesis of polypyrrole
Polypyrrole films were also prepared at room temperature by the electrochemical polymerization method in a conventional three electrode system. The working electrode was a stainless steel electrode for SEM and transport measurements while an ITO electrode was used for voltamperometry and absorbance measurements. A platinum grid acted as the counter-electrode and the Ag/AgCl electrode was used as reference. Polypyrrole was polymerized from a 0.01 M solution of pyrrole and LiClO4 0.1 M in acetonitrile. Electrochemical polymerization was performed at a deposition intensity of 3 mA. The films, grown on the ITO electrode for 1.5 min, were 120–130 nm thick, whereas those obtained on stainless steel electrodes for 4 h were 100 μm. Finally, the deposited PPy films were rinsed several times with water and ethanol to remove the untreated monomer and then dried in air at room temperature. The layer of PPy was detached from the steel surface and transferred onto a glass substrate for measurement purposes.2.4 Electrochemical reduction of polymers
The electrochemical reduction of polymer samples was carried out in an Ivium-n-Stat: multi-channel electrochemical workstation under computer control. The three electrode cell was formed by the ITO coated by the polymer (PEDOT or PPy) as the working electrode, the platinum grid as the counter-electrode, and the Ag/AgCl electrode as the reference electrode in a 0.1 M of LiClO4 in acetonitrile solution. The polymer samples were subjected to several voltages vs. the Ag/AgCl reference electrode, in order to determine the absorbance and the transport properties (electrical conductivity and Seebeck coefficient) at different reduction states.2.5 Characterization
The thickness of the polymer films was determined using a Dektak profilometer. The morphology of the polymers was determined by scanning electron microscopy using a Hitachi 4800 S microscope at an acceleration voltage of 20 kV and at a working distance of 14 mm for gold-coated surfaces. Cyclic voltammetry was performed using an Ivium-n-Stat: multi-channel electrochemical workstation under computer control. The three electrode cell was the same as that for polymerization. The films were equilibrated at −2.0 V and −3.0 V for 2 min for PEDOT and PPy, respectively, before cyclic voltammetric measurements at 20 mV s−1. The absorbance measurements were performed using a Shimadzu UV-2501PC UV-Vis spectrophotometer from 1100 to 300 nm. Thermal conductivity was determined with the aid of modulated differential scanning calorimetry (MDSC) according to the ASTM E1952-11 standard test method. This method is valid to measure thermal conductivities of polymeric materials with values within the range 0.1–1 W m−1 K−1. The MDSC measurements were performed using DSC Q-20 TA Instruments calibrated with indium and sapphire. All MDSC measurements were carried out at 300 K under modulated conditions with a period of 80 s and a temperature amplitude of ±1 K.The electrical conductivity was determined by the van der Pauw method.24 A Keithley 2400 multimeter was used as a driving source and a voltmeter. The conductivity of the sample is obtained by solving the van der Pauw equation:
| e−πdR1σ + e−πdR2σ = 1, | (2) |
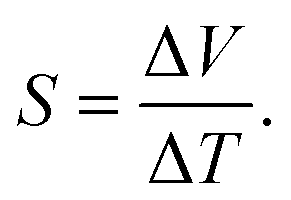 | (3) |
3 Results and discussion
Scanning electron microscopy (SEM) images of the PEDOT and PPy films obtained after electrochemical polymerization are depicted in Fig. 1. The surface morphology of PEDOT (Fig. 1a) shows a homogeneous and compact surface with a certain degree of roughness of nearly interconnected polymer chains and voids in between.19,25 The SEM surface image of PPy (Fig. 1b) shows typical granular (cauliflower) morphologies distributed over the substrate surface and densely packed.26,27 The differences between the morphology found in the SEM images can be attributed to the different interaction between the polymers and the solvent, in our case acetonitrile.28 | ||
| Fig. 1 SEM images of: (a) PEDOT and (b) PPy film surfaces after electrochemical polymerization. | ||
Cyclic voltammetry between −3 and +1.5 V at a rate of 20 mV s−1 was performed for the PEDOT and PPy films (Fig. 2) in order to determine the voltage at which the oxidized/reduced polymer species appear. The shape of the voltammogram with anodic (I) and cathodic (III) peaks related to the oxidation and reduction processes is similar to those reported in the literature.29–36 The peak appearing at positive current values is related to the oxidation of the polymer, while that appearing at negative current is related to the reduction state (Fig. 2). The reduction or cathodic peak appears in the range from −1.0 to +0.2 V, while the oxidation peak is located in a potential range between 0 and 0.5 V (see Fig. 2a) for PEDOT. Polypyrrole films depict a clear oxidation peak in the range from −1.0 to 0 V and a reduction peak in the range from −2.5 to −1.5 V (see Fig. 2b). Fig. 2c shows a scheme of the evolution of the polymer chains depending on the applied voltage. At oxidation potentials, the polymer chains are in a bipolaronic state, at intermediate potentials there is a mixture of bipolaronic, polaronic and neutral states and finally at reduction potentials the chains are in their neutral states.
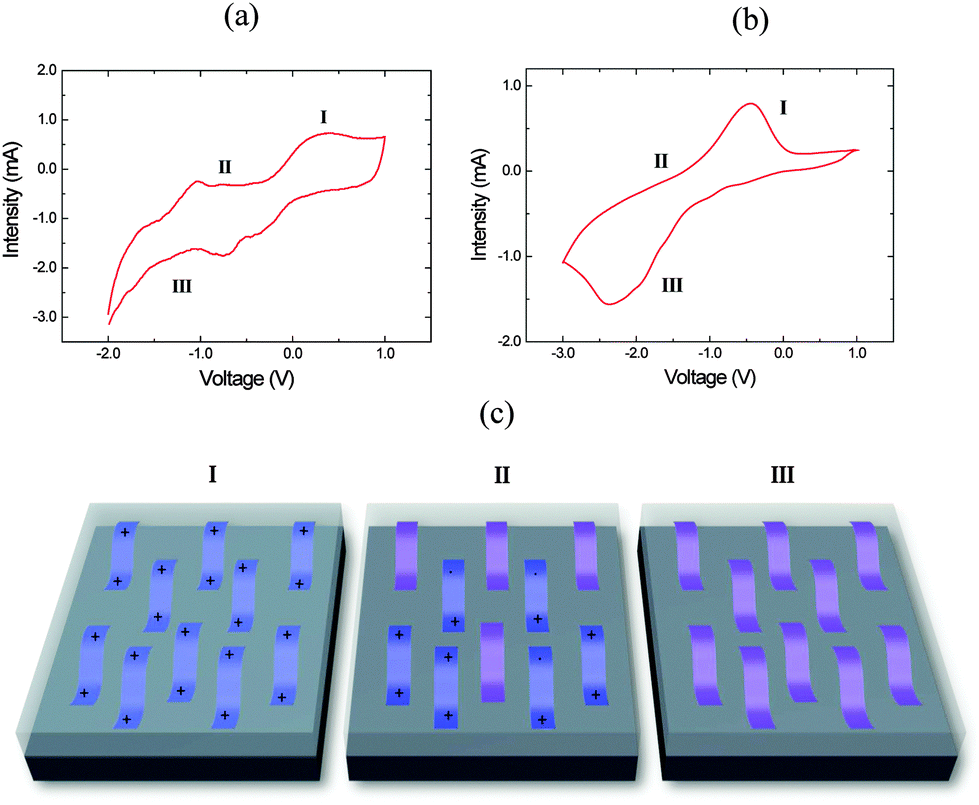 | ||
| Fig. 2 Cyclic voltammetry of: (a) PEDOT and (b) PPy. (c) Scheme of polymer electronic states as a function of the oxidation/reduction level. | ||
The nature of the doping states of conjugated polymers is still a matter of debate. A high doping level is desirable to improve the applications of conducting polymers; in the present case thermoelectric devices. Cyclic voltammetry was performed to determine the voltage values at which the oxidized and reduced species appear. These voltages will change the doping state of the polymer and, as expected, the thermoelectric efficiency. In order to change the doping level of PEDOT and PPy, different voltages (V vs. Ag/AgCl, taken as reference voltage) have been applied to the films, until the current remained unchanged. In this way, we have different doped polymers from neutral to bipolaron. Optical absorption, electrical and thermal conductivity, and the Seebeck coefficient have been determined for those doped films.
Ultraviolet-visible (UV-Vis) spectrochemical curves are shown in Fig. 3 for PEDOT and PPy films deposited on an indium tin oxide (ITO) coated glass. The spectra depict different bands related to different oxidation states due to electrochemical doping. A broad absorption band centered at 900 nm related to the oxidized PEDOT state (0.0 < V < 1.0 V) appears during the doping process and it is related to polaron and bipolaron states.36 However, an absorption band centered at 600 nm appears in the PEDOT reduced state (V = −1.0 V), which decreases its intensity as the applied voltage increases, that is, as the polymer changes from a reduced to an oxidized state, related to the π–π* transition.33–38 Thus, changing the doping states of the PEDOT chains from bipolarons/polarons to neutral states results in changes in the optical properties of the polymer. The colour of the polymer changes from dark blue in the reduced state to light blue in the oxidized (bipolaron) state. Fig. 3b shows the UV-Vis spectrum of PPy films obtained at different doping levels. The absorption band between 500–900 nm at high applied voltages is related to the polaronic and bipolaronic (oxidized) states of PPy.35 The band at 300–500 nm, related to the absorption of the neutral or reduced state of PPy,35 cannot be observed since it overlaps with the substrate signal (PET-ITO). The colour of PPy changes from a neutral yellow form to blue-green for the fully oxidized, bipolaron state.
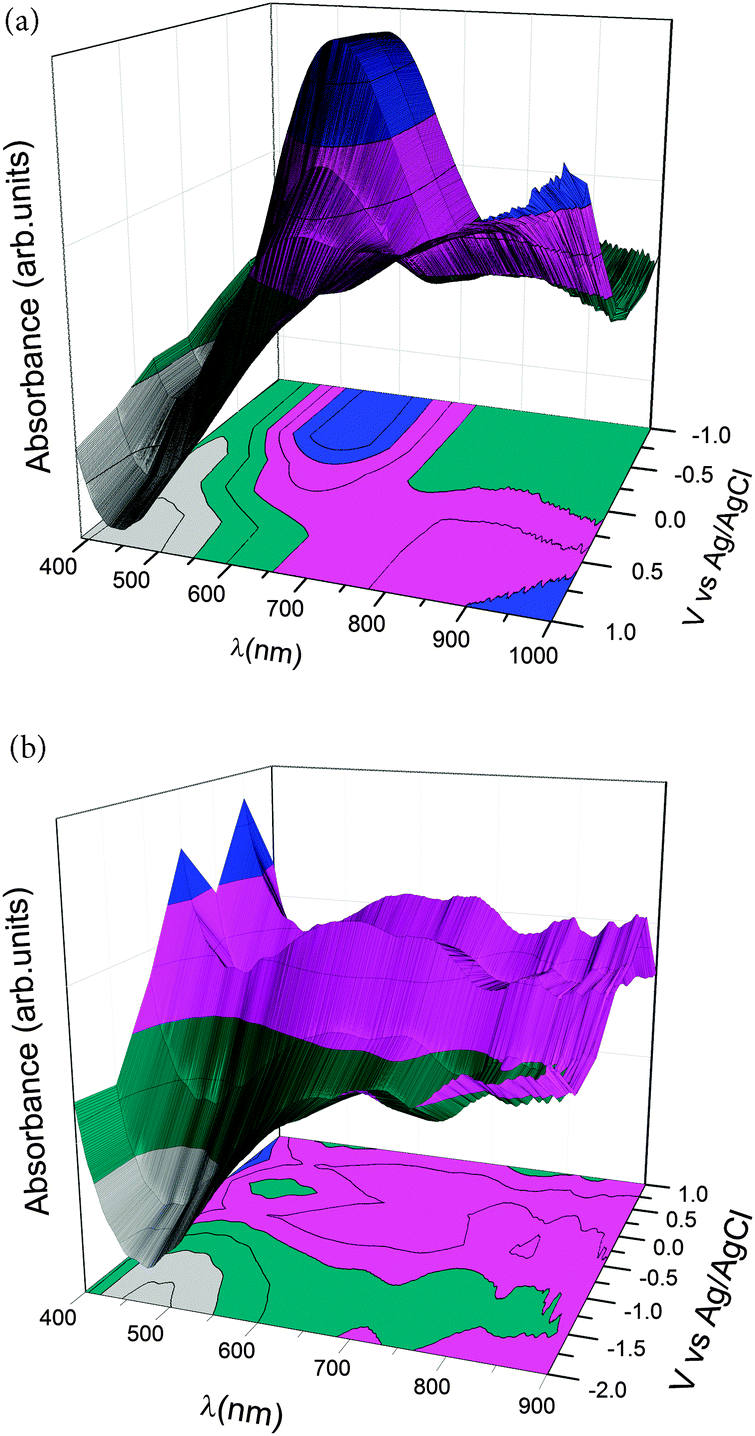 | ||
| Fig. 3 UV-Vis spectra of: (a) PEDOT and (b) polypyrrole as a function of different applied voltages vs. Ag/AgCl on ITO coated glass electrodeposited polymer films. | ||
Fig. 4 shows the electrical conductivity, the Seebeck coefficient and the power factor of PEDOT (Fig. 4a) and PPy (Fig. 4b) films as a function of the applied voltage (vs. Ag/AgCl). In both cases, the electrical conductivity increases with the applied voltage or the oxidation state of the polymers. As the applied voltage changes from negative to positive, the polymers change from the reduced to the neutral and to the oxidized state, i.e. a bipolaron state as corroborated by cyclic voltammetry and UV-Vis measurements. The electrical conductivity of PEDOT changes from 80 S cm−1 at −1.0 V to 766 S cm−1 at +1.0 V. These values are of the same order of magnitude as those found in the literature. For example PEDOT:PSS (PEDOT:poly(styrenesulfonate)) doped with dimethyl sulfoxide (DMSO) has an electrical conductivity between 500 and 1000 S cm−1.20,25,39,40 The electrical conductivity of PPy increases from 15 S cm−1 at −2.0 V to 160 S cm−1 at +1.0 V. The small depth present at 0.5 V is probably due to the existence of some oscillation in the oxidation process. At the highest oxidation level, we obtain similar values to that reported for PPy:PF6 (100–600 S cm−1),41 but higher than those obtained for PPy composites such as PPy/MWCNTs, 70 S cm−1,42 or PPy/graphene nanosheets, 40 S cm−1.43 The opposite trend has been observed in the variation of the Seebeck coefficient (Fig. 4). The Seebeck coefficient of PEDOT decreases from 37.0 μV K−1 at −1.0 V to 9.6 μV K−1 at +1.0 V, whereas the Seebeck coefficient of PPy changes from 51.0 μV K−1 at −2.0 V to 6.7 μV K−1 at +1.0 V (always referenced to the Ag/AgCl reference electrode). The values of S obtained for the PEDOT films are similar to those reported in ref. 19. However, the values of the Seebeck coefficient obtained in this work for PPy are the highest values ever measured.18
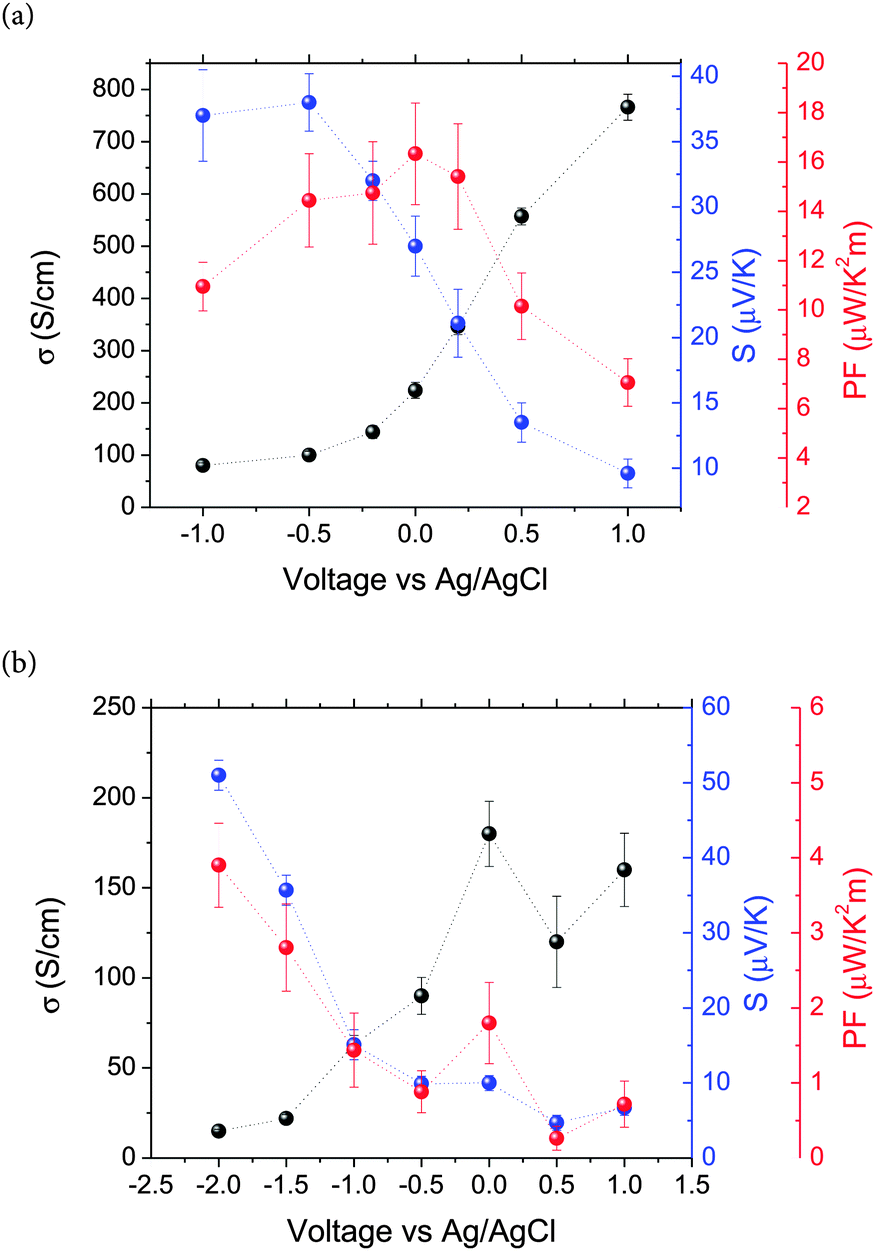 | ||
| Fig. 4 Electrical conductivity, Seebeck coefficient and power factor of (a) PEDOT and (b) PPy as a function of voltage referred to the Ag/AgCl electrode. | ||
The PF corresponding to PEDOT and PPy has also been plotted in Fig. 4. The maximum PF is 16.3 μV K−1 m−2 for PEDOT at 0.01 V, while for the case of PPy the maximum value has been reached at −2.0 V, 3.9 μV K−1 m−2. These values are related to an intermediate polaron structure for PEDOT and a reduced, neutral state for PPy due to the relationship between the Seebeck coefficient and the electrical conductivity.
In order to obtain the values of ZT, thermal conductivity was determined for the different samples, being independent of the doping level of the samples. The values obtained at 300 K were 0.35 ± 0.02 W m−1 K−1 for PEDOT and 0.17 ± 0.02 W m−1 K−1 for PPy. We have checked that the thermal conductivity does not change with the applied voltage. Two recent papers reported a small anisotropy in the thermal conductivity of PEDOT44,45 and a certain dependence on the electrical conductivity. The in-plane thermal conductivity increases with increasing σ, while the perpendicular thermal conductivity remains constant. This can be interpreted from the configuration of the polymeric chains (the amount of chains in-plane as compared to off-plane). In a very thin film, as it is the case here, the chains remain in plane and the thermal conductivity actually corresponds to the in plane conductivity, even if we produce a pellet crashing the film. The values measured in the present work using a DSC are in agreement with previous literature results.19,21,46
The efficiency of a thermoelectric device, ZT, calculated from eqn (1) has been plotted in Fig. 5 for the different samples. The highest values of ZT were 6.8 × 10−3 for PPy and 1.4 × 10−2 for PEDOT, using the values of thermal conductivity obtained from our DSC measurements. These results are of the same order of magnitude as other recently reported results on conducting polymers.15,18
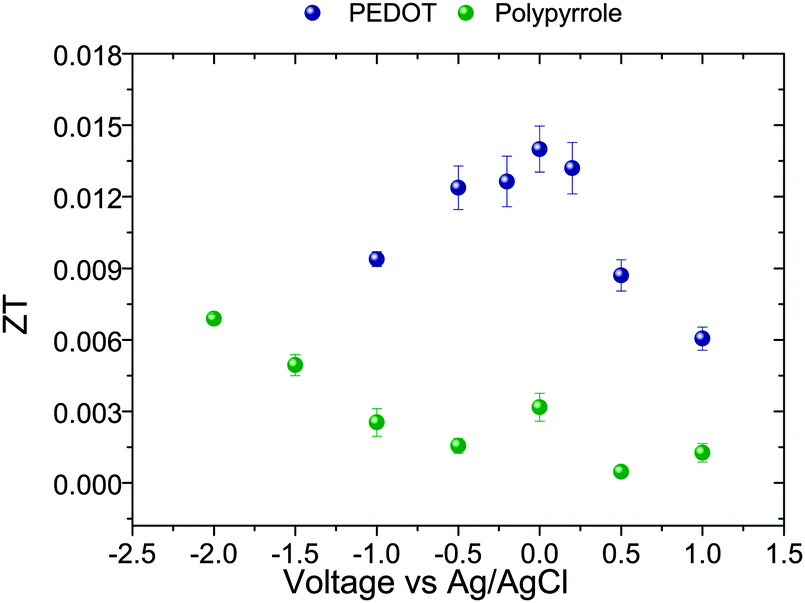 | ||
| Fig. 5 ZT of PEDOT and PPy as a function of applied potential at 300 K. | ||
4 Conclusions
In conclusion, free-standing films of PEDOT and PPy with high electrical conductivity have been prepared by electrochemical polymerization in a three electrode cell using a Ag/AgCl reference electrode. The electrical conductivity and the Seebeck coefficient of the polymer films can be controlled by applying different potential differences to the films. The electrical conductivity is maximum at oxidation potentials of 0.5–1.0 V for PEDOT and 0.0–1.0 V for PPy, while the Seebeck coefficient is maximum at reduction potentials of −1.0 V for PEDOT and −2.0 V for PPy. The maximum thermoelectric efficiency has been obtained at 0.01 V for PEDOT and at −2.0 V for PPy. The results show a very simple method to control and optimize the ZT of conducting polymers.References
- R. Ahiska and H. Mamur, Int. J. Renew. Energy Res., 2014, 4, 128–136 Search PubMed.
- D.-H. Lee, J.-U. Lee, S.-J. Jung, S.-H. Baek, J.-H. Kim, D.-I. Kim, D.-B. Hyun and J.-S. Kim, J. Electron. Mater., 2014, 43, 2255–2261 CrossRef CAS PubMed.
- Y. Gelbstein and J. Davidow, Phys. Chem. Chem. Phys., 2014, 16, 20120–20126 RSC.
- T. C. Hasapis, S. N. Girard, E. Hatzikraniotis, K. M. Paraskevopoulos and M. G. Kanatzidis, J. Nano Res., 2012, 17, 165–174 CrossRef CAS.
- P. H. Le, C.-N. Liao, C. W. Luo and J. Leu, J. Alloys Compd., 2014, 615, 546–552 CrossRef CAS PubMed.
- B. Poudel, Q. Hao, Y. Ma, Y. Lan, A. Minnich, B. Yu, X. Yan, D. Wang, A. Muto, D. Vashaee, X. Chen, J. Liu, M. S. Dresselhaus, G. Chen and Z. Ren, Science, 2008, 320, 634–638 CrossRef CAS PubMed.
- R. Venkatasubramanian, E. Siivola, T. Colpitts and B. O’Quinn, Nature, 2001, 413, 597–602 CrossRef CAS PubMed.
- N. Stein, N. Petermann, R. Theissmann, G. Schierning, R. Schmechel and H. Wiggers, J. Mater. Res., 2011, 26, 1872–1878 CrossRef CAS.
- E. Rausch, B. Balke, S. Ouardi and C. Felser, Phys. Chem. Chem. Phys., 2014, 16, 25258–25262 RSC.
- J. R. Salvador, J. Y. Cho, Z. Ye, J. E. Moczygemba, A. J. Thompson, J. W. Sharp, J. D. Koenig, R. Maloney, T. Thompson, J. Sakamoto, H. Wang and A. A. Wereszczak, Phys. Chem. Chem. Phys., 2014, 16, 12510–12520 RSC.
- L. D. Hicks and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 12727–12731 CrossRef CAS.
- Z. Guo, D. Lee, Y. Liu, F. Sun, A. Sliwinski, H. Gao, P. C. Burns, L. Huang and T. Luo, Phys. Chem. Chem. Phys., 2014, 16, 7764–7771 RSC.
- R. C. Chiechi and J. C. Hummelen, ACS Macro Lett., 2012, 1, 1180–1183 CrossRef CAS.
- S. K. Yee, N. E. Coates, A. Majumdar, J. J. Urban and R. A. Segalman, Phys. Chem. Chem. Phys., 2013, 15, 4024–4032 RSC.
- M. Culebras, C. M. Gomez and A. Cantarero, Materials, 2014, 7, 6701–6732 CrossRef PubMed.
- Q. Wei, M. Mukaida, K. Kirihara and T. Ishida, ACS Macro Lett., 2014, 3, 948–952 CrossRef CAS.
- D. Wang, W. Shi, J. Chen, J. Xi and Z. Shuai, Phys. Chem. Chem. Phys., 2012, 14, 16505–16520 RSC.
- Y. Du, S. Z. Shen, K. Cai and P. S. Casey, Prog. Polym. Sci., 2012, 37, 820–841 CrossRef CAS PubMed.
- M. Culebras, A. Cantarero and C. M. Gómez, J. Mater. Chem. A, 2014, 2, 10109–10115 CAS.
- S. H. Lee, H. Park, S. Kim, W. Son, I. W. Cheong and J. H. Kim, J. Mater. Chem. A, 2014, 2, 7288–7294 CAS.
- O. Bubnova, Z. U. Khan, A. Malti, S. Braun, M. Fahlman, M. Berggren and X. Crispin, Nat. Mater., 2011, 10, 429–433 CrossRef CAS PubMed.
- T. Park, C. Park, B. Kim, H. Shin and E. Kim, Energy Environ. Sci., 2013, 6, 788–792 CAS.
- O. Bubnova, M. Berggren and X. Crispin, J. Am. Chem. Soc., 2012, 134, 16456–16459 CrossRef CAS PubMed.
- L. J. Van Der Pauw, Philips Tech. Rev., 1958, 20, 220–224 Search PubMed.
- J. Feng-Xing, X. Jing-Kun, L. Bao-Yang, X. Yu, H. Rong-Jin and L. Lai-Feng, Chin. Phys. Lett., 2008, 25, 2202–2205 CrossRef.
- D. Ge, J. Mu, S. Huang, P. Liang, O. U. Gcilitshana, S. Ji, V. Linkov and W. Shi, Synth. Met., 2011, 161, 166–172 CrossRef CAS PubMed.
- J. V. Thombare, M. C. Rath, S. H. Han and V. J. Fulari, J. Semicond., 2013, 34, 103002 CrossRef.
- E. Poverenov, M. Li, A. Bitler and M. Bendikov, Chem. Mater., 2010, 22, 4019–4025 CrossRef CAS.
- H. Randriamahazaka, J. Phys. Chem. C, 2007, 111, 4553–4560 CAS.
- A. Aydin and I. Kaya, Org. Electron., 2013, 14, 730–743 CrossRef CAS PubMed.
- P. Anjaneyulu, V. Varade, C. S. S. Sangeeth, K. P. Ramesh and R. Menon, J. Phys. D: Appl. Phys., 2014, 47, 505106 CrossRef.
- A. Diaz, K. Kanazawa and G. Gardini, J. Chem. Soc., Chem. Commun., 1979, 635–636 RSC.
- S. Garreau, G. Louarn, J. Buisson, G. Froyer and S. Lefrant, Macromolecules, 1999, 32, 6807–6812 CrossRef CAS.
- H. Ahonen, J. Lukkari and J. Kankare, Macromolecules, 2000, 33, 6787–6793 CrossRef CAS.
- P. Rapta, A. Neudeck, A. Petr and L. Dunsch, J. Chem. Soc., Faraday Trans., 1998, 94, 3625–3630 RSC.
- S. Garreau, J. Duvail and G. Louarn, Synth. Met., 2001, 125, 325–329 CrossRef.
- X. Chen and O. Inganas, J. Phys. Chem., 1996, 100, 15202–15206 CrossRef CAS.
- J. Wang, K. Cai and S. Shen, Org. Electron., 2015, 17, 151–158 CrossRef CAS PubMed.
- G.-H. Kim, L. Shao, K. Zhang and K. P. Pipe, Nat. Mater., 2013, 12, 719–723 CrossRef CAS PubMed.
- K.-C. Chang, M.-S. Jeng, C.-C. Yang, Y.-W. Chou, S.-K. Wu, M. A. Thomas and Y.-C. Peng, J. Electron. Mater., 2009, 38, 1182–1188 CrossRef CAS.
- K. Sato, M. Yamaura, T. Hagiwara, K. Murata and M. Tokumoto, Synth. Met., 1991, 40, 35–48 CrossRef CAS.
- J. Wang, K. Cai, S. Shen and J. Yin, Synth. Met., 2014, 195, 132–136 CrossRef CAS PubMed.
- S. Han, W. Zhai, G. Chen and X. Wang, RSC Adv., 2014, 4, 29281–29285 RSC.
- A. Weathers, Z. U. Khan, R. Brooke, D. Evans, M. T. Pettes, J. W. Andreasen, X. Crispin and L. Shi, Adv. Mater., 2015, 27, 2101–2106 CrossRef CAS PubMed.
- J. Liu, X. Wang, D. Li, N. E. Coates, R. A. Segalman and D. G. Cahill, Macromolecules, 2015, 48, 585–591 CrossRef CAS.
- J. Wu, Y. Sun, W. B. Pei, L. Muang, W. Xu and Q. Zhang, Synth. Met., 2014, 196, 173–177 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2015 |