
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nonseparable exchange–correlation functional for molecules, including homogeneous catalysis involving transition metals†
Haoyu S.
Yu
 a,
Wenjing
Zhang
ab,
Pragya
Verma
a,
Xiao
He
ac and
Donald G.
Truhlar
*a
a,
Wenjing
Zhang
ab,
Pragya
Verma
a,
Xiao
He
ac and
Donald G.
Truhlar
*a
aDepartment of Chemistry, Chemical Theory Center, Inorganometallic Catalyst Design Center, and Supercomputing Institute, University of Minnesota, Minneapolis, Minnesota 55455-0431, USA. E-mail: truhlar@umn.edu
bThe College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou, Henan 450001, China
cState Key Laboratory of Precision Spectroscopy, East China Normal University, Shanghai, China
First published on 9th April 2015
Abstract
The goal of this work is to develop a gradient approximation to the exchange–correlation functional of Kohn–Sham density functional theory for treating molecular problems with a special emphasis on the prediction of quantities important for homogeneous catalysis and other molecular energetics. Our training and validation of exchange–correlation functionals is organized in terms of databases and subdatabases. The key properties required for homogeneous catalysis are main group bond energies (database MGBE137), transition metal bond energies (database TMBE32), reaction barrier heights (database BH76), and molecular structures (database MS10). We also consider 26 other databases, most of which are subdatabases of a newly extended broad database called Database 2015, which is presented in the present article and in its ESI. Based on the mathematical form of a nonseparable gradient approximation (NGA), as first employed in the N12 functional, we design a new functional by using Database 2015 and by adding smoothness constraints to the optimization of the functional. The resulting functional is called the gradient approximation for molecules, or GAM. The GAM functional gives better results for MGBE137, TMBE32, and BH76 than any available generalized gradient approximation (GGA) or than N12. The GAM functional also gives reasonable results for MS10 with an MUE of 0.018 Å. The GAM functional provides good results both within the training sets and outside the training sets. The convergence tests and the smooth curves of exchange–correlation enhancement factor as a function of the reduced density gradient show that the GAM functional is a smooth functional that should not lead to extra expense or instability in optimizations. NGAs, like GGAs, have the advantage over meta-GGAs and hybrid GGAs of respectively smaller grid-size requirements for integrations and lower costs for extended systems. These computational advantages combined with the relatively high accuracy for all the key properties needed for molecular catalysis make the GAM functional very promising for future applications.
1 Introduction
Kohn–Sham (KS) density functional theory has been very successful for electronic structure calculations in both physics and chemistry.1 The accuracy of KS calculations depends on the quality of the exchange–correlation functional. The quest for quantum mechanical methods that can be accurately applied to study atomic, molecular, and material properties has resulted in the design of exchange–correlation functionals with a variety of ingredients, costs, and accuracies, where the accuracy may depend strongly on the kind of property that is calculated. Exchange–correlation functionals that depend only on spin-up and spin-down electronic densities (ρα and ρβ) are known as local spin density approximations (LSDAs), and ones that depend on both the spin densities and spin density gradients are called gradient approximations (GAs, in particular GGAs and NGAs). More complicated functionals include ingredients calculated from the orbitals (which are functionals of the density), in particular spin-up and spin-down local kinetic energy densities (as in meta-GGAs and meta-NGAs), nonlocal Hartree–Fock exchange (as in hybrid functionals), and/or nonlocal correlation (as in doubly hybrid functionals, which have both nonlocal exchange and nonlocal correlation.). (One can also include nonlocal correlation without including nonlocal exchange.) Functionals depending only on local variables, such as spin densities, their gradients, and spin-specific local kinetic energy densities, are often called local (especially in the chemistry literature, while the physics literature often labels them as semilocal if they include density gradients or spin kinetic energy densities).Even though the meta and nonlocal functionals can give more accurate results than GGAs and LSDAs, GAs are still of great interest for four reasons. First, GAs are widely implemented in many programs because of their ease of coding. Second, GAs often have better self-consistent field (SCF) convergence and smaller grid requirements than meta functionals. Third, calculations employing GAs are less expensive than calculations involving nonlocal functionals, with the difference being more pronounced for extended and large systems and when geometries are optimized.
The fourth reason for special interest in GAs is that local functionals often have better performance than hybrid functionals, on average, for systems with high multi-reference character. Multi-reference character is the extent to which a wave function is inherently multi-configurational so that a single Slater determinant does not provide a good starting point (reference function) for approximating the complete wave function. Although KS theory does not calculate the wave function of the interacting system, it does use a Slater determinant to represent the density, and calculating the exchange from the Slater determinant, as in Hartree–Fock exchange, can introduce static correlation error, a result of which is that it is often more challenging to obtain good approximations for multi-reference systems when Hartree–Fock exchange is included. (The unknown exact exchange–correlation energy functional includes nonlocal effects and does not have static correlation error, but the problem just mentioned is not completely solved by currently available functionals.) Multi-reference systems are sometimes called strongly correlated systems. Many open-shell systems and transition-metal systems have multi-reference character, and hence the ability to treat multi-reference systems is critical to the ability to treat many catalytic reaction mechanisms. Systems without high multi-reference character are called single-reference systems.
Most GAs have a form that separately approximates exchange and correlation, as first introduced by Langreth and Mehl2 and usually called a generalized gradient approximation3 (GGA); however, it has been shown that a nonseparable gradient approximation4 (which has more flexibility at the cost of satisfying less exact constraints) is capable of performing well for a broader set of properties. The original NGA, called N12, was designed to give good predictions both of solid-state lattice constants and of cohesive energies and molecular atomization energies; it also gives good predictions of molecular bond lengths.4 Here we show that we can get improved performance for barrier heights (which are important for studies of both uncatalyzed and catalyzed reactions) by relaxing the accuracy for lattice constants, which are not needed for molecular (as opposed to solid-state) processes. By diminishing the emphasis on obtaining good lattice constants we can obtain an exchange–correlation functional that may be more useful for treating many large and complex homogeneous and enzymatic catalysts that do not require the calculations on solid-state material.
A second goal of this work is to obtain improved results for compounds containing metal atoms, including transition metal compounds with high multi-reference character, by incorporating a greater amount of representative data for metal–ligand bond energies in the training set of a density functional. A third goal of the present work is to obtain a very smooth exchange–correlation functional by enforcing an unsmoothness penalty as part of the optimization process.
Combining these three goals, we have designed a new exchange–correlation functional called gradient approximation for molecules, or GAM, and this new functional is presented here. The GAM functional is an NGA, and so it depends only on spin densities and spin density gradients. The parameters of the GAM functional are optimized against a broad set of molecular and solid-state data in a new database called Database 2015, which is also presented here. We will show that the resulting GAM functional yields good results for main group bond energies, chemical reaction barrier heights, transition-metal bond energies, weak interaction energies between noble gas atoms, and bond lengths of diatomic molecules.
Section 2 describes the computational details. Section 3 describes Database 2015, for which complete information is given in the ESI.† Section 4 describes previously available functionals to be used for comparison. Section 5 describes the design and optimization of the GAM functional. Section 6 gives results; Section 7 provides discussion; and Section 8 summarizes the main conclusions.
2 Computational details
All the calculations in this paper were performed by a locally modified version5 of the Gaussian 09 program.6 Ultrafine grids (“99,590”) are used to evaluate the exchange–correlation energies of our new GAM functional. We use the stable = opt keyword in Gaussian 096 to find the stationary solution to the Kohn–Sham equations by allowing symmetry breaking in the wave function if the symmetry-constrained solution is unstable. The periodic boundary condition (PBC) algorithm7 in Gaussian 096 is used to calculate the lattice constants, cohesive energies, and semiconductor band gaps in our new Database 2015, which will be explained in the next section.Besides testing the new functional on the training subset of Database 2015, we made several tests outside the training set. First we tested the new functional against subdatabases for semiconductor band gaps (SBG31) and solid-state cohesive energies (SSCE8), which are in Database 2015 but outside the training set. We also tested our functional against other data that is not in the training set. This data includes a recently published database WCCR for transition metal coordination reactions56 (renamed here as WCCR10 for consistency with our general naming scheme), the enthalpies of binding of O2 and N2 to the metal–organic framework Fe2(dobdc), the binding of C2H4 to Pd(PH3)2, Ag2 dimer and FeC bond dissociation energies, transition metal dimer bond distances, and the Ar2 potential energy curve.
For the WCCR10 database we use the same basis set (def2-QZVPP) and geometries as used in the original paper; these geometries, which were optimized by functional BP86,33,34 are provided in the ESI† of the WCCR paper.56
For calculating the binding enthalpies of O2 and N2 bound to Fe2(dobdc), we used an 88-atom cluster model of the experimental structure of Fe2(dobdc) containing three iron centers. The details of this cluster and rationale for its design are described in our earlier work.8 This cluster has three iron atoms, and here we studied binding at the central iron, which best represents the immediate environment around iron in the actual MOF. During optimization, the cluster of the MOF was frozen and the guest molecules (O2 or N2) were allowed to relax. The binding enthalpies were calculated using the formula given in eqn (1) of ref. 8.
The binding energy of the Pd(PH3)2C2H4 complex were computed using four basis sets. In all four basis sets, Pd atom has 18 active electrons and 28 core electrons that are replaced by an effective core potential. Basis set BS1 denotes the Stuttgart–Dresden–Dunning (SDD) basis set for Pd9 and the cc-pVTZ basis set for P,10 C, and H.11 Basis set BS2 denotes the def2-TZVP basis set for Pd12 and the cc-pVTZ basis set for P, C, and H. Basis set BS3 denotes the def2-TZVP basis set for Pd, the cc-pV(T+d)Z basis set for P,13,14 and the cc-pVTZ basis set for C and H. Basis set BS4 denotes the def2-TZVP basis set for Pd, the maug-cc-pV(T+d)Z basis set for P,15 the maug-cc-pVTZ basis set for C,15,16 and the cc-pVTZ basis set for H.
One basis set was used for Ag dimer, namely jun-cc-pVTZ-PP,17–19 one basis set was used for homonuclear transition metal bond distance, namely LanL2DZ,20–23 and two basis sets were used for Ar dimer, namely the aug-cc-pVQZ10,24 and aug-cc-pV6Z25 basis sets.
3 Database 2015
Database 2015 is our new database for optimizing and testing density functionals. Compared to Database 2.0 that we used in previous work26 the following changes are made:We divide the previous bond energy databases according to two types of classification: (i) whether the molecule contains only main-group nonmetal atoms or it also contains main-group-metal atoms or transition-metal atoms; (ii) whether the molecule has singe-reference character, i.e., can be well described by a single configuration wave function, or multi-reference character, i.e., cannot be so described. Then we added additional data to the underpopulated classes. Accordingly we have six new subdatabases for bond energies. Theses subdatabases are as follows (their shorthand names are in parentheses, where the final number in the shorthand name of a subdatabase is the number of data):
• single-reference main-group-metal bond energies (SR-MGM-BE9),
• single-reference main-group-nonmetal bond energies (SR-MGN-BE107),
• single-reference transition-metal bond energies (SR-TM-BE17),
• multi-reference main-group-metal bond energies (MR-MGM-BE4),
• multi-reference main-group nonmetal bond energies (MR-MGN-BE17),
• multi-reference transition-metal bond energies (MR-TM-BE15).
A new subdatabase called NGDWI21 has been added for noble-gas-dimer weak interactions. It comprises both homodimers and heterodimers.
We have added three new subdatabases for atomic excitation energies, namely
• 3d transition metal atomic excitation energies (3dAEE7),
• 4d transition metal atomic excitation energies (4dAEE5),
• p-block excitation energies (pEE5).
Two new subdatabases for p-block isomerization energies are added:
• 2p isomerization energies (2pIsoE4),
• 4p isomerization energies (4pIsoE4).
A new subdatabase for molecular geometries has been added; it is called diatomic geometries for heavy atoms (DGH4).
The above points summarize the main changes made to our previous database,26 called Database 2.0. A complete list of the subdatabases included in Database 2015 is given in Table 1, which also shows the number of data in each category (the inverse weight column of this table will be explained in Section 5). The database is divided into primary subdatabases, and some of the primary subdatabases are further divided into secondary subdatabases. Complete details of the new database and its layers of subdatabases, including geometries and references for the included data and also the basis sets we use for calculations on the various subdatabases, are given in the ESI.†
| n | Primary subset | Secondary | Description |
I
n
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c c |
Ref. |
|---|---|---|---|---|---|
| a Databases 1–27 were used with various inverse weights in training, and databases 1–29 constitute Database 2015. Database 30 is from T. Weymuth et al. (ref. 56), and – like databases 28 and 29 – it was used only for testing. b In the name of a database or subdatabase, the number at the end of the name or before the solidus is the number of data. For example, ME417, SR-MGM-BE9, IsoL6/11, and DGH4 contain respectively 417, 9, 6, and 4 data. c Inverse weights with units of kcal mol−1 per bond for databases 1–7, kcal mol−1 for databases 8–24, and Å for databases 25–27. d TM denotes transition metal. e NA denotes not applicable. | |||||
| ME417 | |||||
| 1 | SR-MGM-BE9 | Single-reference main-group metal bond energies | 2.00 | ||
| SRM2 | Single-reference main-group bond energies | 26 | |||
| SRMGD5 | Single-reference main-group diatomic bond energies | 26 and 65 | |||
| 3dSRBE2 | 3d single-reference metal–ligand bond energies | 66 | |||
| 2 | SR-MGN-BE107 | Single-reference main-group nonmetal bond energies | 0.20 | 26 | |
| 3 | SR-TM-BE17 | Single-reference TMd bond energies | 3.15 | ||
| 3dSRBE4 | 3d single-reference metal–ligand bond energies | 66 | |||
| SRMBE10 | Single-reference metal bond energies | 26 | |||
| PdBE2 | Palladium complex bond energies | 67 | |||
| FeCl | FeCl bond energy | 68 | |||
| 4 | MR-MGM-BE4 | Multi-reference main-group metal bond energies | 4.95 | 65 | |
| 5 | MR-MGN-BE17 | Multi-reference main-group nonmetal bond energies | 1.25 | 26 | |
| 6 | MR-TM-BE13 | Multi-reference TM bond energies | 0.76 | ||
| 3dMRBE6 | 3d multi-reference metal–ligand bond energies | 66 | |||
| MRBE3 | Multi-reference bond energies | 26 | |||
| Remaining | Bond energies of remaining molecules: CuH, VO, CuCl, NiCl | 68 | |||
| 7 | MR-TMD-BE2 | Multi-reference TM dimer bond energies (Cr2 and V2) | 10.00 | 26 | |
| 8 | IP23 | Ionization potentials | 5.45 | 26 and 69 | |
| 9 | NCCE30 | Noncovalent complexation energies | 0.10 | 26 and 70–73 | |
| 10 | NGDWI21 | Noble gas dimer weak interactions | 0.01 | 26 and 74 | |
| 11 | 3dAEE7 | 3d TM atomic excitation energies | 0.40 | 69 and 75 | |
| 12 | 4dAEE5 | 4d TM atomic excitation energies | 6.90 | 76 | |
| 13 | pEE5 | p-block excitation energies | 1.74 | 77 | |
| 14 | 4pIsoE4 | 4p isomerization energies | 8.00 | 78 | |
| 15 | 2pIsoE4 | 2p isomerization energies | 7.81 | 78 | |
| 16 | IsoL6/11 | Isomerization energies of large molecules | 2.00 | 26 | |
| 17 | EA13/03 | Electron affinities | 2.96 | 26 | |
| 18 | PA8 | Proton affinities | 2.23 | 26 | |
| 19 | πTC13 | Thermochemistry of π systems | 5.75 | 26 | |
| 20 | HTBH38/08 | Hydrogen transfer barrier heights | 0.25 | 26 | |
| 21 | NHTBH38/08 | Non-hydrogen transfer barrier heights | 0.80 | 26 | |
| 22 | AE17 | Atomic energies | 10.22 | 26 | |
| 23 | HC7/11 | Hydrocarbon chemistry | 6.48 | 26 | |
| 24 | DC9/12 | Difficult cases | 10.00 | 26 | |
| MS10 | |||||
| 25 | DGL6 | Diatomic geometries of light-atom molecules | 0.01 | 26 | |
| 26 | DGH4 | Diatomic geometries of heavy-atom molecules: ZnS, HBr, NaBr | 0.01 | 79 | |
| Diatomic geometry of Ag2 | 0.0013 | 80 | |||
| SS17 | |||||
| 27 | LC17 | Lattice constants | 0.013 | 26 | |
| SSE39 | |||||
| 28 | SBG31 | Semiconductor band gaps | NAe | 26 | |
| 29 | SSCE8 | Solid-state cohesive energies | NA | 26 | |
| WCCR10 | |||||
| 30 | WCCR10a | Ligand dissociation energies of large cationic TM complexes | NA | 56 | |
4 Functionals for comparison
We compare our results to 22 previously available exchange–correlation functionals. Since GAM depends only on spin densities and spin density gradients, we compare our results mainly to GAs, in particular to 14 GGAs and the one previously available NGA. In a practical sense, three of the GGAs are corrected to second order in the density gradient expansion for exchange, and the other 11 are not. Altogether we compare to 20 local functionals of four types and to two hybrid functionals. The local functionals are an LSDA, namely GKSVWN5;27–29 14 GGAs, namely SOGGA,30 PBEsol,31 PBE,32 BP86,33,34 PW91,35 BLYP,34,36 mPWPW,37 revPBE,38 BPW91,34,35 RPBE,39 HCTH407,40 SOGGA11,41 OLYP,36,42 and OreLYP;36,42,43 an NGA, namely N12;4 and four meta-GGAs, namely TPSS,44 revTPSS,45 M06-L,46 and M11-L.47 For context we also compare to two popular hybrid functionals, namely a global-hybrid GGA, B3LYP;42,48,49 and a range-separated hybrid GGA, HSE06.50,51 All these functionals are listed in Table 2 with the type, the percentage of Hartree–Fock exchange, the year, and the original reference. A more complete comparison of gradient approximations to more advanced functionals of the meta-GGA, meta-NGA, and hybrid type is found elsewhere26 and will not be repeated here, where our emphasis is on gradient approximations for the four reasons stated in the introduction, so comparisons to more advanced functionals are limited here to providing context.| Category | X | Type | Year | Method | Ref. |
|---|---|---|---|---|---|
| a X is the percentage of nonlocal Hartree–Fock exchange. When a range is given, the first value is for small interelectronic distances, and the second value is for large interelectronic distances. Details of the functional form that joins these regions of interelectronic separation are given in the references. b GVWN5 denotes the Gáspár approximation for exchange and the VWN5 fit to the correlation energy; this is an example of the local spin density approximation (LSDA), and it has the keyword SVWN5 in the Gaussian 09 program. Note that Kohn–Sham exchange is the same as Gáspár exchange, but Slater exchange (not tested here) is greater by a factor of 1.5. c PW91 formally satisfies the gradient expansion for exchange to second order but only at such small values of the gradient that for practical purposes it should be grouped with functionals that do not satisfy the gradient expansion to second order. | |||||
| Local | 0 | LSDA | 1980 | GKSVWN5b | 27–29 |
| 0 | GGA – correct to 2nd order in exchange | 2008 | SOGGA | 30 | |
| 0 | 2008 | PBEsol | 31 | ||
| 0 | 2011 | SOGGA11 | 41 | ||
| 0 | GGA – other | 1988 | BP86 | 33 and 34 | |
| 0 | 1988 | BLYP | 34 and 36 | ||
| 0 | 1991 | PW91c | 35 | ||
| 0 | 1991 | BPW91 | 34 and 35 | ||
| 0 | 1996 | PBE | 32 | ||
| 0 | 1997 | mPWPW | 37 | ||
| 0 | 1997 | revPBE | 38 | ||
| 0 | 1999 | RPBE | 39 | ||
| 0 | 2000 | HCTH407 | 40 | ||
| 0 | 2001 | OLYP | 36 and 42 | ||
| 0 | 2009 | OreLYP | 36, 42 and 43 | ||
| 0 | NGA | 2012 | N12 | 26 and 74 | |
| 0 | 2015 | GAM | Present | ||
| 0 | Meta-GGA | 2003 | TPSS | 44 | |
| 0 | 2006 | M06-L | 46 | ||
| 0 | 2009 | revTPSS | 45 | ||
| 0 | 2011 | M11-L | 47 | ||
| Nonlocal | 20 | Global hybrid GGA | 1994 | B3LYP | 42 and 48 |
| 0–25 | Range-separated hybrid GGA | 2009 | HSE06 | 50 and 51 | |
5 Design and optimization of the GAM functional
The general functional form of GAM is the same as N12,4 which has the flexibility to approximate both exchange and correlation effects in terms of spin density ρσ and reduced spin density gradient xσ. In order to design a good functional, we use a broad molecular and solid-state database to optimize the parameters of the functional, and we also add smoothness constraints to our optimization. We will discuss the functional form in Sections 5.1–5.3 and the optimization of the functional in Section 5.4.5.1 Functional form
The exchange–correlation energy Exc of the GAM functional is the sum of nonseparable exchange–correlation component ENSGAnxc and an additional term that is nominally treated as a correlation energy Ec. Typically one writes the first component as Ex, however, we label it as ENSGAnxc to show that it is a nonseparable approximation involving both exchange and correlation. Since we optimize the functional empirically and do not enforce the factorizable form on the first term, the first term also represents part of correlation energy, and similarly the second term is not purely correlation. Both terms must also include an empirical contribution required to account for the difference of the exact electronic kinetic energy from that computed from the orbitals of the Kohn–Sham determinant. The philosophy used in designing the functional form is consistent with the statement of Tozer and Handy that “The functionals represent exchange and correlation effects in a combined manner. Individual exchange and correlation terms cannot be isolated”.52 Our total exchange–correlation functional is| Exc = ENSGAnxc + Ec | (1) |
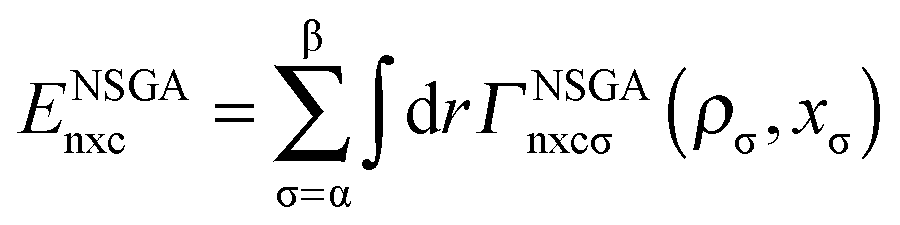 | (2) |
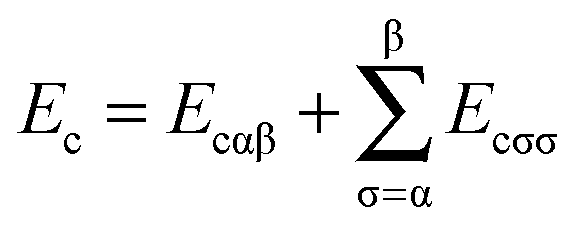 | (3) |
5.2 Nonseparable exchange–correlation functional form
In eqn (2), the nonseparable energy density is written as| ΓNSGAnxcσ = εUEGxσ(ρσ)Fx(ρσ, xσ) | (4) |
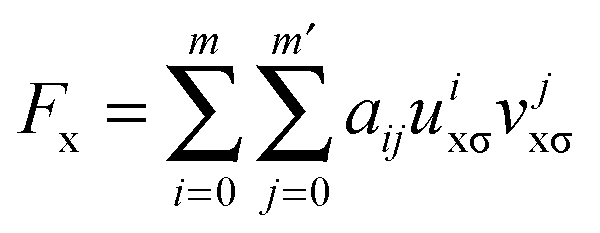 | (5) |
| uxσ = γxσxσ2/(1 + γxσxσ2) | (6) |
| vxσ = ωxσρ1/3σ(1 + ωxσρ1/3σ) | (7) |
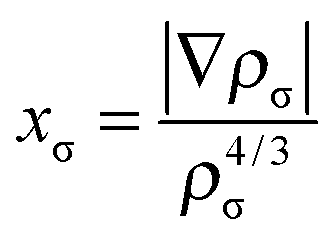 | (8) |
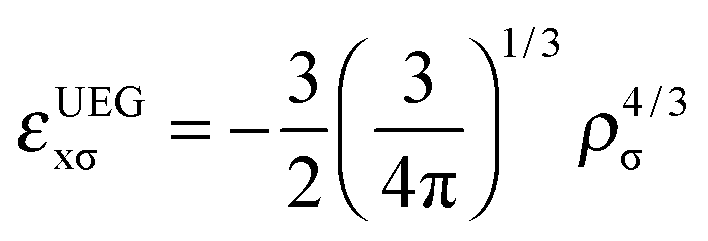 | (9) |
A GGA exchange functional can be written like eqn (4) but where the enhancement factor Fx depends only on the reduced spin density gradient xσ. For an NGA we allow the enhancement factor to depend also on the spin density ρσ.
5.3 Additional correlation functional form
In eqn (3), the correlation functional has two parts. One is the contribution Ecαβ from opposite spins, and the other is the contribution Ecσσ from same spins. These two contributions are defined by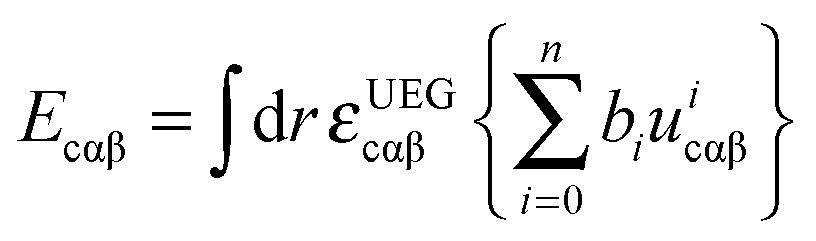 | (10) |
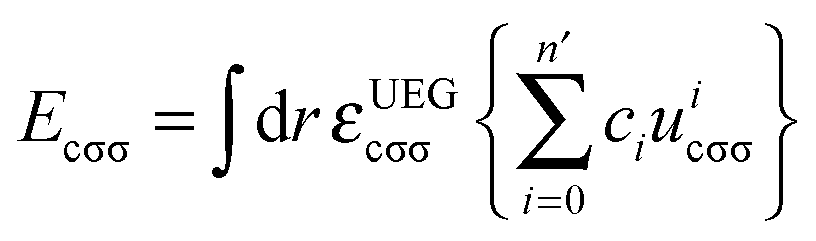 | (11) |
 | (12) |
 | (13) |
5.4 Functional optimization
In eqn (5), (10), and (11) above, we see that aij, bi, and ci are linear parameters of the functionals, which will be optimized. We do not force the uniform-electron-gas limit to hold when we optimize the functional. In order to make our functional smooth, smoothness constraints are added to the optimization, which will be explained in detail in the last paragraph of this section. The values of m, m′, n, and n′ are chosen as 3, 3, 4, and 4 respectively. We found that the performance of the functional is not significantly improved by increasing these values, which shows that one cannot obtain improved functionals simply by adding more parameters. Therefore, in order to design good density functionals we must pay more attention to the mathematical form of the functional and the diversity of the database we are optimizing against, instead of concentrating on the number of parameters.We optimize our functional against 27 primary databases, including 24 molecular energy databases, two molecular structure databases, and one solid-state structure database. We optimize the GAM functional self-consistently by minimizing the following unfitness function:
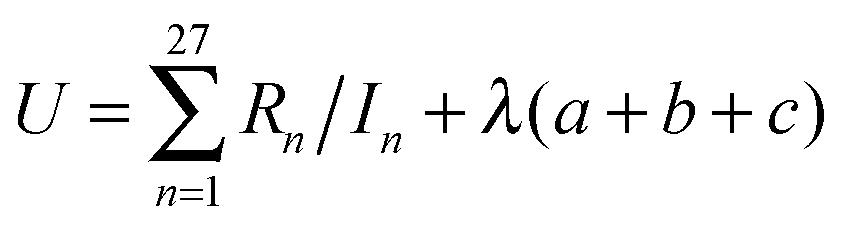 | (14) |
 | (15) |
 | (16) |
 | (17) |
 . We made fits with various values of λ, and we monitored the smoothness of the resulting exchange–correlation functionals by plotting them, by examining the magnitudes of the linear coefficients of the exchange–correlation functional (they should not be too large in magnitude or having severely oscillating signs), and by checking whether there is any difficulty in achieving self-consistent-field convergence on difficult cases (we had made a list of cases where previous functionals sometimes showed SCF convergence difficulties). After balancing the performance of the functional and the smoothness of the enhancement factor (as judged by the three criteria just mentioned), we finally chose λ to be 0.001, which gives what we judged to be the best combination of overall accuracy, convergence, and smoothness of the exchange–correlation functional.
. We made fits with various values of λ, and we monitored the smoothness of the resulting exchange–correlation functionals by plotting them, by examining the magnitudes of the linear coefficients of the exchange–correlation functional (they should not be too large in magnitude or having severely oscillating signs), and by checking whether there is any difficulty in achieving self-consistent-field convergence on difficult cases (we had made a list of cases where previous functionals sometimes showed SCF convergence difficulties). After balancing the performance of the functional and the smoothness of the enhancement factor (as judged by the three criteria just mentioned), we finally chose λ to be 0.001, which gives what we judged to be the best combination of overall accuracy, convergence, and smoothness of the exchange–correlation functional.
In order to design a functional with good across-the-board performance, we include various molecular and solid-state properties in our training set; these properties include main-group bond energies, transition metal bond energies, transition metal atomic excitation energies, barrier heights, ionization potentials, proton affinities, electron affinities, lattice constants, etc. In Table 1, the inverse weight of each primary database is given. The smaller the inverse weight is, the more emphasis we put on that primary database. The inverse weights were chosen as follows: first we calculated the mean unsigned errors (MUEs) of 80 exchange–correlation functionals (previously published functionals developed in many different groups) for all the molecular subdatabases in Database 2015; this shows how well previous exchange–correlation functionals typically perform for each kind of data. The average of these MUEs for a given subdatabase were used as our initial inverse weights. Then we modified the inverse weights iteratively to improve performance on the various subdatabases where we wished to reduce the error. Our goal in this process was to obtain good across the board performance for as many subdatabases as possible, not to simply reduce the overall mean unsigned error.
Whereas the N124 functional involved 20 optimized linear coefficients and the constraint that it reduced to PBEsol at low density, the new GAM functional involves optimizing 26 linear coefficients in eqn (5), (10), and (11) with no constraints. We use the same values as N12 for the nonlinear parameters ωxσ, γxσ, γcαβ, and γcσσ. A key element in the optimization is the choice of weights. We do not choose them to minimize the overall error but rather to try to get small errors across the board, i.e., relatively small errors for each of the subdatabases, to the greatest extent possible. The final choice of weights was determined after considerable trial and error and is a subjective decision that cannot be justified by any numerical argument.
Table 3 lists the values for the optimized and inherited parameters of the GAM functional.
| Exchange | Correlation | ||
|---|---|---|---|
| Optimized parameters | |||
| a 00 | 1.32730 | b 0 | 0.860548 |
| a 01 | 0.886102 | b 1 | −2.94135 |
| a 02 | −5.73833 | b 2 | 15.4176 |
| a 03 | 8.60197 | b 3 | −5.99825 |
| a 10 | −0.786018 | b 4 | −23.4119 |
| a 11 | −4.78787 | c 0 | 0.231765 |
| a 12 | 3.90989 | c 1 | 0.575592 |
| a 13 | −2.11611 | c 2 | −3.43391 |
| a 20 | 0.802575 | c 3 | −5.77281 |
| a 21 | 14.4363 | c 4 | 9.52448 |
| a 22 | 8.42735 | ||
| a 23 | −6.21552 | ||
| a 30 | −0.142331 | ||
| a 31 | −13.4598 | ||
| a 32 | 1.52355 | ||
| a 33 | −10.0530 | ||
| Inherited parameters | |||
| ω xσ | 2.5 | γ cαβ | 0.006 |
| γ xσ | 0.004 | γ cσσ | 0.2 |
6 Tables of results
In Tables 4 and 5 we compare the performance of the new functional to that of 22 existing functionals for molecular energetic data, and in Table 6 we do the same for molecular bond distances.| Type | LSDA | GGA | GGA | GGA | GGA | GGA | GGA | GGA | GGA | GGA | GGA | GGA | GGA | GGA | GGA | NGA | NGA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Functional | GKSVWN5 | SOGGA | PBEsol | PBE | BP86 | PW91 | BLYP | mPWPW | revPBE | BPW91 | RPBE | HCTH407 | SOGGA11 | OLYP | OreLYP | N12 | GAM |
| a The MGBE137 database consists of SR-MGM-BE9, SR-MGN-BE107, MR-MGM-BE4, and MR-MGN-BE17. b The TMBE32 database consists of SR-TM-BE17, MR-TM-BE13, and MR-TMD-BE2. c The BH76 database consists of HTBH38/08 and NHTBH38/08. d The ME417 database consists all the 24 subdatabases above and the ME400xAE consists all the subdatabases except AE17. | |||||||||||||||||
| SR-MGM-BE9 | 11.64 | 4.43 | 4.47 | 2.72 | 3.10 | 2.57 | 5.07 | 2.87 | 4.26 | 3.20 | 4.57 | 3.52 | 8.79 | 4.67 | 4.06 | 5.92 | 2.00 |
| SR-MGN-BE107 | 16.21 | 7.27 | 7.28 | 3.40 | 4.06 | 3.51 | 2.78 | 2.80 | 2.99 | 2.49 | 3.35 | 2.55 | 2.77 | 2.32 | 2.56 | 2.38 | 2.27 |
| SR-TM-BE17 | 20.89 | 11.59 | 11.33 | 7.20 | 7.39 | 8.76 | 6.52 | 6.73 | 6.22 | 7.34 | 6.24 | 8.36 | 11.44 | 9.32 | 7.15 | 8.31 | 6.31 |
| MR-MGM-BE4 | 24.56 | 14.48 | 15.81 | 9.31 | 9.49 | 10.26 | 8.75 | 9.02 | 6.24 | 8.03 | 6.43 | 10.11 | 7.44 | 8.39 | 8.35 | 9.10 | 7.76 |
| MR-MGN-BE17 | 36.89 | 21.29 | 23.16 | 14.80 | 13.87 | 14.80 | 6.67 | 12.45 | 5.94 | 10.74 | 5.51 | 5.24 | 8.57 | 5.15 | 4.25 | 6.93 | 4.22 |
| MR-TM-BE13 | 34.07 | 22.03 | 21.24 | 12.73 | 12.11 | 13.25 | 10.64 | 11.67 | 8.55 | 10.81 | 7.73 | 19.70 | 18.79 | 5.77 | 5.10 | 12.54 | 4.94 |
| IsoL6/11 | 2.05 | 1.89 | 1.55 | 1.98 | 2.28 | 1.92 | 3.73 | 2.16 | 2.82 | 2.38 | 2.99 | 3.02 | 1.73 | 3.44 | 3.39 | 1.73 | 1.96 |
| IP23 | 9.59 | 4.84 | 5.82 | 6.19 | 8.44 | 7.29 | 6.52 | 6.85 | 5.00 | 6.30 | 4.92 | 6.81 | 5.92 | 3.12 | 3.03 | 4.36 | 4.53 |
| EA13/03 | 5.70 | 2.70 | 2.16 | 2.27 | 4.21 | 2.60 | 2.68 | 2.31 | 2.40 | 2.26 | 2.37 | 3.70 | 5.23 | 3.60 | 2.32 | 4.12 | 4.49 |
| PA8 | 5.07 | 2.33 | 2.10 | 1.34 | 1.41 | 1.30 | 1.58 | 1.52 | 2.00 | 1.88 | 1.98 | 2.84 | 2.11 | 2.40 | 1.70 | 1.35 | 3.84 |
| πTC13 | 4.80 | 4.06 | 4.20 | 5.59 | 5.85 | 5.73 | 6.07 | 6.41 | 7.15 | 7.08 | 7.20 | 8.23 | 7.41 | 8.26 | 7.27 | 8.61 | 8.59 |
| HTBH38/08 | 17.56 | 12.88 | 12.69 | 9.31 | 9.16 | 9.60 | 7.52 | 8.43 | 6.58 | 7.38 | 6.43 | 5.48 | 6.57 | 5.63 | 6.28 | 6.94 | 5.35 |
| NHTBH38/08 | 12.42 | 9.68 | 9.86 | 8.42 | 8.72 | 8.80 | 8.53 | 8.03 | 6.82 | 7.26 | 6.82 | 6.29 | 4.32 | 5.25 | 5.57 | 6.86 | 5.15 |
| NCCE30 | 3.61 | 2.12 | 2.07 | 1.46 | 1.53 | 1.60 | 1.64 | 1.42 | 1.71 | 1.74 | 1.61 | 1.32 | 1.48 | 2.52 | 2.68 | 1.38 | 1.29 |
| AE17 | 421.13 | 283.06 | 245.90 | 47.24 | 16.92 | 4.63 | 8.68 | 12.55 | 10.88 | 11.95 | 9.39 | 16.80 | 10.06 | 10.13 | 2.37 | 14.21 | 10.18 |
| HC7/11 | 21.45 | 17.88 | 13.31 | 3.97 | 9.95 | 4.55 | 27.39 | 8.08 | 13.65 | 10.77 | 14.96 | 14.97 | 6.26 | 17.01 | 16.34 | 4.27 | 6.24 |
| 3dAEE7 | 11.86 | 10.87 | 10.77 | 9.80 | 10.36 | 10.47 | 10.27 | 10.63 | 10.05 | 10.84 | 9.78 | 12.00 | 12.50 | 11.56 | 10.98 | 18.51 | 9.82 |
| 4dAEE5 | 14.10 | 4.77 | 8.48 | 4.70 | 5.07 | 4.73 | 5.73 | 4.89 | 4.49 | 5.03 | 4.27 | 7.75 | 7.60 | 5.94 | 6.42 | 10.24 | 5.23 |
| pEE5 | 4.36 | 6.30 | 5.15 | 3.96 | 3.46 | 4.14 | 5.10 | 5.22 | 4.37 | 6.33 | 3.51 | 4.27 | 5.01 | 2.09 | 3.25 | 14.86 | 2.99 |
| DC9/12 | 17.35 | 14.61 | 13.34 | 14.99 | 15.11 | 13.94 | 17.88 | 14.76 | 20.35 | 16.21 | 21.48 | 19.74 | 16.65 | 21.71 | 22.57 | 10.20 | 23.07 |
| 2pIsoE4 | 2.05 | 1.44 | 1.71 | 2.73 | 3.21 | 2.87 | 5.45 | 3.20 | 3.59 | 3.43 | 3.70 | 4.59 | 1.72 | 3.95 | 3.72 | 3.41 | 5.02 |
| 4pIsoE4 | 3.05 | 2.29 | 2.28 | 2.43 | 2.87 | 2.58 | 4.00 | 2.50 | 2.16 | 2.41 | 2.16 | 3.29 | 3.27 | 2.15 | 2.22 | 1.73 | 3.57 |
| NGDWI21 | 0.212 | 0.082 | 0.081 | 0.102 | 0.528 | 0.165 | 0.385 | 0.220 | 0.282 | 0.587 | 0.179 | 0.246 | 0.650 | 0.323 | 0.389 | 0.387 | 0.019 |
| MR-TMD-BE2 | 51.28 | 33.08 | 30.87 | 28.10 | 24.40 | 27.97 | 42.70 | 29.43 | 28.40 | 30.96 | 26.79 | 20.09 | 35.20 | 25.18 | 12.74 | 27.97 | 10.67 |
| MGBE137a | 18.72 | 9.04 | 9.31 | 4.94 | 5.38 | 5.05 | 3.58 | 4.18 | 3.54 | 3.73 | 3.79 | 3.17 | 4.02 | 3.00 | 3.04 | 3.37 | 2.65 |
| TMBE32b |
28.14 | 17.18 | 16.58 | 10.75 | 10.37 | 11.79 | 10.45 | 10.15 | 8.55 | 10.22 | 8.13 | 13.70 | 15.91 | 8.87 | 6.66 | 11.26 | 6.03 |
| BH76c | 14.99 | 11.28 | 11.27 | 8.87 | 8.94 | 9.20 | 8.02 | 8.23 | 6.70 | 7.32 | 6.62 | 5.88 | 5.44 | 5.44 | 5.92 | 6.90 | 5.25 |
| ME417d | 30.67 | 19.55 | 18.04 | 7.45 | 6.68 | 5.98 | 5.89 | 5.80 | 5.27 | 5.60 | 5.26 | 5.90 | 5.74 | 5.01 | 4.56 | 5.57 | 4.51 |
| ME400xAEd | 14.07 | 8.36 | 8.36 | 5.76 | 6.25 | 6.03 | 5.77 | 5.51 | 5.03 | 5.33 | 5.09 | 5.44 | 5.56 | 4.79 | 4.66 | 5.20 | 4.27 |
| Type | NGA | Meta | Meta | Meta | Meta | Hybrid | Hybrid |
|---|---|---|---|---|---|---|---|
| Functional | GAM | TPSS | revTPSS | M06-L | M11-L | B3LYP | HSE06 |
| a The MGBE137, TMBE32, BH76, ME417, and ME400xAE notations are explained in footnotes to Table 4. | |||||||
| SR-MGM-BE9 | 2.00 | 2.55 | 2.91 | 3.40 | 7.24 | 4.58 | 3.47 |
| SR-MGN-BE107 | 2.27 | 2.43 | 2.24 | 2.03 | 1.76 | 2.45 | 2.08 |
| SR-TM-BE17 | 6.31 | 6.11 | 6.13 | 6.24 | 5.73 | 5.48 | 4.96 |
| MR-MGM-BE4 | 7.76 | 6.69 | 5.98 | 6.15 | 13.50 | 7.76 | 8.52 |
| MR-MGN-BE17 | 4.22 | 4.25 | 4.62 | 3.11 | 4.02 | 5.09 | 5.30 |
| MR-TM-BE13 | 4.94 | 8.87 | 6.81 | 4.40 | 4.44 | 5.33 | 4.87 |
| IsoL6/11 | 1.96 | 3.66 | 3.96 | 2.76 | 1.57 | 2.61 | 1.25 |
| IP23 | 4.53 | 4.29 | 4.07 | 3.91 | 4.77 | 5.51 | 4.06 |
| EA13/03 | 4.49 | 2.35 | 2.59 | 3.83 | 5.54 | 2.33 | 2.77 |
| PA8 | 3.84 | 2.66 | 2.79 | 1.88 | 2.17 | 1.02 | 1.10 |
| πTC13 | 8.59 | 8.12 | 7.85 | 6.69 | 5.14 | 6.03 | 6.20 |
| HTBH38/08 | 5.35 | 7.71 | 6.96 | 4.15 | 1.44 | 4.23 | 4.23 |
| NHTBH38/08 | 5.15 | 8.91 | 9.07 | 3.81 | 2.86 | 4.55 | 3.73 |
| NCCE30 | 1.29 | 1.34 | 1.33 | 0.90 | 0.81 | 1.09 | 0.95 |
| AE17 | 10.18 | 18.04 | 23.81 | 7.04 | 21.81 | 18.29 | 32.82 |
| HC7/11 | 6.24 | 10.48 | 6.42 | 3.35 | 2.42 | 16.80 | 7.34 |
| 3dAEE7 | 9.82 | 10.78 | 10.47 | 7.84 | 14.03 | 8.47 | 10.62 |
| 4dAEE5 | 5.23 | 5.19 | 5.11 | 6.58 | 11.04 | 5.67 | 5.07 |
| pEE5 | 2.99 | 2.25 | 2.31 | 7.50 | 10.39 | 2.87 | 5.70 |
| DC9/12 | 23.07 | 14.20 | 14.94 | 10.67 | 5.90 | 12.02 | 9.08 |
| 2pIsoE4 | 5.02 | 3.54 | 2.53 | 3.16 | 3.32 | 4.69 | 2.44 |
| 4pIsoE4 | 3.57 | 2.60 | 3.27 | 2.88 | 5.03 | 4.24 | 2.64 |
| NGDWI21 | 0.019 | 0.171 | 0.174 | 0.125 | 0.568 | 0.276 | 0.102 |
| MR-TMD-BE2 | 10.67 | 26.21 | 26.59 | 7.22 | 22.18 | 31.21 | 45.13 |
| MGBE137a | 2.65 | 2.79 | 2.69 | 2.37 | 2.74 | 3.07 | 2.76 |
| TMBE32 | 6.03 | 8.49 | 7.68 | 5.55 | 6.24 | 7.03 | 7.43 |
| BH76 | 5.25 | 8.31 | 8.01 | 3.98 | 2.15 | 4.39 | 3.98 |
| ME417 | 4.51 | 5.40 | 5.42 | 3.55 | 4.15 | 4.68 | 4.83 |
| ME400xAE | 4.27 | 4.86 | 4.64 | 3.41 | 3.40 | 4.10 | 3.64 |
| Functional | Type | DGL6 | DGH4 | MS10a |
|---|---|---|---|---|
| a The MS10 database consists of DGL6 and DGH4 subdatabases. The functionals are listed in the same order as in Tables 4 and 5. | ||||
| GKSVWN5 | LSDA | 0.011 | 0.031 | 0.019 |
| SOGGA | GGA | 0.009 | 0.013 | 0.010 |
| PBEsol | GGA | 0.010 | 0.007 | 0.009 |
| PBE | GGA | 0.013 | 0.020 | 0.016 |
| BP86 | GGA | 0.015 | 0.021 | 0.018 |
| PW91 | GGA | 0.012 | 0.019 | 0.015 |
| BLYP | GGA | 0.019 | 0.037 | 0.026 |
| mPWPW | GGA | 0.012 | 0.021 | 0.016 |
| revPBE | GGA | 0.015 | 0.034 | 0.023 |
| BPW91 | GGA | 0.013 | 0.022 | 0.017 |
| RPBE | GGA | 0.016 | 0.038 | 0.025 |
| HCTH407 | GGA | 0.004 | 0.033 | 0.015 |
| SOGGA11 | GGA | 0.008 | 0.053 | 0.026 |
| OLYP | GGA | 0.009 | 0.036 | 0.020 |
| OreLYP | GGA | 0.011 | 0.034 | 0.020 |
| N12 | NGA | 0.008 | 0.007 | 0.008 |
| GAM | NGA | 0.007 | 0.034 | 0.018 |
| TPSS | Meta | 0.010 | 0.015 | 0.012 |
| revTPSS | Meta | 0.011 | 0.009 | 0.010 |
| M06-L | Meta | 0.006 | 0.018 | 0.011 |
| M11-L | Meta | 0.012 | 0.033 | 0.021 |
| B3LYP | Hybrid | 0.009 | 0.027 | 0.016 |
| HSE06 | Hybrid | 0.003 | 0.015 | 0.008 |
Table 7 gives the performance for solid-state databases, but since B3LYP calculations with periodic boundary conditions are very expensive, we only compare 21 density functionals for the solid-state lattice constant and energetic data of Table 7. Table 8 compares the performance of GAM to that of eight density functionals for the WCCR10 database of Weymuth et al.56Table 9 is a test for the binding of dioxygen and dinitrogen to Fe2(dobdc), which is also called Fe-MOF-74, where we compare to experiments of Bloch et al.57Table 10 presents results for the binding of ethylene to Pd(PH3)2, where we compare the results of GAM to the best estimate computed using BCCD(T)58 in our earlier work.67Table 11 presents results for the bond distance of homonuclear transition metal dimers, where we compare the results of GAM and N12 with 5 functionals in a recent paper.80
| Functionala | Type | LC17 | NNDb | SBG31 | SSCE8 |
|---|---|---|---|---|---|
| a The functionals are listed in the same order as in Tables 4 and 5. b The values in this column are obtained by dividing the previous column by 2.15 (a standard factor determined in previous work – see text) so that the results may be compared more physically to errors in molecular bond lengths. | |||||
| GKSVWN5 | LSDA | 0.069 | 0.032 | 1.14 | 0.70 |
| SOGGA | GGA | 0.022 | 0.010 | 1.14 | 0.31 |
| PBEsol | GGA | 0.023 | 0.011 | 1.14 | 0.27 |
| PBE | GGA | 0.068 | 0.031 | 0.98 | 0.11 |
| BP86 | GGA | 0.073 | 0.034 | 1.12 | 0.12 |
| PW91 | GGA | 0.065 | 0.030 | 1.11 | 0.50 |
| BLYP | GGA | 0.111 | 0.052 | 1.14 | 0.37 |
| mPWPW | GGA | 0.075 | 0.035 | 1.11 | 0.10 |
| revPBE | GGA | 0.110 | 0.051 | 1.08 | 1.12 |
| BPW91 | GGA | 0.083 | 0.038 | 1.10 | 0.20 |
| RPBE | GGA | 0.119 | 0.055 | 1.07 | 0.61 |
| HCTH407 | GGA | 0.120 | 0.056 | 0.89 | 0.30 |
| SOGGA11 | GGA | 0.125 | 0.058 | 0.89 | 0.07 |
| OLYP | GGA | 0.118 | 0.055 | 0.90 | 0.36 |
| OreLYP | GGA | 0.113 | 0.053 | 0.92 | 0.20 |
| N12 | NGA | 0.027 | 0.012 | 0.99 | 0.13 |
| GAM | NGA | 0.092 | 0.046 | 0.99 | 0.13 |
| TPSS | Meta | 0.055 | 0.025 | 0.85 | 0.22 |
| revTPSS | Meta | 0.039 | 0.018 | 1.00 | 0.13 |
| M06-L | Meta | 0.080 | 0.037 | 0.73 | 0.17 |
| M11-L | Meta | 0.073 | 0.034 | 0.54 | 0.24 |
| HSE06 | Hybrid | 0.041 | 0.019 | 0.26 | 0.11 |
a
| M S(Fe, X2)c | ΔHb | ||
|---|---|---|---|
| GAM | Expt.e | ||
| a The basis set is def2-TZVP. b The binding enthalpy (a positive value indicates exothermic binding). c This column has the MS values for the central Fe and the guest molecule in the initial iteration of self-consistent field calculations. The two peripheral Fe centers where no guest is bound were taken to have MS = 2 for all the calculations. d This column is calculated by eqn (1) of ref. 8. e The most recent experimental value is shown, as discussed in the text. f NA denotes not applicable. | |||
| Fe–N2 | 2, 0 | 3.9 | 5.5 |
| Fe–O2 | 2, 1 | 10.8 | 9.8 |
| 2, 0 | 7.8 | NAf | |
| 2, −1 | 5.0 | NA | |
| Basis seta | GAM | Best estimateb |
|---|---|---|
| a The various basis sets used are: BS1 = SDD (Pd), cc-pVTZ (P, C, H); BS2 = def2-TZVP (Pd), cc-pVTZ (P, C, H); BS3 = def2-TZVP (Pd), cc-pV(T+d)Z (P), cc-pVTZ (C, H); BS4 = def2-TZVP (Pd), maug-cc-pV(T+d)Z (P), maug-cc-pVTZ (C), cc-pVTZ (H). b The best estimate was calculated in an earlier work using BCCD(T) and is described in ref. 67. | ||
| BS1 | 11.0 | 17.6 |
| BS2 | 11.1 | |
| BS3 | 11.1 | |
| BS4 | 11.1 | |
| Cu2 | Au2 | Ni2 | Pd2 | Pt2 | Ir2 | Os2 | MUE | |
|---|---|---|---|---|---|---|---|---|
| a The experimental bond length is taken from ref. 80. | ||||||||
| LSDA | 2.215 | 2.495 | 2.118 | 2.373 | 2.353 | 2.271 | 2.354 | 0.038 |
| PBE | 2.278 | 2.552 | 2.135 | 2.397 | 2.391 | 2.302 | 2.384 | 0.062 |
| B3LYP | 2.292 | 2.577 | 2.099 | 2.411 | 2.392 | 2.301 | 2.387 | 0.071 |
| B3PW91 | 2.288 | 2.552 | 2.095 | 2.367 | 2.375 | 2.287 | 2.373 | 0.068 |
| mPWPW | 2.293 | 2.549 | 2.088 | 2.359 | 2.369 | 2.282 | 2.369 | 0.068 |
| N12 | 2.224 | 2.543 | 2.110 | 2.501 | 2.366 | 2.262 | 2.282 | 0.026 |
| GAM | 2.306 | 2.543 | 2.189 | 2.536 | 2.408 | 2.283 | 2.292 | 0.050 |
| Exp.a | 2.219 | 2.472 | 2.155 | 2.480 | 2.333 | 2.270 | 2.280 | 0.000 |
6.1 Molecular energy database
Tables 4 and 5 show the mean unsigned error (MUE) for molecular energy database ME417 and its subdatabases. Note that we always compute MUEs without weighting the data; it is a straight average over the absolute deviations from the reference data of the database. In order to compare the properties that are especially important for molecular catalysis, we also combine some of existing subdatabases to form three larger subdatabases, in particular main-group bond energy (MGBE137), which includes the SR-MGM-BE9, SR-MGN-BE107, MR-MGM-BE4, and MR-MGN-BE17 subdatabases, transition metal bond energy (TMBE32), which includes the SR-TM-BE17, MR-TM-BE13, and MR-TMD-BE2 subdatabases; and barrier heights (BH76), which includes the HTBH38/08 and NHTBH38/08 subdatabases.6.2 Molecular structure database
Table 6 shows the mean unsigned error for DGL6 and DGH4 subdatabases. The last column shows the mean unsigned error for MS10, which is the overall mean unsigned error of these two subdatabases.6.3 Solid-state databases
Table 7 shows the mean unsigned errors for solid-state databases, which include LC17, SBG31, and SSCE8. Lattice constants are related to nearest neighbor distances (NNDs), but the ratio of the lattice constant to the nearest-neighbor distance depends on the crystal structure. For our larger lattice constant database, SSS47,26 we calculated an average value for this ratio of 2.15, and we use this as a typical conversion factor for discussion purposes. Therefore we also report the results for LC17 by dividing by 2.15 so that the reader can more easily compare the errors to the errors in molecular bond distances. These results are labeled NND and are discussed as mean unsigned errors in nearest neighbor distances, but the reader should keep in mind that a slightly different result would be obtained if we first converted to NND and then averaged. The rationale of having both columns in Table 7 is that the LC17 column can be directly compared to physics literature papers that report average errors in lattice constants, while the NND column allows a more physical comparison to average errors in molecular bond lengths.6.4 WCCR10 database
We show mean unsigned errors for the WCCR10 database of transition metal coordination reactions in Table 8. The mean unsigned error of GAM against WCCR10 is 6.60 kcal mol−1. This is larger than the mean unsigned error of GAM against TMBE32 subdatabase, i.e. 6.03 kcal mol−1, but still reasonable since it is the second best among the functionals tested in Table 8.6.5 Separation of O2 and N2 on Fe2(dobdc)
The performance of the newly designed functional, GAM was also tested for its ability to predict the separation of a mixture of O2 and N2 on a metal–organic framework (MOF), in particular Fe2(dobdc). The binding enthalpies for O2 and N2 bound to Fe2(dobdc) were computed using the GAM exchange–correlation functional with the def2-TZVP basis set, and then compared to experimental values57 of isosteric heat of adsorption. The values for the Fe–O2 and Fe–N2 interacting systems in their ground spin states computed at 201 K were 10.8 and 3.9 kcal mol−1, respectively, while the corresponding experimental values reported in ref. 57 are 9.8 kcal mol−1 and 8.4 kcal mol−1, respectively. Later, a more accurate experimental adsorption enthalpy for N2 bound to Fe2(dobdc) under different experimental conditions and with different procedures was reported to be 5.5 kcal mol−1.59Table 9 also presents the binding enthalpies of higher energy spin states of the Fe–O2 interacting system, which are predictions for which no experimental data is available (these calculations were necessary to be sure that the reported binding energy corresponds to lowest-energy spin state of the system).6.6 Binding energy of Pd(PH3)2C2H4
The binding of C2H4 to Pd(PH3)2 was computed using GAM and compared to the best estimate of binding energy performed in our earlier work.67 The binding energies calculated using various basis sets are reported in Table 10.6.7 Bond length of homonuclear transition metal dimer
The equilibrium bond lengths of seven transition metal dimers are calculated by GAM and six other density functionals with the LanL2DZ basis set.20–23Table 11 shows the bond length and mean unsigned error calculated by each functional.7 Other results and discussion
7.1 Convergence
In order to design a smooth functional, we add a smoothness constraint to the parameter optimization of the GAM functional. The linear coefficients optimized for GAM all have magnitudes in the range 0.1–23, which is a reasonably narrow range so there is no excessive cancellation between terms. The convergence of the new functional has been tested against our common Database 2015. In the common Database 2015, there are 483 data including ME417, MS10, SBG31, SSCE8, and LC17. If we also count the fragments that are used to calculate these data, there are more than one thousand data calculated by the GAM functional. Among the over-a-thousand data, only FeCl shows some SCF convergence issues; all other calculations converged without any problems. The smoothness of the exchange–correlation enhancement factor of the GAM functional has also been examined in plots, and it will be discussed in Section 7.3.7.2 Performance of the GAM functional
Tables 4 and 5 show that the GAM functional gives especially good results for main group bond energies, transition metal bond energies, reaction barrier heights, molecular structures, and noble gas weak interactions. Furthermore, the GAM functional provides reasonably good results for the test sets including semiconductor band gaps, solid-state cohesive energies, and transition metal coordination reactions.Table 4 shows that among LSDA, all the GGAs, and the previous NGA, the new functional GAM gives the smallest overall mean unsigned error for the entire molecular energy database ME417; the mean unsigned error is only 4.51 kcal mol−1. We also show the overall error of ME400xAE, which is the average error for the molecular energy database when we exclude absolute atomic energies, and in this case too, the GAM functional gives the smallest error among GGAs, LSDA, and NGA. We emphasize that we could reduce these total errors more, if that were our goal, but that is not our goal. Our goal is rather to obtain good performance across a broad range of databases. In order to have a functional that is especially good for studying molecular catalysis, the functional should be good for main-group bond energy (MGBE137), which includes the SR-MGM-BE9, SR-MGN-BE107, MR-MGM-BE4, and MR-MGN-BE17 subdatabases, for transition metal bond energy (TMBE32), which includes the SR-TM-BE17, MR-TM-BE13, and MR-TMD-BE2 subdatabases, for barrier heights (BH76), which includes the HTBH38/08 and NHTBH38/08 subdatabases, and for molecular structure (MS10), which includes DGL6 and DGH4 subdatabases. In Tables 4 and 5 we calculate the average error for each of these four categories by averaging the errors from each subdatabase. Among LSDA and all GGAs and NGAs, the GAM functional ranked the best for the MGBE137, TMBE32, and BH76 subdatabases. If we consider all the functionals in Tables 4 and 5, the GAM functional ranks the second best for TMBE32 subdatabase, for which M06-L is the best with an error 0.48 kcal mol−1 smaller than the GAM; the GAM functional ranks the second best for the MGBE137 subdatabase, for which M06-L is the best with an error 0.28 kcal mol−1 smaller than the GAM; and the GAM functional ranks the fifth best for BH76 subdatabase, for which M11-L is the best followed by M06-L, B3LYP, and HSE06. We note that M06-L is a meta functional, and therefore it should be better than a simpler gradient approximation, but we gave several reasons for optimizing a gradient approximation in the introduction.
In addition to the databases mentioned above, the GAM functional also provides good results for 3d transition metal atomic excitation energies, which are very hard for most available density functionals, but which we have recently shown60 can be very important for understanding metal–metal bonding. The GAM functional ranks the fifth best for the 3dAEE7 subdatabase, behind M06-L, B3LYP, PBE, and RPBE.
Next we consider noble-gas weak interactions. From Tables 4 and 5 we can see that all the functionals tested except GAM give a mean unsigned error larger than 0.081 kcal mol−1 for the NGDWI21 subdatabase, for which GAM only gives 0.019 kcal mol−1. The average value of all the noble gas weak interaction energies in our database is 0.160 kcal mol−1, which means that most functionals give an average error that is larger than 50% of the average of the reference values. The GAM functional gives the best results for NCCE30 subdatabase as compared to all tested GGAs and N12.
The GAM functional also provides the second best results for MR-TMD-BE2 (Cr2 and V2, which are known to be very hard cases for density functional theory) among all functionals tested.
Table 6 shows that the relative performance of GAM for molecular structures is not quite as good as for energies. The GAM functional ranks the 13th for MS10 subdatabase with an MUE of 0.018 Å, which is 0.002 Å larger than the average MUE of all functionals tested in Table 6. However, a more fair comparison in this case is to compare to the 12 GGAs excluding PBEsol and SOGGA (we exclude PBEsol and SOGGA at this point since their design is understood to make them better for structures than for energies, and so we do not consider them to be general-purpose functionals). As compared to the remaining group of 12 GGAs, only HCTH407 does better for DGL6 and only four of the 12 do better for MS10. The less than stellar performance of GAM on MS10 is primarily due to a large overestimation of the bond length of Ag dimer; this bond length behaves differently than other bond lengths in MS10, and success for this bond length is highly correlated to performance on lattice constants, which we downplayed. This downplay is evidenced in Table 7, which shows that the GAM functional does not give good results for the solid-state lattice constant database with a mean unsigned error of 0.046 Å for the quantities we nominally call nearest neighbor distances (NND – see Section 6.3); this error is 0.010 Å larger than the average mean unsigned error for NND. As discussed in the introduction, this results from a strategic decision to emphasize molecular energies over lattice constants in the creation of GAM. The “M” (for “molecules”) at the end of GAM is primarily to indicate our awareness that we still do not have a universally good functional, which is so far unattainable by any functional containing only density and density gradient ingredients. Nevertheless, despite not being universal, the performance of the new functional developed here is very good if we consider molecules rather than solid-state lattice constants.
Next we turn to data not used for training.
Table 7 shows that the GAM functional also shows reasonably good results for the solid-state energies databases. Among the 17 LSDA, GGAs, and NGAs, the GAM functional ranks the sixth best for the SBG31 database and fifth best for the SSCE8 database. These databases were not used for training.
In Table 8, the GAM functional ranks the second best among all functionals being tested, where the functionals tested are those chosen by the previous56 authors. The WCCR10 database includes ten transition metal coordination reactions. The molecules involved in these reactions are very large and very different from the training sets in Database 2015. The performance against these large molecules is slightly worse than that for the transition metal molecules in our training set, but within a reasonable range.
Table 9 presents the results for the performance of GAM on MOFs. We find that GAM gives good results when compared to experiments for the separation of O2 and N2 on Fe2(dobdc), with a magnitude of the deviation from experimental adsorption enthalpies of 1.0 kcal mol−1 for O2 and 1.6 kcal mol−1 for N2. It should be noted here that our training set has no data on MOFs or any other type of nanoporous materials. This average deviation from the latest experimental values is under 3 kcal mol−1 and is within experimental error. This indicates that the GAM functional shows good agreement with experimental data that are not used for training.
In Table 10, results for the binding of C2H4 to Pd(PH3)2 are presented. This datum is outside the training set. This is a difficult case for functionals; for example, BLYP gives a binding energy of 10.2 kcal mol−1 as compared to the best estimate of 17.6 kcal mol−1. Table 10 shows good stability with respect to changes in the basis set and that the GAM functional deviates from the best estimate by 6.5 kcal mol−1 with the largest basis set used. This is comparable to the 6.3 kcal mol−1 mean unsigned error for single-reference transition metal bond energies of molecules in the training set, and therefore it is an example where we obtain comparable performance inside and outside of the training set.
A very recent paper, which we considered only after our training set weights were final, reported bond distance for eight transition metal dimers, only one of which (Ag2) is in our training set. We therefore use the bond distances of the seven others as a test against data quite different from that used for training. These seven dimers, Cu2, Au2, Ni2, Pd2, Pt2, Ir2, and Os2, include two 3d metals, one 4d metals, and four 5d metals. (No 5d data were used for training.) The GAM functional gives the third best results among all the functionals tested in Table 11, with an MUE of 0.05 Å; the only functionals that do better are LSDA, which is much better for bond lengths than for molecular energies, and N12, the only previous NGA. This is very encouraging performance well outside the training set.
We also tested our new functional against the experimental bond dissociation energies of Ag2 and FeC, which are 38.0 kcal mol−1 and 88.32 kcal mol−1 respectively.61,62 The GAM functional predicts these bond dissociation energies to be 39.21 kcal mol−1 and 86.49 kcal mol−1. Li et al.62 have tested the bond dissociation energy of FeC with various functionals, and in Table 12 we add our new result to their comparison. The results in Table 12 show that the GAM functional is the second best among all 18 functionals being tested, and that many of the previous functionals have large errors for this difficult case.
| M11-L | SOGGA11 | τ-HCTHhyb | M06-L | BLYP | B3LYP | M05 | M06 | |
|---|---|---|---|---|---|---|---|---|
| FeC | −4.60 | 10.81 | −7.13 | −7.36 | 12.88 | −1.38 | 5.75 | −20.93 |
| ωB97 | ωB97X | ωB97X-D | M08-SO | M08-HX | M11 | SOGGA11-X | GAM | |
|---|---|---|---|---|---|---|---|---|
| FeC | −38.87 | −20.01 | 21.39 | −26.68 | −35.65 | −37.03 | −67.16 | 1.83 |
Recent studies pointed out that some density functionals give unstable results for large basis sets.63Fig. 1 shows the potential energy curve of Ar2 with our new GAM functional and the aug-cc-pVQZ and aug-cc-pV6Z basis sets. Fig. 1 shows that our results are very close to the reference values64 and there is no slow convergence issue with respect to the basis sets. Moreover, the excellent agreement with the reference plot shows that the GAM functional provides good results for noble gas weak interactions. This is consistent with Tables 4 and 5 showing that the GAM functional is the best for the NGDWI21 subdatabase among all the functionals tested in the present paper.
 | ||
| Fig. 1 Ar–Ar potential curve, the bonding energies are calculated with GAM/aug-cc-pVQZ and GAM/aug-cc-pV6Z level of theory. The reference is from the Tang–Toennies model. | ||
7.3 Exchange–correlation enhancement factor of the GAM functional
The enhancement factor is defined by the following equations:| Exc = ∫drεUEGx(ρ)Fxc(rs, s) | (18) |
 | (19) |
 | (20) |
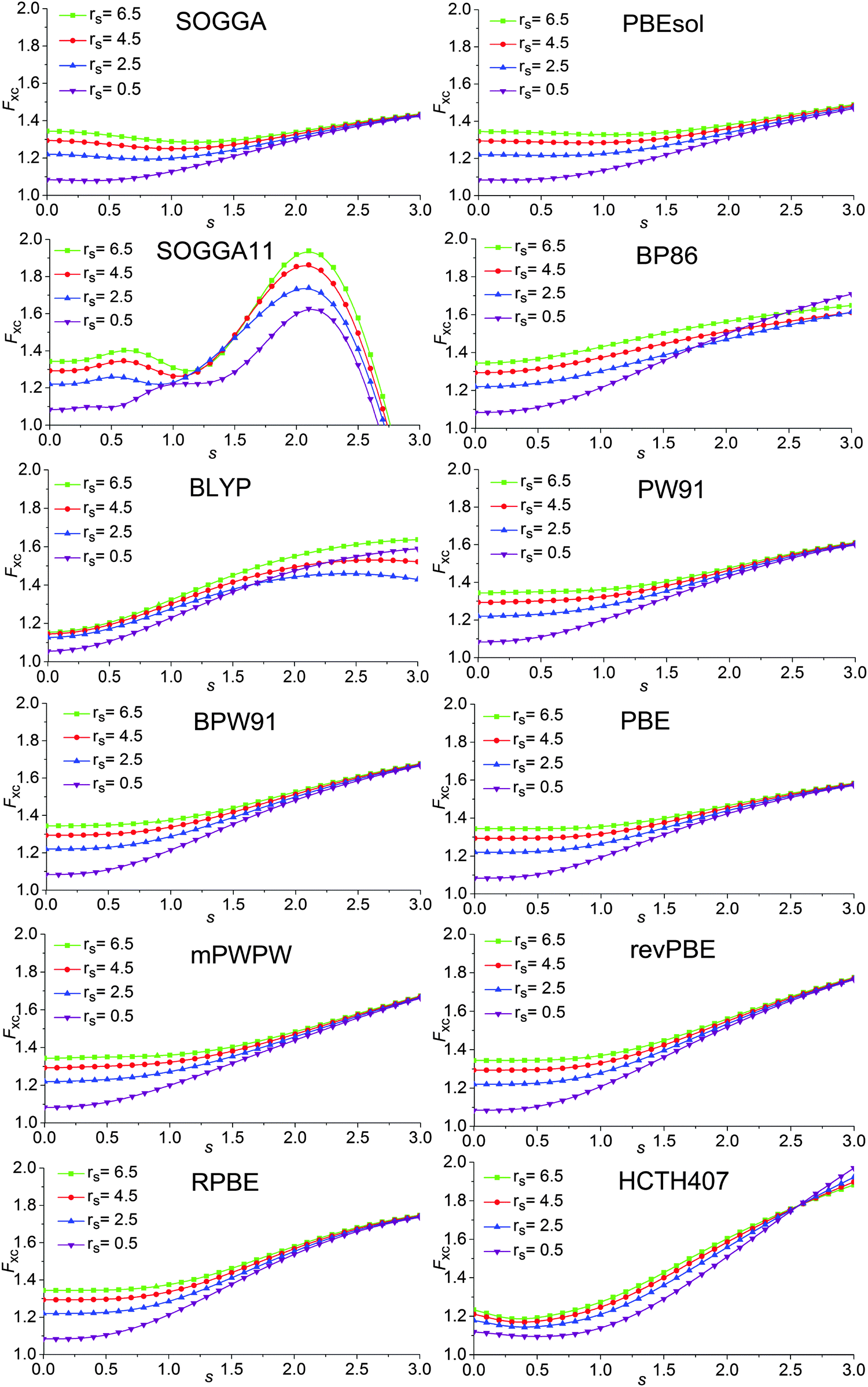 | ||
| Fig. 2 Enhancement factors of 12 GGAS. | ||
 | ||
| Fig. 3 Enhancement factor of OLYP, OreLYP, N12, and GAM. | ||
A key design element of the NGA functional form is that, unlike GGAs, we do not attempt to separately fit exchange and correlation. Therefore, unlike a GGA, we do not have a pure-exchange enhancement factor that depends only on s. However, Fig. 2 and 3 show that after we add correlation to exchange, the extent of dependence on ρ for closed-shell systems is not qualitatively different in GAM and in the GGAs.
8 Conclusions
The GAM functional has the following advantages over current GGAs and N12:(1) The GAM functional gives the smallest mean unsigned error for main group bond energies (MGBE137), transition metal bond energies (TMBE32), and reaction barrier heights (BH76).
(2) The GAM functional gives the smallest mean unsigned error of 0.019 kcal mol−1 for the noble gas dimer weak interaction energies (NGDWI21), with all the other functionals tested here giving a mean unsigned error larger than 0.081 kcal mol−1, which is about 50% of the reference value.
(3) GAM is best of any LSDA, GGA, or NGA for both the overall mean unsigned error for molecular energies, either including total atomic energies (ME417) or excluding them (ME400xAE). OreLYP (which has not previously been widely tested) and OLYP are the second and the third best.
The GAM functional gives an MUE of 0.018 Å for the molecular structure subdatabase (MS10), which is reasonable, although not outstanding.
Besides the training sets tested in the paper, we also tested the performance of the GAM functional against band gaps (SBG31), solid-state cohesive energies (SSCE8), transition metal coordination reactions (WCCR10), the bond energies of Ag2 and FeC, adsorption enthalpies of gases on MOFs, the binding of C2H4 to Pd(PH3)2, and the bond distances of homonuclear transition metal dimers (HTMD7). The last-named test includes four 5d transition metals, although no 5d transition metal data was used for training. The GAM functional does acceptably well in these tests. We conclude that the GAM functional we designed is transferable to molecular problems outside our training sets.
The linear coefficients optimized for GAM are in a narrow range of magnitude so there is no excessive cancellation between terms. The self-consistent-field convergence of the GAM functional has been tested against more than one thousand data; only one of them shows some convergence problems. The enhancement factor plot of the GAM functional is reasonably smooth.
With all these advantages over the GGAs and the previous NGA, with the advantage of an NGA requiring smaller grids than meta-GGAs or meta-NGAs, and with the advantage of an NGA requiring considerably less computation time for extended systems than hybrid functionals, we expect the GAM functional to be very useful for molecular catalysis and a wide variety of other applications to large and complex molecular systems.
Acknowledgements
The authors are grateful to Roberto Peverati, Sijie (Andy) Luo, Shaohong Li, Wei-Guang Liu, and Kaining Duanmu for helpful contributions to various stages of this work. This work was supported in part by the U.S. Department of Energy, Office of Basic Energy Sciences, and Division of Chemical Sciences under award DE-SC0012702. This work was supported in part by a Molecular Science Computing Facility Computational Grand Challenge grant at the Environmental Molecular Sciences Laboratory, supported by DOE at Pacific Northwest National Laboratory. HY acknowledges partial support by a Graham N. Gleysteen Excellence Fellowship. PV acknowledges partial support by a Phillips Excellence Fellowship and a Doctoral Dissertation Fellowship.References
- W. Kohn, A. D. Becke and R. G. Parr, J. Phys. Chem., 1996, 100, 12974–12980 CrossRef CAS.
- D. C. Langreth and M. J. Mehl, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 1809–1834 CrossRef CAS.
- J. P. Perdew and Y. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8800–8802 CrossRef.
- R. Peverati and D. G. Truhlar, J. Chem. Theory Comput., 2012, 8, 2310–2319 CrossRef CAS.
- Y. Zhao, R. Peverati, K. Yang, S. Luo and D. G. Truhlar, MN-GFM, version 6.4: Minnesota Gaussian Functional Module, University of Minnesota, Minneapolis, MN, 2012 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision A.1, Gaussian, Inc., 2009 Search PubMed.
- K. Kudin and G. E. Scuseria, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 16440–16453 CrossRef CAS.
- P. Verma, X. Xu and D. G. Truhlar, J. Phys. Chem. C, 2013, 117, 12648–12660 CAS.
- D. Andrae, U. Häussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- D. E. Woon and T. H. Dunning Jr., J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS PubMed.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- T. H. Dunning, Jr., K. A. Peterson and A. K. Wilson, J. Chem. Phys., 2001, 114, 9244 CrossRef PubMed.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045 CrossRef CAS PubMed.
- E. Papajak, H. R. Leverentz, J. Zheng and D. G. Truhlar, J. Chem. Theory Comput., 2009, 5, 1197–1202 CrossRef CAS.
- E. Papajak and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 597 CrossRef CAS.
- K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283–296 CrossRef CAS.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227–244 CrossRef CAS PubMed.
- E. Papajak and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 10–18 CrossRef CAS.
- T. H. Dunning Jr. and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum, New York, 1977, vol. 3, pp. 1–28 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 284–298 CrossRef PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS PubMed.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef PubMed.
- T. van Mourik and T. H. Dunning, Jr., Int. J. Quantum Chem., 2000, 76, 205–221 CrossRef CAS.
- R. Peverati and D. G. Truhlar, Philos. Trans. R. Soc., A, 2014, 372, 20120476 CrossRef PubMed.
- W. Kohn and L. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- (a) R. Gáspár, Acta Phys. Hung., 1954, 3, 263–286 CrossRef; (b) R. Gáspár, Acta Phys. Hung., 1974, 35, 213–218 CrossRef.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2008, 128, 184109 CrossRef PubMed.
- J. P. Perdew, A. Ruzsinsky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, in Electronic Structure of Solids ’91, ed. P. Ziesche and H. Eschrig, Akademie Verlag, Berlin, 1991, pp. 11–20 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1997, 108, 664–675 CrossRef PubMed.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1997, 80, 890 CrossRef.
- B. Hammer, L. Hansen and J. Norskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- A. D. Boese and N. C. Handy, J. Chem. Phys., 2000, 114, 5497–5503 CrossRef PubMed.
- R. Peverati, Y. Zhao and D. G. Truhlar, J. Phys. Chem. Lett., 2011, 2, 1991–1997 CrossRef CAS.
- N. Handy and A. Cohen, Mol. Phys., 2001, 99, 403–412 CrossRef CAS.
- A. J. Thakkar and S. P. McCarthy, J. Chem. Phys., 2009, 131, 134109 CrossRef PubMed.
- J. M. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, L. A. Constantin and J. Sun, Phys. Rev. Lett., 2009, 103, 026403 CrossRef.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- R. Peverati and D. G. Truhlar, J. Phys. Chem. Lett., 2011, 3, 117–124 CrossRef.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8027–8215 CrossRef PubMed.
- T. M. Henderson, A. F. Izmaylov, G. Scalmani and G. E. Scuseria, J. Chem. Phys., 2009, 131, 044108 CrossRef PubMed.
- D. J. Tozer and N. C. Handy, J. Phys. Chem. A, 1988, 102, 3162–3168 CrossRef.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef.
- H. Stoll and C. Pavlidou, Theor. Chim. Acta, 1978, 149, 143–149 CrossRef.
- H. Stoll and E. Golka, Theor. Chim. Acta, 1980, 55, 29–41 CrossRef CAS.
- T. Weymuth, E. P. A. Couzijn, P. Chen and M. Reiher, J. Chem. Theory Comput., 2014, 10, 3092–3103 CrossRef CAS.
- E. D. Bloch, L. J. Murray, W. L. Queen, S. Chavan, S. N. Maximoff, J. P. Bigi, R. Krishna, V. K. Peterson, F. Grandjean, G. J. Long, B. Smit, S. Bordiga, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14814–14822 CrossRef CAS PubMed.
- N. C. Handy, J. A. Pople, M. Head-Gordon, K. Raghavachari and G. W. Trucks, Chem. Phys. Lett., 1989, 164, 185–192 CrossRef CAS.
- K. Lee, W. C. Isley III, A. L. Dzubak, P. Verma, S. J. Stoneburner, L.-C. Lin, J. D. Howe, E. D. Bloch, D. A. Reed, M. R. Hudson, C. M. Brown, J. R. Long, J. B. Neaton, B. Smit, C. J. Cramer, D. G. Truhlar and L. Gagliardi, J. Am. Chem. Soc., 2014, 136, 698–704 CrossRef CAS PubMed.
- W. Zhang, D. G. Truhlar and M. Tang, J. Chem. Theory Comput., 2014, 10, 2399–2409 CrossRef CAS.
- V. Beutel, H. G. Krämer, G. L. Bhale, M. Kuhn, K. Weyers and W. Demtröder, J. Chem. Phys., 1993, 98, 2699–2708 CrossRef CAS PubMed.
- R. Li, R. Peverati, M. Isegawa and D. G. Truhlar, J. Phys. Chem. A, 2012, 117, 169–173 CrossRef PubMed.
- N. Mardirossian and M. Head-Gordon, J. Chem. Theory Comput., 2013, 9, 4453–4461 CrossRef CAS.
- K. T. Tang and J. P. Toennies, J. Chem. Phys., 2003, 118, 4976–4983 CrossRef CAS PubMed.
- H. S. Yu and D. G. Truhlar, J. Chem. Theory Comput., 2015 Search PubMed , submitted.
- W. Zhang, D. G. Truhlar and M. Tang, J. Chem. Theory Comput., 2013, 9, 3965–3977 CrossRef CAS.
- B. Averkiev, Y. Zhao and D. G. Truhlar, J. Mol. Catal. A: Chem., 2010, 80–88 CrossRef CAS PubMed.
- X. Xu, W. Zhang, M. Tang and D. G. Truhlar, J. Chem. Theory Comput., 2015 DOI:10.1021/acs.jctc.5b00081.
- S. Luo, B. Averkiev, K. R. Yang, X. Xu and D. G. Truhlar, J. Chem. Theory Comput., 2014, 10, 102–121 CrossRef CAS.
- O. A. Vydrov and T. V. Voorhis, J. Chem. Theory Comput., 2012, 8, 1929–1934 CrossRef CAS.
- K. M. Lange and J. R. Lane, J. Chem. Phys., 2011, 134, 034301 CrossRef PubMed.
- J. D. McMahon and J. R. Lane, J. Chem. Phys., 2011, 135, 154309 CrossRef PubMed.
- M. S. Marshall, L. A. Burns and C. D. Sherrill, J. Chem. Phys., 2011, 135, 194102 CrossRef PubMed.
- K. T. Tang and J. P. Toennies, J. Chem. Phys., 2003, 118, 4976–4983 CrossRef CAS PubMed.
- H. S. Yu and D. G. Truhlar, J. Chem. Theory Comput., 2014, 10, 2291–2305 CrossRef CAS.
- S. Luo and D. G. Truhlar, J. Chem. Theory Comput., 2012, 8, 4112–4126 CrossRef CAS.
- K. Yang, R. Peverati, D. G. Truhlar and R. J. Valero, J. Chem. Phys., 2011, 135, 044188 Search PubMed.
- T. Schwabe, Phys. Chem. Chem. Phys., 2014, 16, 14559–14567 RSC.
- http://cccbdb.nist.gov/expbondlengths1.asp, accessed on Oct. 29, 2014.
- A. Posada-Borbón and A. Posada-Amarillas, Chem. Phys. Lett., 2015, 618, 66–71 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A complete description of Database 2015 and the geometries and basis sets used with it. See DOI: 10.1039/c5cp01425e |
| This journal is © the Owner Societies 2015 |