
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of cationic molecules on the oxygen reduction reaction on fuel cell grade Pt/C (20 wt%) catalyst in potassium hydroxide (aq, 1 mol dm−3)†
Ai Lien
Ong
*,
Kenneth K.
Inglis
,
Daniel K.
Whelligan
,
Sam
Murphy
and
John R.
Varcoe
Department of Chemistry, Faculty of Engineering and Physical Sciences, University of Surrey, Guildford, GU2 7XH, UK. E-mail: ongailien@hotmail.com; Tel: +44 (0)1483 686838
First published on 31st March 2015
Abstract
This study investigates the effect of 1 mmol dm−3 concentrations of a selection of small cationic molecules on the performance of a fuel cell grade oxygen reduction reaction (ORR) catalyst (Johnson Matthey HiSPEC 3000, 20 mass% Pt/C) in aqueous KOH (1 mol dm−3). The cationic molecules studied include quaternary ammonium (including those based on bicyclic systems) and imidazolium types as well as a phosphonium example: these serve as fully solubilised models for the commonly encountered head-groups in alkaline anion-exchange membranes (AAEM) and anion-exchange ionomers (AEI) that are being developed for application in alkaline polymer electrolyte fuel cells (APEFCs), batteries and electrolysers. Both cyclic and hydrodynamic linear sweep rotating disk electrode voltammetry techniques were used. The resulting voltammograms and subsequently derived data (e.g. apparent electrochemical active surface areas, Tafel plots, and number of [reduction] electrons transferred per O2) were compared. The results show that the imidazolium examples produced the highest level of interference towards the ORR on the Pt/C catalyst under the experimental conditions used.
Introduction
Growing concerns about climate change and energy security has progressively become the driver for the development of sustainable renewable energy technologies (such as fuel cells).1 Alkaline anion-exchange membranes (AAEM) and anion-exchange ionomers (AEI) are rapidly being developed2 for use as the ionic conducting separators and binders in technologies such as alkaline polymer electrolyte fuel cells (APEFC),3 electrolysers,4 and in various batteries.5 To minimise ionic resistances in the catalyst layer (to improve electrode performances by optimising the triple phase boundaries), the AEIs and catalysts are in intimate contact. It is therefore essential to evaluate the effect of the AEI chemistry on the catalyst performance.The chemistries of the cationic (anion-exchange) groups being evaluated for use in AAEMs and AEI are diverse. Reports on the development of AAEMs with OH− ion conductivities of ca. 0.1 S cm−1 are becoming increasingly common. Examples include: the most commonly studied quaternary ammonium types with conductivities of up to 111 mS cm−1 at 50 °C6 and 65 mS cm−1 at room temperature,7 imidazolium examples with conductivities up to 100 mS cm−1 at 80 °C,8 and quaternised 1,4-diazabicyclo[2.2.2]octane (DABCO) types9 giving 88 mS cm−1 at room temperature. Other AAEM/AEI head-group chemistries of interest include stabilised quaternary phosphoniums10 and quinuclidiums.11
A preceding (published) study investigated the effect of model cationic molecules (such as tetramethylammonium [TMA] and benzyltrimethylammonium [BTMA]) on the oxygen reduction reaction (ORR) on a model polycrystalline platinum (Ptpc) rotating disk electrodes (RDE).12 This ideal system is far removed from fuel cells, which use high surface area nanocatalysts (e.g. carbon supported Pt nanoparticles) but it did produce interesting initial findings. For example, a cationic molecule containing imidazolium chemistry fundamentally changed the mechanism of ORR on Ptpc, while the presence of benzene rings in close proximity to quaternary ammonium groups also had a negative effect on the ORR. These effects need to be studied in more detail with fuel cell relevant catalysts (for scientific completeness). A prior study by Shao et al. investigated the effect of 1-methylimidazole and triethylamine on the ORR and hydrogen oxidation reaction (HOR) on Pt/C in aqueous KOH:13 however, they did not investigate the effect of the related charged imidazolium or quaternary ammonium species. A more relevant prior study14 showed that BTMA had a stronger negative affect towards the methanol oxidation reaction (MOR) in aqueous KOH (0.1 mol dm−3 containing 0.05 mol dm−3 methanol) than TMA over the additive concentration range 1–120 mmol dm−3.
In this follow-up ex situ study, an extended range of cationic model small molecules (Scheme 1, all with Cl− anions) were investigated for their effects on the ORR on the more fuel cell relevant Pt/C (20 mass%) catalyst: (a) quaternary trimethylammonium types [TMA, BTMA, MBTMA = 1-(4-methylbenzyl)trimethylammonium, and BOTMHA = 6-(benzyloxy)-N,N,N-trimethylhexan-1-aminium], (b) caged quaternary ammonium types [BAABCO = 1-benzyl-4-aza-1-azoniabicyclo[2.2.2]octane, MBAABCO = 1-(4-methylbenzyl)-4-aza-1-azoniabicyclo[2.2.2]octane, and MBABCO = 1-(4-methylbenzyl)-1-azoniabicyclo[2.2.2]octane (quinuclidinium)], (c) imidazolium types [BMI = 1-benzyl-3-methylimidazolium and MBMI = 1-(4-methylbenzyl)-3-methylimidazolium], and (d) a phosphonium exemplar [BTTMPP = benzyltris(2,4,6-trimethoxyphenyl)phosphonium]. The para-methyl examples were studied with a mind on the fact that pendent cationic head-groups in AAEMs/AEIs involve benzene rings that are bound to both the polymer backbone and the anion-exchange groups (i.e. the benzene rings are bound to other carbon atoms via >1 benzene-ring-carbons).
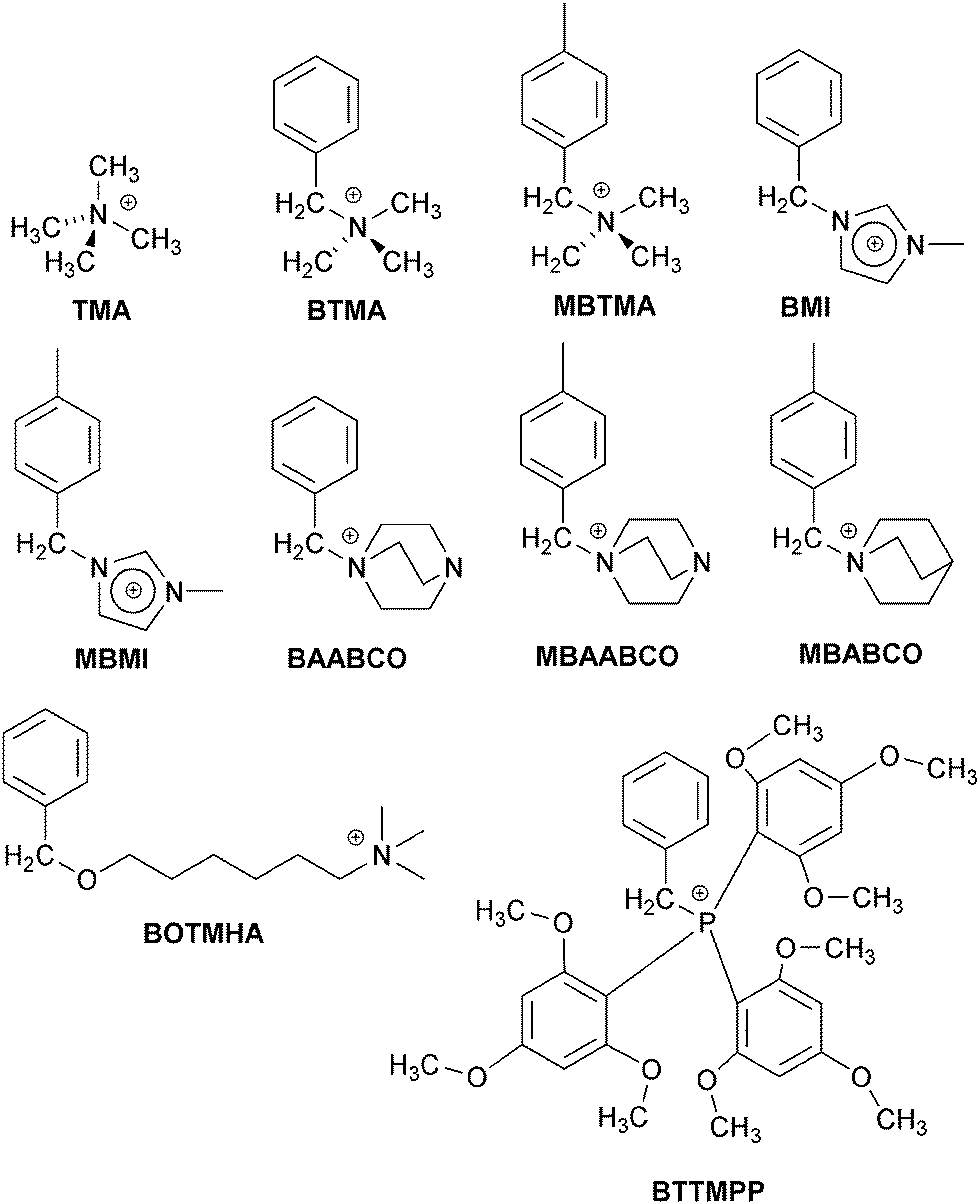 | ||
| Scheme 1 The structures of the model cationic molecules studied: TMA = tetramethylammonium, BTMA = benzyltrimethylammonium, MBTMA = 1-(4-methylbenzyl)trimethylammonium, BMI = 1-benzyl-3-methylimidazolium, MBMI = 1-(4-methylbenzyl)-3-methylimidazolium, BAABCO = 1-benzyl-4-aza-1-azoniabicyclo[2.2.2]octane, MBAABCO = 1-(4-methylbenzyl)-4-aza-1-azoniabicyclo[2.2.2]octane, MBABCO = 1-(4-methylbenzyl)-1-azoniabicyclo[2.2.2]octane, BOTMHA = 6-(benzyloxy)-N,N,N-trimethylhexan-1-aminium, and BTTMPP = benzyltris(2,4,6-trimethoxyphenyl)phosphonium. Cl− counter anions were used throughout (KCl was used in control experiments). | ||
Experimental
Chemicals and materials
Aqueous KOH solution (1 mol dm−3), KCl, TMACl, BTMACl, and BMICl were obtained from Sigma-Aldrich and used as received. The BOTMHACl and BAABCOCl were synthesised in the previous study.12 The MBTMACl, MBAABCOCl, MBMICl, MBABCOCl, and BTTMPPCl were also synthesised in-house (see ESI†). The electrolyte solutions used in electrochemical experiments contained aqueous KOH (1 mol dm−3) with and without the addition of the cationic molecules under study (1 mmol dm−3 unless otherwise stated): control experiments involved aqueous KOH (1 mol dm−3) containing KCl (1 mmol dm−3) to check for any Cl− anion effects. N2 and O2 purging of the electrolytes was conducted where appropriate and as detailed below. Grade I deionised water (resistivity = 18.2 MΩ cm) was used for electrode cleaning and all rinsing steps.Electrochemical instrumentation and electrode preparation
Voltammetric measurements were performed in a conventional three-electrode water-jacketed electrochemical cell (Princeton Applied Research, supplied by Ametek). Reference electrodes (RE) were HydroFlex Reversible Hydrogen Electrodes (RHE) obtained from Gaskatel (Germany) [i.e. the potential scale stays the same even with shifts in pH]. The potential of the RHE was continuously checked (before and after each experiment) using a saturated calomel laboratory master reference electrode (Gamry Instruments USA). An annealed Pt wire (Advent Research Materials) with diameter ∅ = 0.5 mm and length = 0.5 m was coiled and used as a counter electrode.Prior to ink deposition, a commercial glassy-carbon rotating disk electrode (GC-RDE) with a geometrical surface area = 0.196 cm2 (Princeton Applied Research, supplied by Ametek) was prepared by gently polishing with 0.05 µm alumina slurry (BASi) for at least 4 min, rinsing intensively with water, removing the excess water via shaking, and drying in the atmosphere (at room temperature and pressure). The catalyst ink was prepared by ultrasonically dispersing 10 mg Pt/C catalyst (20 mass%, Johnson Matthey HiSPEC 3000) for 1 h in 5 mL of an aqueous solution consisting of: 3.98 mL water (ρ = 18.2 MΩ cm), 1 mL iso-propanol, and 20 µL of a 5 mass% Nafion® dispersion (Sigma-Aldrich, dispersion in a mixture of lower aliphatic alcohols and water).15
The catalyst ink was ultrasonicated for 2 min and a 10 µL droplet of the ink was subsequently pipetted onto the prepared GC-RDE. The catalyst ink coated GC-RDE was then carefully mounted on an inverted shaft of the rotator (Ring-Disk Electrode System model 636 rotator of Princeton Applied Research, supplied by Ametek) and rotationally dried (in the atmosphere) at 400 rpm.16 The platinum (Pt) loading on the GC-RDE working electrode (WE) was calculated as 20 µg cmgeo−2. In this study of the relative effects of the addition of the different cationic species, each set of data (blank run [aqueous KOH only] or each experiment with an added cationic Cl−species) was collected on a freshly catalysed GC-RDE where the base GC-RDE used was the same (cleaned and polished in between each experiment after removal of the prior experiment's catalyst coating).
For electrochemical cleaning, each WE was wetted with deionised water (18.2 MΩ cm) and then subjected to cyclic voltammetry (CV) cycling in N2-saturated aqueous KOH (1 mol dm−3) over a potential range of 0.05–1.2 V vs. RHE at a sweep rate of 50 mV s−1 for ca. 15 cycles (until a stable CV response was obtained). Before data collection, the repeatability response of each catalyst coated GC-RDE was strictly ensured by repeating the above electrochemical cleaning procedure and monitoring the final stable CV responses [in additive-(cationic species)-free aqueous KOH (1 mol dm−3)].
The potentials were controlled using an IviumStat potentiostat (Ivium Technologies Netherlands, supplied by Alvatek UK). The rotation speed of RDE working electrode was varied in the range 400–2000 rpm. All experiments were conducted at a controlled temperature of 25 °C. Measurements were iR-corrected for the uncompensated ohmic drops by means of recording electrochemical impedance spectra from 100 → 0.1 kHz with a voltage perturbation of 10 mVrms. The high frequency x-axis intercept (real part) of the Nyquist plot was taken as the uncompensated resistance.
Cyclic voltammetry (CV)
The aqueous electrolytes being used were deaerated by thorough purging with N2 gas and ensuring the N2 atmosphere above the electrolytes was maintained throughout the experiments. CV curves were recorded by cycling in the potential region +0.05 to +1.20 V vs. RHE at a scan rate of 50 mV s−1; wider potential windows were also probed by increasing the anodic (upper) potential up to +1.50 V in steps of 0.05 V. The electrochemical active surface area (ECSA cm−2) of each WE was determined from the integrated charge required for hydrogen underpotential adsorption/desorption in the CVs in additive-free aqueous KOH (1 mol dm−3):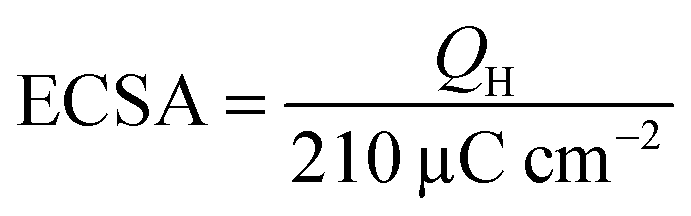 | (1) |
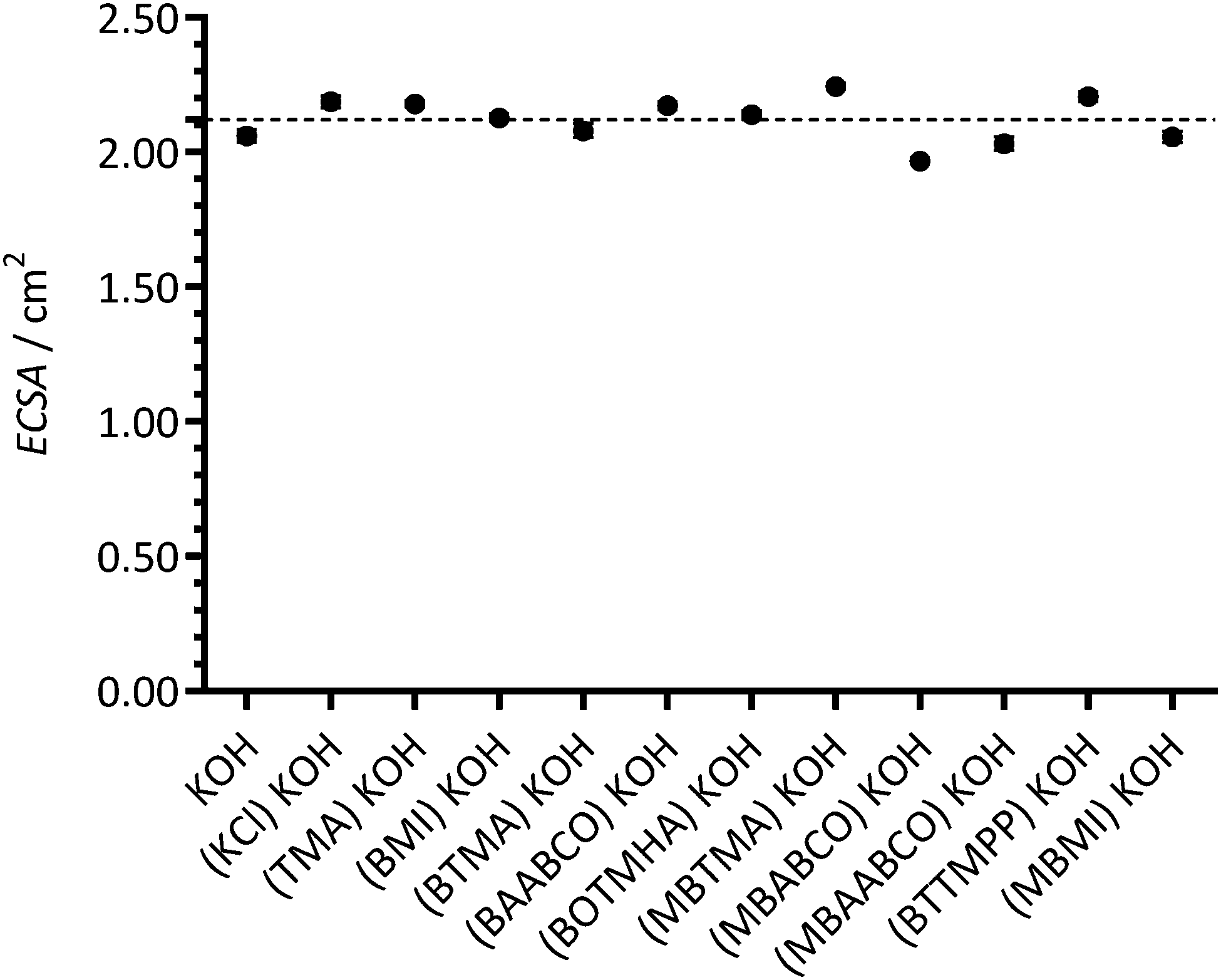 | ||
| Fig. 1 The ECSAs of each Pt/C-coated GC-RDE WE in N2-saturated aqueous KOH (1 mol dm−3) electrolytes at 25 °C before addition of 1 mmol dm−3 of the bracketed model cationic molecules (or KCl). Scan rate = 50 mV s−1. Error bars are the confidence intervals at the 95% confidence level (n = 3 repeated CVs on each GC-RDE WE). The dashed line gives the mean value of all ECSAs collected with all WEs. | ||
The apparent ECSAs were also calculated by using CV data collected with N2-purged electrolytes containing the organic cationic species (or KCl) being studied with each WE: this was to investigate the effect of the additive species under study on the experimentally measured ECSA (with the assumption that the hydrogen desorption under such conditions [in the presence of such species] is a reliable measure of ECSA). Note: these apparent ECSAs measured with the presence of additive species were not used to normalise current density data.
ORR hydrodynamic RDE linear sweep voltammetry (LSV)
The aqueous KOH electrolytes (with and without the addition of cationic species [and KCl]) were initially deaerated by thorough purging with N2 gas followed by a thorough O2 purge in order to obtain O2-saturation. An O2 atmosphere was maintained above the electrolyte surface throughout the subsequent measurements. The ORR activities of the catalyst-coated RDE were evaluated using LSV: the potentials were swept from +1.03 V to +0.20 V vs. RHE at a scan rate of 5 mV s−1 and the experiments were repeated with rotation rates increasing from 400 to 2000 rpm in 200 rpm steps. Kinetic currents were determined using the Koutecký–Levich equation (eqn (2))17 for processes under mixed kinetic (activation) and mass-transport control: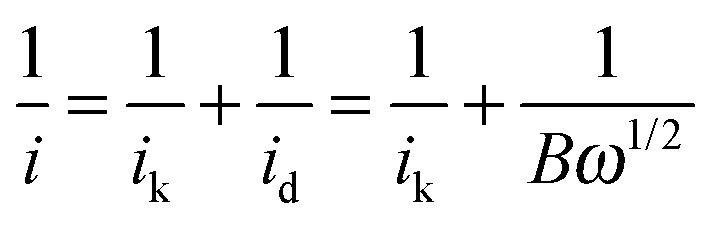 | (2) |
The overall number of electrons transferred (n) per O2 for the ORR was calculated from the eqn (3) by taking the Levich slope B from the straight line of idvs. ω1/2 (where ω is in rpm) at potentials in diffusion-controlled region:
| B = 0.2nAGEOFCoDo2/3ν−1/6 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), Co is the bulk concentration of O2 dissolved in electrolyte (taken here as = 8.60 × 10−7 mol cm−3), Do is the O2 diffusion coefficient (taken here as = 2.04 × 10−5 cm2 s−1), and ν is the kinematic viscosity (taken here as = 0.93 cm2 s−1); these values are taken from literature.18 The assumption used in this study for this simplistic analysis is that the added species did not affect the properties of bulk KOH (1 mol dm−3): this may not be the case (see in particular the data for the bulky [high molecular weight] phosphonium additive).
485 C mol−1), Co is the bulk concentration of O2 dissolved in electrolyte (taken here as = 8.60 × 10−7 mol cm−3), Do is the O2 diffusion coefficient (taken here as = 2.04 × 10−5 cm2 s−1), and ν is the kinematic viscosity (taken here as = 0.93 cm2 s−1); these values are taken from literature.18 The assumption used in this study for this simplistic analysis is that the added species did not affect the properties of bulk KOH (1 mol dm−3): this may not be the case (see in particular the data for the bulky [high molecular weight] phosphonium additive).
The kinetic currents were used to produce Tafel plots and perform Tafel analyses viaeqn (4):17
| E = Eo − blogik | (4) |
| Eo = Er + blogi0 | (5) |
Results and discussion
Cyclic voltammetry (CV)
The effect of 1 mmol dm−3 concentrations of the cationic model molecules (Scheme 1) in aqueous KOH (1 mol dm−3) electrolyte on the electrochemical behaviour of the Pt/C-coated GC-RDE was initially studied using cyclic voltammetry (Fig. 2). The changes in the CVs in the N2-purged electrolytes for the following potential regions are discussed below: (1) Pt-hydrogen adsorption/desorption region (+0.05 to +0.45 V vs. RHE); (2) double-layer region (+0.45 to +0.60 V vs. RHE); and (3) Pt-oxide region (>+0.60 V vs. RHE).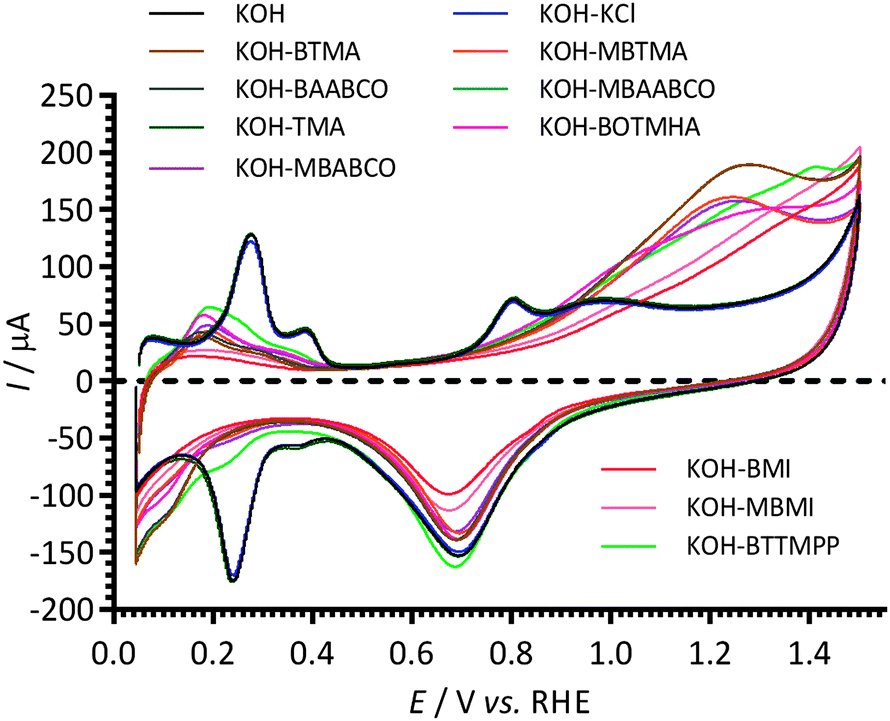 | ||
| Fig. 2 Cyclic voltammograms of a Pt/C-coated GC disk electrode in N2-saturated aqueous KOH (1 mol dm−3) electrolytes at 25 °C with and without addition of 1 mmol dm−3 of cationic molecules and KCl (control experiment for Cl− spectator anions). Scan rate = 50 mV s−1. | ||
The Pt-hydrogen peaks at +0.27 V and +0.38 V vs. RHE with the KCl and TMA containing electrolytes were similar to those recorded for the blank KOH (no additives); however, they were poorly defined with the presence of BTTMPP, BOTMHA, MBABCO, BTMA, MBTMA, BAABCO and MBAABCO, and totally suppressed with the electrolytes containing BMI and MBMI. There was also suppression of the anodic peak at +0.80 V vs. RHE on addition of all organic cationic species.
With the imidazolium-containing electrolytes, larger anodic currents were observed above +1.1. V vs. RHE (above the background levels for the oxidation of the Pt catalyst surfaces observed with the additive-free aqueous KOH blank) and there was also a suppression of the Pt oxidation features above +0.88 V vs. RHE (Pt oxidation features that were observed in the KOH blank experiment). Even higher anodic currents were observed above +0.9 V vs. RHE with the other organic cationic species. There was no apparent correlation between the intensity of these additional oxidation anodic peaks (0.6–1.5 V vs. RHE) with the related cathodic peak intensities in the reverse scan direction (at ca. 0.7 V vs. RHE, Pt-oxide-related with additive-free aqueous KOH). However, when the CV potentials on the forward (anodic) sweep were terminated at lower potentials, such that the Pt surface oxidation had initiated but not plateaued, the intensities of the oxide reduction peaks were smaller with the presence of BMI, and MBMI; this indicates that the surface coverage of Pt-oxide species was less in the presence of imidazolium species.
To study the above in more detail, Fig. 3 compares CVs of the Pt/C-coated GC-RDE WEs cycled to progressively higher anodic potentials (up to +1.50 V vs. RHE) with the different electrolytes. These anodic currents peak at ca. +1.25 V to +1.30 V vs. RHE with the electrolytes containing MBTMA, MBAABCO, MBABCO, BTMA, BOTMHA, and BAABCO: a peak was not observed until +1.40 V vs. RHE with the P-based BTTMPP and no peaks were observed at all with BMI and MBMI. In contrast to the other cationic species, the anodic currents observed with the hexyloxy-spacer-chain species (BOTMHA [Fig. 3D]) and the para-methylbenzyl quaternary ammonium examples (MBTMA [Fig. 3F], MBAABCO [Fig. 3G], and MBABCO [Fig. 3H]) superimposed at +1.5 V vs. RHE with the oxygen evolution currents observed with the additive-free KOH blanks. In addition, the reduction peaks in the reverse scans (potential range 0.9–0.45 V vs. RHE) all increased in intensity when the upper anodic potential limits were increased. These cathodic peaks also shift to less positive peak potentials on increased upper anodic limits (most significantly with the BMI and MBMI species and less significantly with the N-free exemplar [BTTMPP], TMA, and the KCl control). BTTMPP and TMA were also the only examples to yield cathodic peak currents that match or exceeded those observed with the additive-free KOH blanks: in contrast, the most suppressed cathodic peak currents were observed with the imidazolium examples.
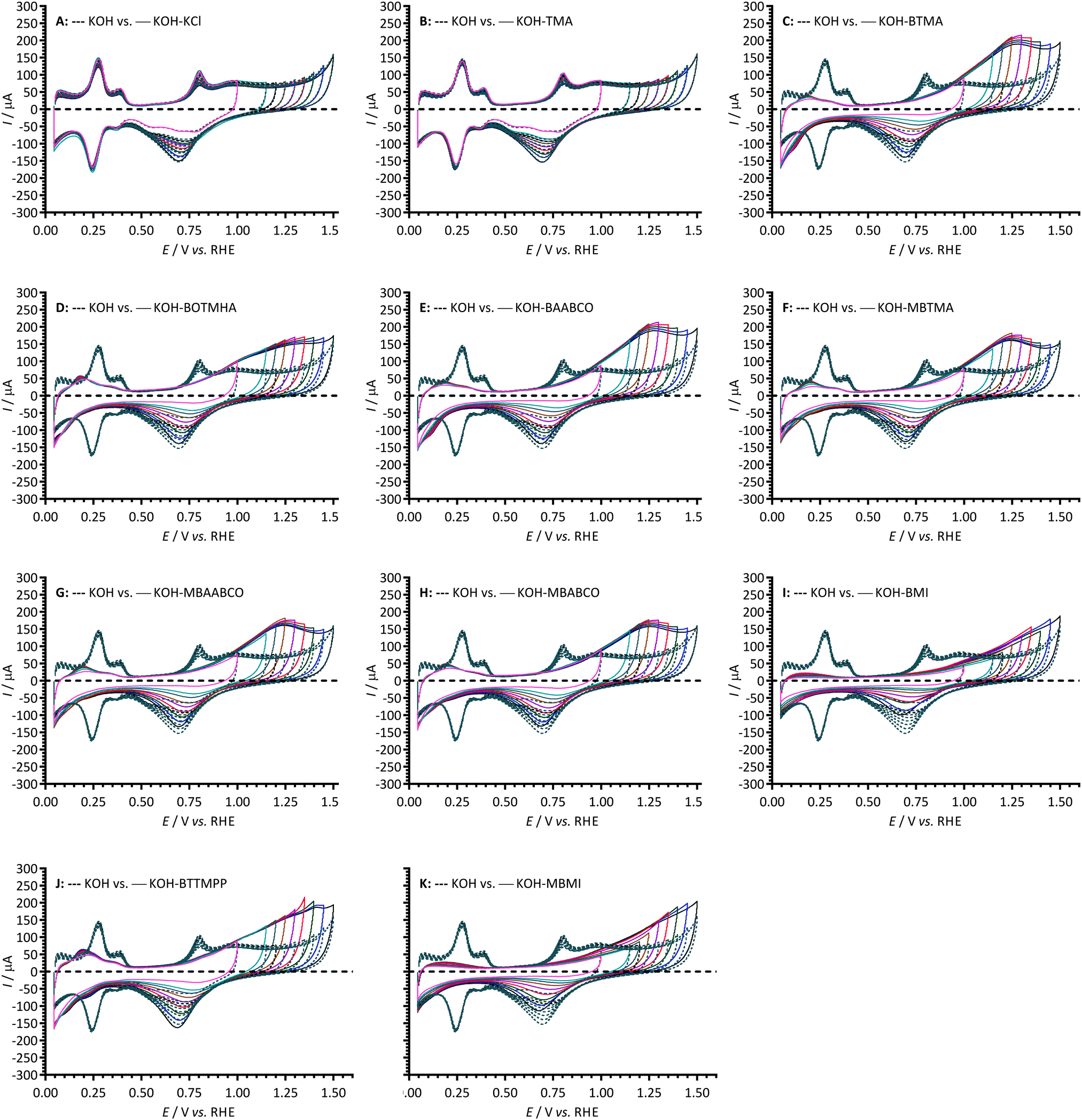 | ||
| Fig. 3 Cyclic voltammograms of Pt/C-coated GC disk electrodes in N2-saturated aqueous KOH (1 mol dm−3) electrolyte at 25 °C without (--- dashed lines) and with addition (— solid lines) of 1 mmol dm−3 of: (A) KCl, (B) TMA, (C) BTMA, (D) BOTMHA, (E) BAABCO, (F) MBTMA, (G) MBAABCO, (H) MBABCO, (I) BMI, (J) BTTMPP and (K) MBMI. The CVs were recorded with progressively higher upper potential limits (Ef) up to 1.5 V vs. RHE. Scan rate = 50 mV s−1. | ||
For more in-depth analysis, the cathodic charge densities (QR) were calculated from the integration of the cathodic peaks in Fig. 3 (potential range +0.9 to +0.45 V vs. RHE) with the assumption that they reflect the surface coverage of oxygen-containing species on the Pt surfaces (normalisation was to the ECSAs measured for each WE in additive-free aqueous KOH): Fig. 4 presents this data as a function of Ef (the upper anodic potential limit). Inflection points in the plots were observed in the Ef range +1.15 to +1.3 vs. RHE (reduction charge density range of 0.02–0.08 mC cm−2) for all the benzene-containing species but not with the additive-free aqueous KOH blank and the electrolytes containing TMA and KCl. The data indicates that more than one oxidation process is occurring on the forward sweep with the presence of the benzene-containing species: this affects the coverage of oxygen-species on the active surfaces of Pt catalysts. Significantly lower QR values (and assumed oxide coverages) were observed in the electrolytes containing BMI and MBMI compared to the other benzene-containing species.
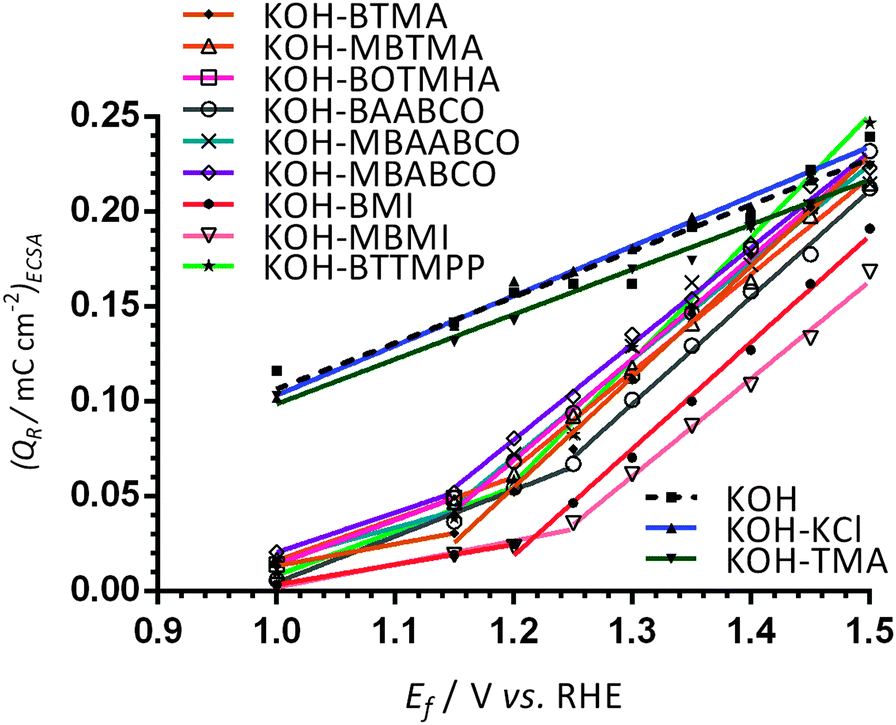 | ||
| Fig. 4 The charge densities QR at 25 °C obtained by integration of the reduction peaks (potential range +0.9 to +0.45 V vs. RHE) in the reverse scans of CVs presented in Fig. 3 (with increasing upper anodic potential limits [Ef]). The Pt/C-coated GC disk electrodes were submerged in N2-saturated aqueous KOH (1 mol dm−3) electrolytes with and without the addition of 1 mmol dm−3 of organic cationic species (and KCl). | ||
All of this CV data suggests that the imidazolium species are affecting the Pt/C catalysts in a fundamentally different way to the benzene-containing quaternary ammonium species (the benzene-ring-free TMA-containing electrolyte does not produce a significant change in behaviour compared to the KCl-containing and KOH only electrolytes). Taking into account the prior study that used a model PtPc disk electrode instead of Pt/C coated GC-RDEs, these results confirm that the benzene-containing cationic species are (not unexpectedly) being oxidised by Pt catalysts at high potentials, including at fuel cell relevant potentials (0.9–1.1 V) with a number of the cationic species. Therefore it must be kept in mind that, as well as the cationic species added to the electrolytes, the presence of electro-oxidation (degradation) products could affect the ORR on Pt catalysts. Future studies should consider this when evaluating the effect on the catalysts of actual AEIs, i.e. polymer bound cationic groups, rather than small molecule fully dissolved cationic species (see the discussion at the end of this paper). This is because there is a risk that polymer-bound cationic head-groups may also be electro-oxidised in the presence of catalyst on the fuel cell cathodes (especially at higher cathode potentials). These results may well be telling us that Pt-based catalysts are the worst option to use with systems containing anion-exchange polymer electrolytes with such cationic head-groups (as Pt is very good at catalysing a very broad range of species); however, later results in this paper, with a simple model Ag/C catalyst, indicate that lower performances can result from the use of alternative (non-Pt-group) metal catalysts.
Fig. 5 presents the apparent percentage ECSA remaining (%ECSARemain) when the Pt/C-coated GC disk electrodes were studied in the electrolytes containing the additives (normalised to the initial ECSAs measured in the additive-free blank KOH electrolytes). The assumption made here is that the suppression of the H2 adsorption/desorption CV features is due to the additives causing a lowering of the ECSA. The reductions in ECSAs were highest with BMI and MBMI (the reductions in ECSA were already significant with the other benzene-containing species). These results suggest that the benzene-containing cationic species significantly interfere with the electrochemical activities of the Pt/C surfaces when present in 1 mmol dm−3 concentrations. Apart from this, no other clear trends can be established: however, reduced losses in ECSA may be occurring with BOTMHA (the example where the benzene ring is separated from the charged N atom) when compared to BTMA and also with the caged quaternary ammoniums examples (BAABCO and [more significantly] the para-methylbenzene-containing MBAABCO and MBABCO).
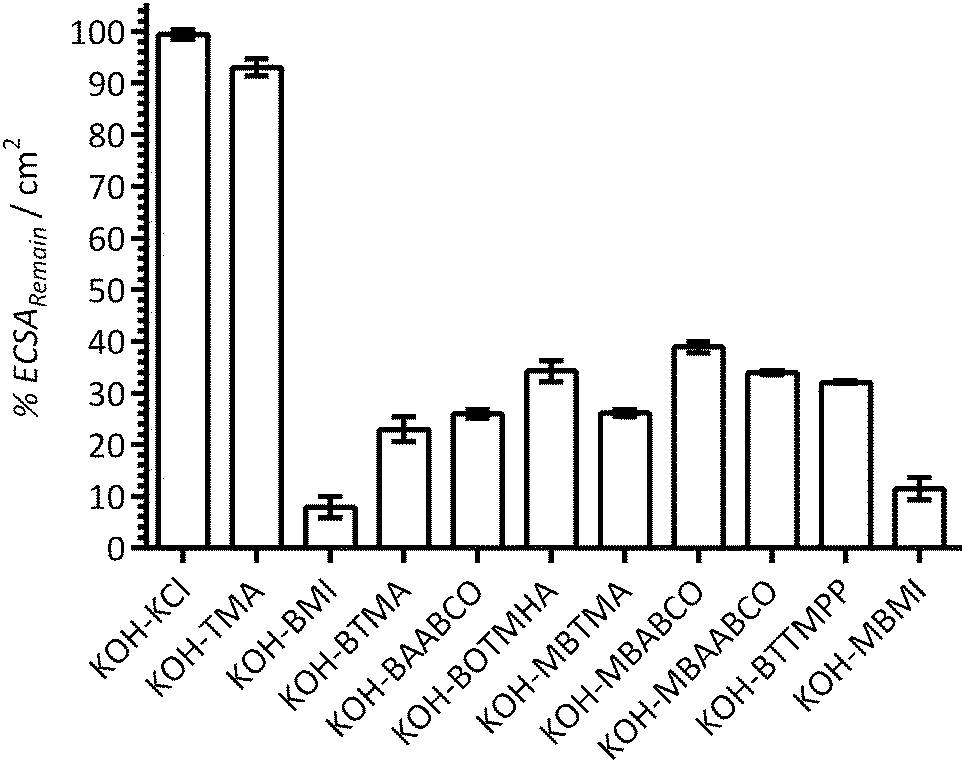 | ||
| Fig. 5 The ECSA remaining (%ECSARemain) at 25 °C for the a Pt/C-coated GC disk electrodes calculated from the ECSAs derived from the data in Fig. 2 (from the hydrogen desorption peaks in the CVs with the aqueous KOH (1 mol dm−3) electrolytes containing additives) and the ECSAs calculated from the [immediately] prior recorded CVs with additive-free aqueous KOH (1 mol dm−3). Error bars are the confidence intervals at the 95% confidence level (n = 3 repeated experiments). | ||
These results again suggest that imidazolium-based cationic groups have the biggest negative effect on Pt catalysts in high pH aqueous electrolytes (compared to the other cationic chemistries that were used in this study). Additionally, the presence of KCl (at the 1 mmol dm−3 concentration used in this study) did not lead to a measureable reduction in ECSA (i.e. it is not the Cl− spectator anions that are interfering with the Pt catalyst). So far, these results with fuel cell grade Pt/C are in a good agreement with the results obtained in our previous published study (that used a polycrystalline-platinum-disk RDE along with a narrower selection of organic cationic species).12
Oxygen reduction reaction hydrodynamic voltammetry
Fig. 6 presents the ORR RDE LSVs collected at 1600 rpm with the different additives. Similar onset potentials of ca. +0.98 V vs. RHE were observed for all of the electrolytes studied, with the exception of the electrolytes containing imidazolium species: MBMI and BMI gave less positive on-set potentials. Since the Pt ECSAs are consistent with each experiment (as the data in Fig. 1 highlights), the currents were assumed to be primarily affected by the nature of the electrolytes.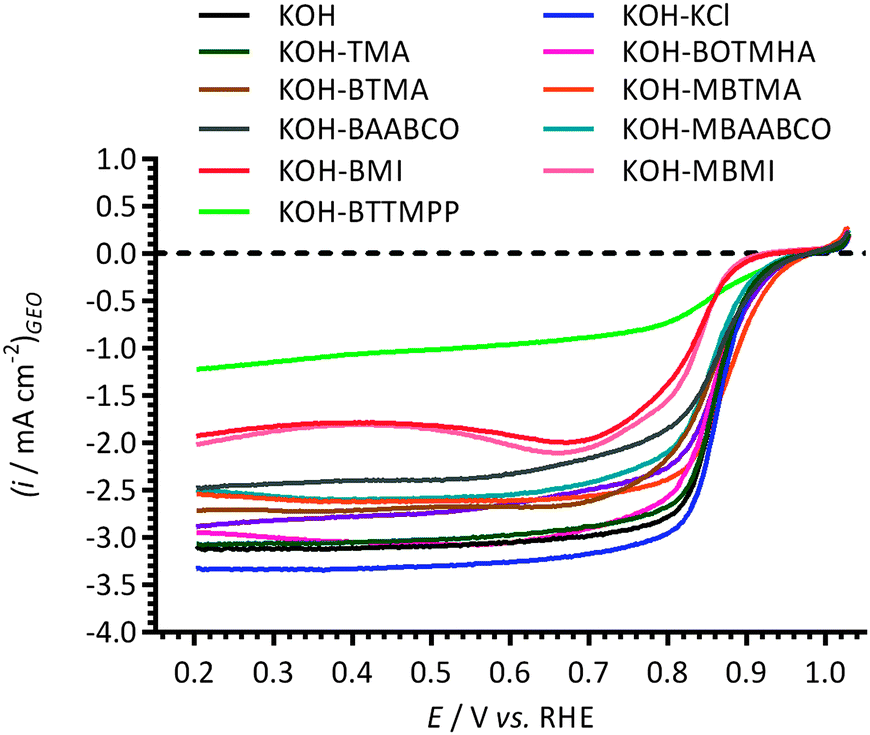 | ||
| Fig. 6 The LSVs at 25 °C for the Pt/C-coated GC-RDEs in O2-saturated aqueous KOH (1 mol dm−3) electrolytes with and without addition of 1 mmol dm−3 of each of the cationic molecules studied. Scan rate = 5 mV s−1 and RDE rotation rate = 1600 rpm. The currents presented here were normalised to the RDE geometric surface area. | ||
In the kinetic and diffusion mix-controlled region (potential range +0.85 to +0.9 V vs. RHE), a relatively comparable ORR activity was observed for the blank KOH electrolyte and those containing the additives, with the exception of the electrolytes containing BMI, MBMI, and BTTMPP (MBTMA may be giving a slightly improved [more positive] onset potential compared to the KOH blank). However, more distinguishable differences in the LSVs were seen with the diffusion-limiting currents observed (in potential region <+0.80 V vs. RHE). The diffusion limited currents at +0.40 V vs. RHE decreased in the order: KCl > KOH-only [blank] ≈ TMA ≈ MBABCO ≈ BOTMHA > MBTMA ≈ MBAABCO ≈ BTMA > BAABCO > MBMI > BMI > BTTMPP. Again, the imidazolium groups had a significantly negative influence on the ORR performances of the Pt/C. The results show a reasonable correlation with the suppression of QR values (defined and presented in Fig. 4) with the exception of the P-example: the presence of BTTMPP (the most bulky species tested) led to undesirably large mass transport correction errors, possibly by blocking a larger number of surface sites and diffusion pathways compared to the smaller ammonium species.
The ORR electrocatalytic performances of the Pt/C in the different electrolytes were further analysed using Tafel plots (Fig. 7) that were derived from ORR RDE LSVs. The kinetic current densities (ik) values were calculated using Koutecký–Levich analysis (eqn (2)). The ik were normalised to the ECSAs presented in Fig. 1 (i.e. the ECSAs recorded with additive-free aqueous KOH solutions). The Pt/C in the different electrolytes exhibited reduced ORR current densities (at +0.75 V vs. RHE) in the following order: blank KOH > KCl > TMA ≈ MBTMA ≈ MBABCO > MBAABCO > BOTMHA > BTMA > BAABCO > MBMI > BMI > BTTMPP. It is also suspected that mass transport effects have not been completely eliminated with the data collected with the (large) BTTMPP-containing electrolyte.
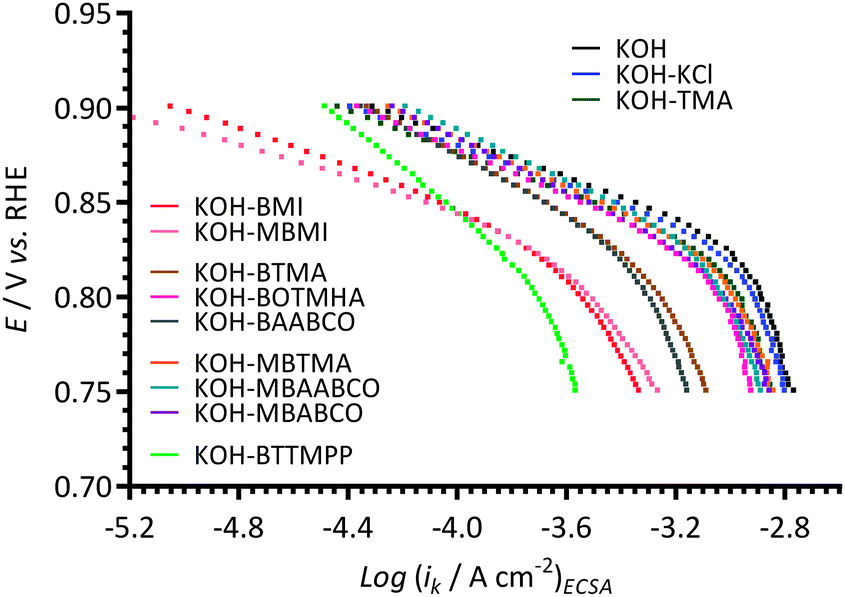 | ||
| Fig. 7 The Tafel plots at 25 °C of the Pt/C-coated GC disk electrode in the O2-saturated aqueous KOH (1 mol dm−3) electrolytes with and without addition of 1 mmol dm−3 of each of the additives (derived from the data in Fig. ESI3 and ESI4†). The kinetic current densities were normalised to the ECSAs presented in Fig. 1. | ||
The ORR activity of Pt/C was improved by adding a hexyloxy spacer chain between the benzyl group and the N-atom (BOTMHA) compared to BTMA (as also observed in the prior study12 with Ptpc instead of Pt/C). More interestingly, the presence of methyl groups on the benzene rings that are para to quaternary ammonium-based –CH2N groups improved the ORR activities (i.e. MBTMA > BTMA and MBAABCO > BAABCO): this may be a hint that cationic molecules such as BTMA are poorer models of polymer-bound cationic head-groups than MBTMA (i.e. BTMA may be providing an underestimation of ORR performances). Note: as stated before (and at the end of this paper), the ultimate goal is to conduct experiments using AEIs (that contain different cationic head-groups) to bind the catalysts to the electrodes, rather than using Nafion binder and fully solubilised model small molecules (as in this study).
Two quasi-linear Tafel regions (with different Tafel slopes) were observed: Table 1 collates these Tafel slopes (derived from the data in Fig. 7). The low overpotential Tafel slopes (above +0.8 V vs. RHE and generally relating to the potential region involving higher surface coverage of oxide species) of the BTMA-, MBTMA-, BAABCO-, MBAABCO-, MBABCO-, BMI-, and BTTMPP-containing electrolytes were observed to be above 60 mV dec−1 (significantly for BTMA, BAABCO, and BTTMPP): in contrast the Tafel slope with the MBMI-containing electrolyte was lower than 60 mV dec−1. In the high overpotential region (0.8–0.75 V vs. RHE), the Tafel slopes increased in order: KOH–MBMI < KOH–MBABCO < KOH–TMA ≈ KOH–BOTMHA < KOH–BMI < KOH–MBTMA < KOH–KCl < KOH–blank < KOH–BTMA < KOH–BAABCO ≈ KOH–MBAABCO < KOH–BTTMPP. The Tafel slopes for both potential regions with the KCl-(57 and 111 mV dec−1), TMA-(57 and 100 mV dec−1) and MBMI-(54 and 89 mV dec−1) containing electrolytes were lower compared to the KOH–blank electrolyte (60 and 120 mV dec−1), while the BTMA-(88 and 126 mV dec−1), BAABCO-(89 and 135 mV dec−1), MBAABCO-(77 and 135 mV dec−1), and BTTMPP-(123 and 142 mV dec−1) containing electrolytes were always relatively higher.
| O2-saturated aqueous electrolyte | Low overpotential Tafel slopes/mV dec−1 | High overpotential Tafel slopes/mV dec−1 |
|---|---|---|
| a Tafel slopes extracted from the Tafel plots presented in Fig. 7 that are based on ECSA-normalised kinetic current densities [normalised to ECSAs presented in Fig. 1 that were measured from the blank N2-purged aqueous KOH (1 mol dm−3) CVs recorded immediately before the addition of each cationic species and prior to O2-purging]. b Caution required as significant mass transport interferences are suspected to be present. | ||
| Blank KOH | −60 | −120 |
| KOH–KCl | −57 | −111 |
| KOH–TMA | −57 | −100 |
| KOH–BMI | −67 | −102 |
| KOH–BTMA | −88 | −126 |
| KOH–BAABCO | −89 | −135 |
| KOH–BOTMHA | −66 | −100 |
| KOH–MBTMA | −69 | −103 |
| KOH–MBABCO | −72 | −98 |
| KOH–MBAABCO | −77 | −135 |
| KOH–BTTMPPb | −123b | −142b |
| KOH–MBMI | −54 | −89 |
The existence of a high coverage of adsorbed O and/or OH related species is known to lower Tafel slopes20 and so this data suggests that there is a high coverage of oxide species with the KCl-, TMA-containing electrolytes (also being observed in Fig. 4). However, we concede that caution is required with this (and the below) interpretations without studies that specifically probe surface coverage with oxide species: this study uses only Tafel slope data as an initial probe, and indication, of the changes in surface oxide species coverage.
With the BTMA-, BAABCO-, and BTTMPP-containing electrolytes, the coverage of surface oxide species on the Pt is relatively low (as interpreted from high Tafel slopes in both overpotentials regions) and this leads to the lower ORR kinetics observed (compared to the blank KOH). Surprisingly, the presence of para-methyl groups on the benzene-ring-containing cation species (MBTMA-, MBABCO, and MBAABCO-) in the electrolytes appears to increase the coverage of Pt-oxide-species (compared to BTMA and BAABCO) and this may go some way towards explaining the improved ORR activities with these para-disubstituted benzene cationic species (as interpreted by the shifts in the Tafel plots in Fig. 7 to more positive potentials).
However, the Tafel data with BMI and MBMI is contradictory as the Tafel slopes in Table 1 suggest high oxide coverages but the Tafel plots indicate low activities. The data (especially with MBMI) also contradicts the data in Fig. 4 when considering the electrolytes containing MBMI. Thus, the assumption made in Fig. 4 (the reduction in the charge densities [QR, calculated from the integration of the cathodic peaks in Fig. 3] that reflect the surface coverage of oxygen-containing species on the Pt surfaces formed during anodic scan) may not be universally valid (e.g. not valid for the MBMI-containing electrolyte). Alternatively, the Tafel slope data may not be giving reliable indications of surface oxide coverage. This data suggests a highly complex situation is present that will require many more experiments to resolve. It also, again, suggests that the behaviour of the Pt/C catalysts is different in the presence of imidazolium species compared to quaternary ammonium species tested.
The ORR specific activities (at +0.90 V vs. RHE and normalised to the ECSAs presented in Fig. 1), apparent exchange current densities (i0), and number of electron transferred (n) per O2, with the Pt/C in the different electrolytes, are presented in Fig. 8. Improved specific activities were obtained with the presence of MBTMA, MBABCO and MBAABCO in KOH electrolyte compared to the blank KOH benchmark: reduced activities were obtained with the presence of the other cationic molecules (most dramatically with the electrolytes containing the imidazolium species).
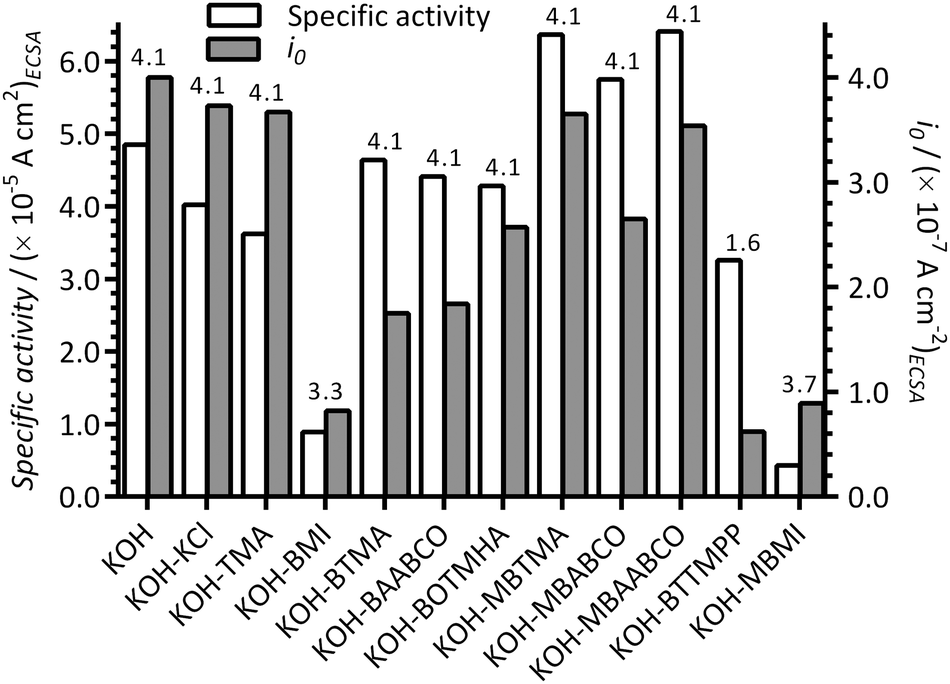 | ||
| Fig. 8 The specific activities (at +0.90 V vs. RHE) and apparent exchange current densities (i0) at 25 °C for the Pt/C-coated GC disk WEs in the O2-saturated aqueous KOH (1 mol dm−3) electrolytes with and without addition of 1 mmol dm−3 of each of the additives. These values were calculated from the data in the Tafel plots in Fig. 7. The ECSAs used are presented in Fig. 1. The number of e− per reduction of an O2 molecule (n values above each pair of bars) were calculated using eqn (3). | ||
However, apparent i0 values were all reduced compared to the blank KOH electrolyte and decreased in the order: KOH-only [blank] > KCl > TMA > MBTMA > MBAABCO > MBABCO > BOTMHA > BAABCO > BTMA ≫ MBMI > BMI > BTTMPP. These results again confirm that the presence of BMI and MBMI (or their electrochemical oxidation products) significantly reduce the performances of the Pt/C catalyst (i.e. specific activities and apparent i0 values that are <50% of those observed with the additive-free KOH blank electrolyte). Comparing the trend in specific activities at +0.90 V vs. RHE and i0, the quaternary ammonium species exhibited higher than expected specific activities: i.e. the i0 values concomitantly decreased with decreases in specific activities for the KOH blank and the KCl and TMA examples but this was not the case with the quaternary ammonium examples.
As commented above, the imidazolium species appear to fundamentally affect the mechanism of the ORR. This was confirmed by examining the number of ORR e− transferred per O2 molecule: n = 3.7 for MBMI and = 3.3 for BMI (cf. n = 4 for the other N-containing molecules). Note, the n < 2 value for BTTMPP is likely affected by the significant mass transport interference in the RDE LSV data and is therefore deemed unreliable. BMI also resulted in a low n = 2.4 with the Ptpc disk electrode used in the prior study.12 The presence of such imidazolium species is therefore leading to an enhancement in the amount of (undesirable) peroxide species being produced with the ORR on the Pt/C catalyst.
The presence of cationic species containing benzene rings (or their electrochemical degradation products) clearly depresses the electrocatalytic behaviour of the Pt/C in aqueous KOH (1 mol dm−3) by affecting both the hydrogen adsorption/desorption (Fig. 2) and oxide formation (Fig. 4) on Pt. The presence of a long alkyloxy link between the quaternary ammonium and benzyl groups (modelled here with BOTMHA) produced a small mitigation towards the reduction in electrocatalytic performance. The impact of the benzene-ring-free TMA was negligible in line with the prior results with the Ptpc disk electrode.12
The cationic groups withdraw π-electrons from the aromatic ring via a negative-inductive effect: the intensity of this effect is diminished with the presence of an aliphatic chain relay (as reported in 1963).21 The positive charges in aliphatic quaternary ammonium species is distributed equally among the N atom and the four α-carbon atoms; i.e. the positive charge in symmetrical molecules such as TMA are distributed over a small volume leading to relatively weak interactions with the surface of the Pt catalyst. In contrast, cationic species containing benzene rings result in the charge delocalization around the aromatic system (spread over to a volume) and the positive charge may be utilised in aromatic-ring stacking π–π interactions (especially demonstrated with the additional presence of aromatic heterocycles such as imidazolium groups).21,22
Thus, future research into APEFCs (and related technologies) should consider developing aromatic-ring-free alkaline anion-exchange polymer electrolytes to further probe for any negative impacts arising from the presence of pendent aromatic rings (benzene rings and imidazolium groups) as long as this does not lead to a concomitant decrease in the chemical (alkaline) stabilities of the aromatic-ring-free cationic groups.
A comparison of the Pt/C data with prior data12 collected using a Ptpc disk electrode
Fig. 9 compares the data obtained with the Pt/C-coated GC electrodes to the data collected using a smooth Ptpc disk electrode (roughness factor < 1.2)12 when using KCl, TMA, BOTMHA, BTMA, and BMI additives (1 mmol dm−3) in the aqueous KOH (1 mol dm−3) electrolytes. Both Pt types yield two distinguishable Tafel slopes (in the two potential regions) and both show the same trend regarding the shifts in ECSA-normalised current densities to lower values (at comparable potentials): BMI > BTMA > BOTMHA > TMA > KCl. However, the Tafel plots consistently show lower ECSA-normalised activities for the Pt/C catalyst compared to the bulk polycrystalline Pt in the Ptpc-disk electrodes for each electrolyte type.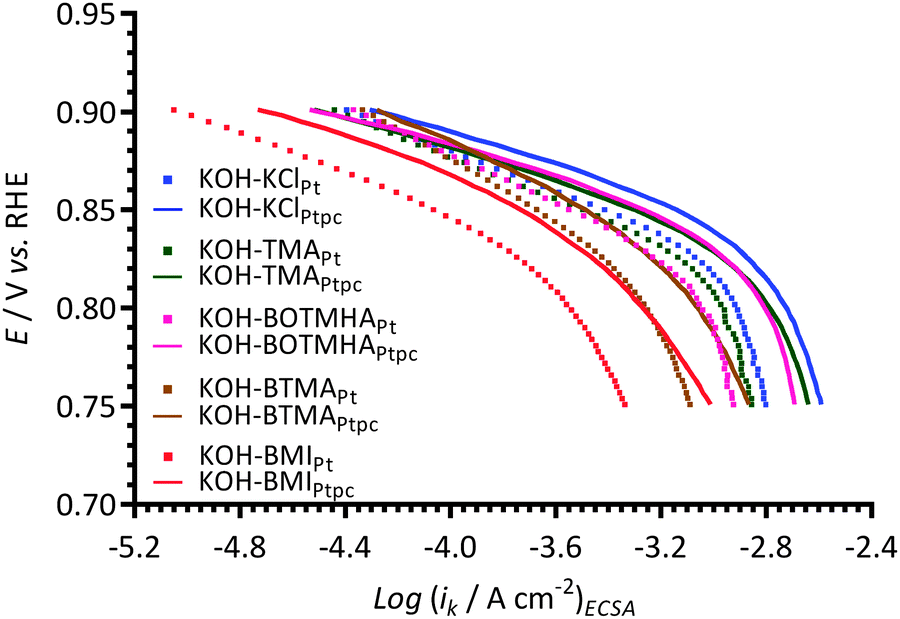 | ||
| Fig. 9 A comparison of Tafel plots with the KCl-, TMA-, BTMA-, BOTMHA-, and BMI-(1 mmol dm−3)-containing aqueous KOH (1 mol dm−3) electrolytes using data from this study (Pt/C-coated GC-disk electrode – squares) and data recorded in the previous study (Ptpc-disk electrode – solid lines).12 | ||
Fig. 10 compares the apparent exchange current densities of the Pt catalyst and Ptpc disk electrode measured in KCl-, TMA-, BOTMHA-, BTMA- and BMI-containing aqueous KOH electrolytes. As expected, the Ptpc (model of bulk Pt) gave enhanced specific exchange current densities (magnitude 10−4 A cm−2) compared to the Pt/C catalyst (magnitude 10−7 A cm−2) for the KOH blank experiments and also with the addition of cationic species (e.g. current densities at the same potential were higher for Ptpc compared to Pt/C for each additive tested.). This is a well-known phenomenon where bulk Pt gives higher specific activities (current per Pt surface reaction site) than Pt nanoparticles (although the latter gives the advantage of much higher specific surface areas).23 However, the trends in ORR activity were the same for the species with both catalysts and generally decreased in the order (compared at 0.8 V vs. RHE, ca. the potential where there is transition between the two different Tafel slopes): KCl > TMA > BOTMHA > BTMA > BMI (although TMA ≈ BOTMHA for the Ptpc electrode).
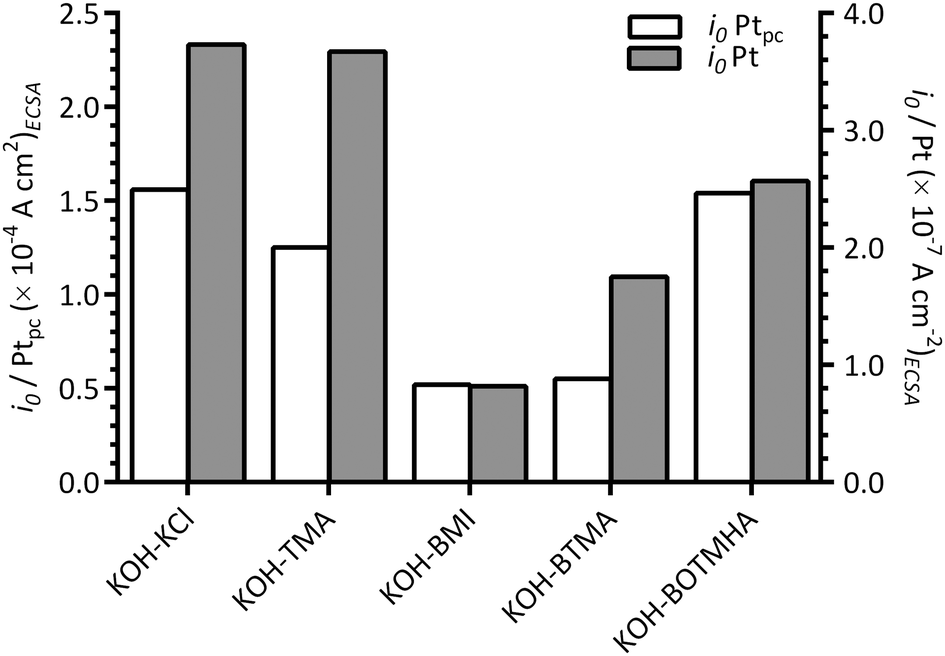 | ||
| Fig. 10 A comparison of apparent exchange current densities (i0) at 25 °C with the KCl-, TMA-, BTMA-, BOTMHA-, and BMI-(1 mmol dm−3)-containing aqueous KOH (1 mol dm−3) electrolytes using data from this study (Pt/C-coated GC-disk electrode – filled bars [y2-axis]) and data recorded in the previous study (Ptpc-disk electrode – unfilled bars [y1-axis]).12 | ||
Initial results collected with different concentrations of TMA and BTMA in aqueous KOH (1 mol dm−3)
In order to further study the effect of the benzene ring in the cationic additives on the activity of ORR on Pt/C, Tafel plots were recorded using different concentrations of TMA (1–20 mmol dm−3) and BTMA (0.01–1 mmol dm−3) in the aqueous KOH (1 mol dm−3) electrolytes (Fig. 11). As can be seen, the presence of TMA in aqueous KOH does not significantly interfere with the ORR on Pt/C even at much higher concentrations (up to 20 mmol dm−3), although these higher concentrations of additive risk introducing additional mass-transfer errors and more significant deviations from the reported properties of the electrolyte (e.g. O2 concentration, kinematic viscosity etc.). However, the negative influence of the presence of BTMA in the electrolyte on the Pt/C catalyst could only be minimised by diluting the concentration to 0.01 mmol dm−3 (i.e. 100 × lower concentration than used in the above sections). This result shows that the presence of the benzene ring in cationic molecules such as BTMA can significantly lower the activity of ORR on Pt/C catalysts.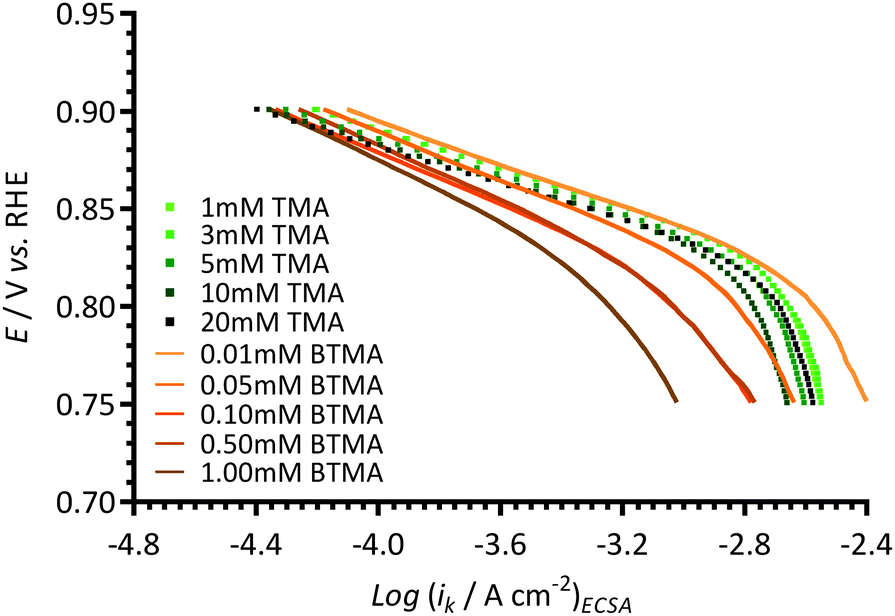 | ||
| Fig. 11 A comparison of Tafel plots of different concentrations of TMA (symbols) and BTMA (lines) in aqueous KOH (1 mol dm−3) electrolytes measured using a Pt/C-coated GC-disk electrode. | ||
Initial results with a Ag/C (20 mass%) catalyst
A primary rationale for studying APEFCs is the potential for utilising non-Pt (especially non-platinum-group metal catalysts). As stated in the discussion above, Pt-based catalysts are especially affected by the presence of positively charged anion-exchange head-groups (analogous to the studied cationic species [and their degradation products]). Therefore, some initial experiments were conducted using Ag/C catalyst instead of Pt/C. The catalyst ink used involved the simple replacement of the Pt/C with Ag/C (20 mass%, Johnson Matthey, 10 mg) while leaving all other experimental parameters unchanged. The Tafel plots with the Ag/C-coated GC measured in KCl-, TMA-, BTMA-, and BMI-containing electrolytes were compared with those recorded using the Pt/C catalyst (Fig. 12). Note that the geometric normalised current densities were used in this simple initial study due to the unknown ECSAs of the Ag/C catalyst on each electrode. The Ag/C catalyst appears to be more resistant to the presence of the BTMA and BMI species in the aqueous KOH electrolyte compared to Pt catalyst (in that the relative shifts to lower current densities appear smaller than when Pt/C is used). However, BMI, again, appears to have the largest negative effect. As expected, significantly lower ORR activities were observed for the Ag/C-coated electrodes (when compared to the electrodes coated with the same mass loading of Pt/C) for all of the additives.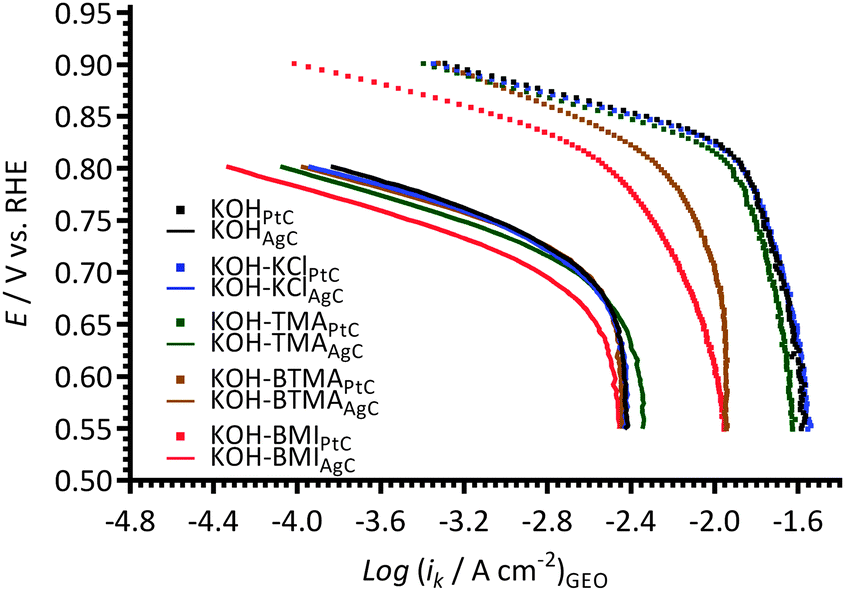 | ||
| Fig. 12 A comparison of Tafel plots with the KCl-, TMA-, BTMA-, and BMI-(1 mmol dm−3)-containing aqueous KOH (1 mol dm−3) electrolytes measured using a Pt/C (symbols) and a Ag/C (lines) coated GC-disk electrodes. | ||
Conclusions and final directions
The effect of cationic molecules on the oxygen reduction reaction (ORR) on Pt/C (20 mass%) catalyst in aqueous electrolytes based on KOH (1 mol dm−3) was investigated. The cationic additives studied (all with Cl− counter anions; KCl was used for control experiments) were: TMA = tetramethylammonium, BTMA = benzyltrimethylammonium, MBTMA = 1-(4-methylbenzyl)trimethylammonium, BOTHMA = 6-(benzyloxy)-N,N,N-trimethylhexan-1-aminium, BAABCO = 1-benzyl-4-aza-1-azoniabicyclo[2.2.2]octane, MBAABCO = 1-(4-methylbenzyl)-4-aza-1-azoniabicyclo[2.2.2]octane, BMI = 1-benzyl-3-methylimidazolium, MBMI = 1-(4-methylbenzyl)-3-methylimidazolium, MBABCO = 1-(4-methylbenzyl)-1-azoniabicyclo[2.2.2]octane, and BTTMPP = 1-benzyltris(2,4,6-trimethoxyphenyl)phosphonium [see Scheme 1]. The suppression of the hydrogen adsorption/desorption features in the cyclic voltammograms decreased in the order: BMI (highest suppression) > MBMI > BTMA > BAABCO ≈ MBTMA > BTTMPP > MBAABCO > BOTMHA > MBABCO > TMA > KCl, and KOH-only (additive free blank electrolyte). The hindrance/interference towards the ORR on Pt/C generally increased in the following trend: blank KOH < KCl < TMA < MBTMA < MBABCO < MBAABCO < BOTMHA < BTMA < BAABCO < MBMI < BMI < BTTMPP.These results demonstrate that small concentrations of such cationic species (e.g. 1 mmol dm−3 and much lower concentrations for species such as BTMA), and/or their electrochemical oxidation products, can seriously inhibit Pt/C catalytic activities in high pH aqueous electrolytes. The presence of imidazolium species (modelled by BMI and MBMI in this study) gave particularly severe reductions in catalytic performances and appeared to significantly alter the ORR pathway. The P-containing example (high molecular weight BTTMPP) gave the worst apparent catalytic inhibition effect, but this was likely due to predominant mass transport errors and interferences [electrolyte viscosity and O2 solubilisation effects]; therefore no comment can be made [with confidence] on the effect of this species on the ORR kinetics with this cationic molecule. The presence of benzene rings always led to a reduction in the ORR kinetics on Pt/C, but this was less pronounced with molecules where the benzene rings contained methyl groups that were para to the –CH2N+RR′R″ substituents. Removing the benzene ring completely (modelled here using TMA) led to almost insignificant losses in ORR performances.
Nafion dispersion was used as the ionomer throughout this study. This is a non-ideal situation as Nafion ionomer contains an anionic polymer electrolyte (cation-exchanging). The next stage of this series of investigations will consider the kinetics on Pt/C and other ORR [and HOR] catalysts when bound using anion-exchange ionomers (AEI) containing different cationic head-groups (quaternary ammonium vs. imidazolium etc.) in aqueous KOH [i.e. Nafion-ionomer-free and without addition of fully solubilised cationic molecules]. After all, Yan et al. concluded that phosphonium cationic groups poison Ag ORR catalysts much more when they are dissolved small molecules than when they are bound to polymers.24 This will add to recent studies that investigate the effect on various reactions at high pH with the use of a commercial quaternary ammonium AEI (Tokuyama AS-4)13,14,25 and a polysulfone-imidazolium-based AEI.13 Note: these prior studies did not compare AEIs containing different head-groups but the same polymer backbone. This is the aim of our future work as an AEI concept is now available, that would facilitate such a study, that uses a selection of bulk producible AEIs containing different cationic head-groups (but with the same ion-exchange capacity and polymer backbone chemistry).26
Acknowledgements
The authors thank the UK's Engineering & Physical Sciences Research Council for funding (EPSRC grant EP/I004882/1). The University of Surrey is also thanked for providing funds towards Kenneth Inglis' undergraduate final year project. Francesca Robertson and Anuska Mann are thanked for HRMS and Qinmin Zhang is thanked for NMR spectroscopy.Notes and references
- (a) M. Gharaie, N. Zhang, M. Jobson, R. Smith and M. Hassan Panjeshahi, Chem. Eng. Res. Des., 2013, 91, 1483 CrossRef CAS; (b) S. A. Didas, A. R. Kulkarni, D. S. Sholl and C. W. Jones, ChemSusChem, 2012, 5, 2058 CrossRef CAS PubMed; (c) N. S. Spinner, J. A. Vega and W. E. Mustain, Catal. Sci. Technol., 2012, 2, 19 RSC.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135 CAS.
- (a) J. G. Verkade, K. Wadhwa, X. Kong and K. Schmidt-Rohr, Anion Exchange Membrane, US 8436057 B1, USA, 2013 Search PubMed; (b) Y. J. Wang, J. Qiao, R. Baker and J. Zhang, Chem. Soc. Rev., 2013, 42, 5768 RSC; (c) L. Zeng and T. S. Zhao, Electrochem. Commun., 2013, 34, 278 CrossRef CAS; (d) J. R. Varcoe, R. C. T. Slade and E. L. H. Yee, Chem. Commun., 2006, 1428 RSC; (e) J. R. Varcoe and R. C. T. Slade, Electrochem. Commun., 2006, 8, 839 CrossRef CAS.
- X. Wu, K. Scott, F. Xie and N. Alford, J. Power Sources, 2014, 246, 225 CrossRef CAS.
- (a) S. Maurya, S. H. Shin, M. K. Kim, S. H. Yun and S. H. Moon, J. Membr. Sci., 2013, 443, 28 CrossRef CAS; (b) N. Fujiwara, M. Yao, Z. Siroma, H. Senoh, T. Ioroi and K. Yasuda, J. Power Sources, 2011, 196, 808 CrossRef CAS; (c) Q. Fan, CO2 Tolerant Alkaline Fuel Cells and Alkaline Batteries, US 0239921 A1, USA, 2010 Search PubMed.
- N. J. Robertson, H. A. Kostalik IV, T. J. Clark, P. F. Mutolo, H. D. Abruña and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 3400 CrossRef CAS PubMed.
- J. Wang, J. Wang, S. Li and S. Zhang, J. Membr. Sci., 2011, 368, 146 Search PubMed.
- A. H. N. Rao, H. J. Kim, S. Nam and T. H. Kim, Polymer, 2013, In, Press Search PubMed.
- B. S. Ko, J. Y. Sohn and J. Shin, Polymer, 2012, 53, 4652 CrossRef CAS.
- L. Jiang, X. Lin, J. Ran, C. Li, L. Wu and T. Xu, Chin. J. Chem., 2012, 30, 2241 CrossRef CAS.
- J. Zhou, K. Joseph, J. M. Ahlfield, D. Y. Park and P. A. Kohl, J. Electrochem. Soc., 2013, 160, F573 CrossRef CAS.
- A. L. Ong, D. K. Whelligan, M. L. Fox and J. R. Varcoe, Phys. Chem. Chem. Phys., 2013, 15, 18992 RSC.
- W. Lu, Z.-G. Shao, G. Zhang, Y. Zhao, J. Li and B. Yi, Int. J. Hydrogen Energy, 2013, 38, 9285 CrossRef CAS.
- M. Ünlü, D. Abbott, N. Ramaswamy, X. Ren, S. Mukerjee and P. A. Kohl, J. Electrochem. Soc., 2011, 158, B1423 CrossRef.
- Y. Garsany, Anal. Chem., 2010, 82, 6321 CrossRef CAS PubMed.
- Y. Garsany, I. L. Singer and K. E. Swider-Lyons, J. Electroanal. Chem., 2011, 662, 396 CrossRef CAS.
- C. H. Hamann, A. Hamnett and W. Vielstich, Electrochemistry, Wiley-VCH, Weinheim, 2nd edn, 2007, p. 1 Search PubMed.
- J. A. Vega and W. E. Mustain, Electrochim. Acta, 2010, 55, 1638 CrossRef CAS.
- S. Srinivasan, E. A. Ticianelli, C. R. Derouin and A. Redondo, J. Power Sources, 1988, 22, 359 CrossRef CAS.
- J. S. Spendelow and A. Wieckowski, Phys. Chem. Chem. Phys., 2007, 2654 RSC.
- J. Thomas and W. Marlow, J. Med. Chem., 1963, 6, 107 CrossRef CAS PubMed.
- T. Zhou, M. Ao, G. Xu, T. Liu and J. Zhang, J. Colloid Interface Sci., 2013, 389, 175 CrossRef CAS PubMed.
- A. Anastasopoulos, J. C. Davis, L. Hannah, B. E. Hayden, C. E. Lee, C. Milhano, C. Mormiche and L. Offin, ChemSusChem, 2013, 6, 1973 CrossRef CAS PubMed.
- S. Gu, W. Sheng, R. Cai, S. M. Alia, S. Song, K. O. Jensen and Y. Yan, Chem. Commun., 2013, 49, 131 RSC.
- A. J. Lemke, A. W. O'Toole, R. S. Phillips and E. T. Eisenbraun, J. Power Sources, 2014, 256, 319 CrossRef CAS.
- S. D. Poynton, R. C. T. Slade, W. E. Mustain, T. J. Omasta, R. Escudero-Cid, P. Ocón and J. R. Varcoe, J. Mater. Chem. A, 2014, 2, 5124 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: This include a selection of relevant raw electrochemical data (RDE voltammograms and Koutecký–Levich plots) as well as details on the synthesis of 1-(4-methylbenzyl)trimethylammonium chloride, 1-(4-methylbenzyl)-3-methylimidazolium chloride, 1-(4-methylbenzyl)-4-aza-1-azoniabicyclo[2.2.2]octane chloride, 1-(4-methylbenzyl)-1-azoniabicyclo[2.2.2]octane chloride, and benzyltris(2,4,6-trimethoxyphenyl)phosphonium chloride. See DOI: 10.1039/c4cp04973j |
| This journal is © the Owner Societies 2015 |