
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiple scale investigation of molecular diffusion inside functionalized porous hosts using a combination of magnetic resonance methods†
Martin
Wessig
,
Martin
Spitzbarth
,
Malte
Drescher
,
Rainer
Winter
and
Sebastian
Polarz
*
Department of Chemistry, University of Konstanz, D-78457 Konstanz, Germany. E-mail: sebastian.polarz@uni-konstanz.de
First published on 13th May 2015
Abstract
Mass transport of molecular compounds through porous solids is a decisive step in numerous, important applications like chromatography or heterogeneous catalysis. It is a multi-scale, hierarchical phenomenon: macrodiffusion (>μm) is influenced, in addition to parameters like grain boundaries and particle packing, by meso-scale (>10 nm, <μm) factors like particle size and the connectivity of pores. More importantly, meso-scale diffusion and macro-scale diffusion are first and foremost determined directly by processes on the molecular scale (<10 nm), which depend on numerous factors like pore-size, interactions of the host with the solid surfaces and with the solvent. Due to the high complexity of the latter and the fact that current analytical techniques enable only limited insights into solvent-filled pores with sufficient spatial and temporal resolution, the knowledge about the molecular origins of diffusive processes in porous materials is still restricted. The main focus of the current paper is on the development of continuous wave (CW) electron paramagnetic resonance (EPR) spectroscopy into a tool shedding some new light on molecular diffusion inside mesoporous silica materials differing systematically in pore size and surface functionalities. The advantages of CW-EPR are that its spatial resolution fits ideally to the size of mesopores (2–10 nm), it is fast enough for spotting molecular processes, and any conventional solvent and the porous matrix are EPR silent. Diffusion coefficients have been calculated considering spin exchange occurring from the diffusive collision of radicals, and are compared to complementary analytical techniques like MAS PFG NMR (sensitive for meso-scale) and EPR-imaging (sensitive to macroscale diffusion). Our results show that the choice of surface bound functional groups influences diffusion much stronger than pore-size. There are indications that this is not only due to different guest–surface interactions but also due to an altered mobility within the solvent under confinement.
1. Introduction
Diffusion inside porous hosts (DIPH) is of utmost relevance for a large number of industrial processes and applications like chromatography and heterogeneous catalysis, to name only two important examples.1 Therefore, one was and still is highly interested in investigating and gathering a deep understanding of DIPH.2–6 There are several reasons why, even after more than 50 years of research, there is still a large demand for studying DIPH (in particular in the liquid phase). First and foremost DIPHs is a highly dynamic process of enormous complexity spanning orders of length and time scales, from ≈10−12 m and ≈10−12 s (determined by the whole range of intermolecular interactions of dissolved molecular species with the solvent and potentially functionalized surfaces of the porous host, see also Scheme 1) to ≈1 m and ≈103 s for the macroscopic mass transport. The multi-scale character of diffusion in porous hosts poses major difficulties with regard to obtaining a comprehensive experimental data basis, because a set of different analytical methods with different probe species could easily give varying results depending on the spatial and temporal resolution of the applied technique. Furthermore, it is often a big problem to conduct physical investigations under conditions close to relevant applications, respectively in solvents and at elevated temperatures. | ||
| Scheme 1 Graphical summary for the multiple scale investigation of diffusion in mesoporous hosts indicated by the red path. The diffusion of TEMPONE (red cylinders), respectively TEMPONE-OH for NMR, was studied on all length scales relevant for DIPH. Obviously diffusion starts on the molecular scale influenced by a complex interplay between probe–surface, probe–solvent, probe–probe, solvent–solvent and solvent–surface interactions. This length- and time scale becomes accessible using cw-EPR. This is the basis to understand diffusion on the ‘middle’, the mesoscale (10 nm–10 μm) as seen by MAS PFG NMR. On an even greater length scale macroscopic diffusion through the whole mesoporous particle was probed by EPR imaging. | ||
The established analytical techniques for studying DIPH can be divided into two complementary categories addressing either macroscopic or microscopic diffusion and transport or self-diffusion.7 Different methods exist for studying macroscopic transport, e.g. uptake rate measurements, zero length columns (ZLC) or macroscopic self-diffusion (tracer ZLC) to name only a few.7 Imaging techniques are in general very suitable for studying diffusion on larger length scales up to the macroscopic one. Techniques are designated as microscopic if the diffusion path length is much smaller than the particle size of the porous host. Quasi-elastic neutron scattering has been used to acquire information about microscopic self-diffusion, and by using interference microscopy or IR microscopy8 one can analyse microscopic transport diffusion.
The most well-known technique to study microscopic diffusion is pulsed field gradient nuclear magnetic resonance (PFG-NMR).9 Despite the fact that NMR is an established technique, an obvious advantage is that countless compounds contain NMR active nuclei. Thus, PFG NMR has been successfully applied to characterize the diffusion properties of pure substances like alkanes, alcohols or water. To date there are quite a few fundamental studies on the transport properties in zeolithes and mesoporous silica as hosts using PFG NMR, and the interested readers are referred to one of the recent, excellent review articles by Kärger et al.9–11 The investigation of compound mixtures or solutions is much more demanding, because now the NMR activity of the surrounding matrix becomes a disadvantage.12 Magic angle spinning (MAS) PFG NMR is needed for observing sufficiently resolved spectra for different components, and so far only few molecules and their diffusion in solvents could be studied in this manner,13–15 in particular dissolved species inside a solid, porous host.16 Guenneau et al. applied MAS PFG NMR to study the diffusion of diluted ibuprofen in ethanol confined to MCM-41 with 3.5 and 11.6 nm pore diameters.17 Other examples for the diffusion of diluted substances are mainly found in the field of chromatography, where PFG NMR was used to characterize diffusion in the presence of porous silica as a stationary phase.18,19 A comprehensive investigation was performed by Pemberton et al., who studied the diffusion of a range of substances dissolved in a mixture of CHCl3 and CH2I2. Sharifi et al. synthesized MCM-41-like materials with different amounts of SO3H groups and probed the proton conductivity using MAS PFG NMR.20
Despite the term “microscopic” diffusion there is still a gap between microscopic diffusion studied by NMR and diffusion at the molecular or the macroscopic level. On the one hand PFG NMR probes diffusion on length scales of micrometers, but it cannot reveal the very initial steps when diffusion starts on the nanometer or molecular scale. But it is obvious that diffusion at a molecular level is extremely important in context of materials with very small pores (<10 nm); see Scheme 1. Furthermore, macroscopic DIPH properties arising from internal barriers like grain boundaries are also not resolved. It can be concluded that it would be highly desirable to develop one analytical method into a tool, which is applicable on several length and time scales relevant for DIPH providing complementary information on PFG NMR.
We propose EPR spectroscopy as a complementary method for NMR diffusion studies on the basis of the following considerations. Continuous wave (CW) EPR spectroscopy in combination with nitroxides is the ideal method to study dynamics inside porous materials since it is very sensitive to the microenvironment and delivers information about rotational dynamics and surface interaction under thermal equilibrium.21–24 Because typically the porous matrix and relevant solvents are EPR-silent, this method is very suitable to track guests and species confined inside porous hosts.22,25–27 CW-EPR has already been used to study zeolithes,28 mesoporous materials,29,30 MOFs31 and PMOs21,22,32 and gave insight into the formation process,33,34 surface polarity and surface interaction32,35 or the dynamics of confined solutions.22,36 Information about translational diffusion can be obtained from the Heisenberg exchange evaluation of the line broadening originating from the random collision of two dissolved radicals.
Because the mutual effect on the spectra is based on the interaction of nearby molecules, the described methodology was successfully applied to study the diffusion through membranes.37–39 Okazaki studied Heisenberg exchange in MCM-41 and SBA-15 materials and developed a qualitative model of collective flow.30,40 Despite its great potential for obtaining insights into diffusion of molecules under confinement at the molecular level, it was not used for the determination of intra-pore diffusion coefficients.
The limited understanding of DIPH processes on the molecular scale is not only due to a lack of a suitable analytical technique, but it is important to note that so far all studies have almost exclusively been performed inside siliceous, non-modified, mesoporous silica materials like MCM-41 or SBA-15.41,42 Concerning advanced applications, materials with functionalized surfaces, for instance organosilicas, are of much greater interest.43–45
Therefore, in the current work, we aim at a more refined knowledge about DIPH in direct proximity to organically functionalized surfaces. First, we will very briefly discuss the used mesoporous materials for our DIPH study. Special emphasis will be on silica materials with chemically functionalized surfaces, in order to probe specific interactions between the probe molecules and the walls of the porous host. In this context periodic mesoporous organosilicas (PMOs) are of particular importance.46–49 The advantage of PMOs is that a high density of tailor-made functional groups can be combined with the superior control over pore-size and material morphology developed in the research on mesoporous materials over the last 20 years.
In addition to MAS PFG NMR we will acquire complementary information using EPR spectroscopy. CW-EPR will be used to study molecular diffusion on the nanometer scale, and EPR imaging will deliver information about macroscopic diffusion. By combining these three techniques, differences between molecular and microscopic diffusion and macroscopic transport will be observed, which cannot be revealed by using only one method.
2. Experimental procedures
2.1. Synthesis of mesoporous materials
Mesoporous silicas (MPS) with varying pore diameters were synthesized according to the literature.50 In a typical synthesis, 2 g of Pluronic 123 were dissolved in 4 g of tetraethylorthosilicate (TEOS) at 45 °C and ethanol was added dropwise until homogenization occurred. Then 2 g of 1 M hydrochloric acid was added and the solution was stirred for a few minutes. The evolving ethanol was removed under vacuum and the viscous gel was aged at 60 °C for 3 days. A variation of pore diameters was reached by adding up to 8 g of mesitylene. The template was removed via calcination at 550 °C for 10 h. For the current paper the materials have been designated as monolithic since the average particle diameter is much greater than the explored range from the MAS PFG NMR experiments.Postmodification of MPS was carried out as followed: to 1.01 g of dried PS-5.4, 17 ml of trimethylchlorosilane in 46 ml of toluene was added. The suspension was heated under reflux overnight, filtered off and the residue material was extracted two times with THF for 1 d.
The precursors for the UKON materials had been synthesized as described in the literature.51
UKON1 was synthesized by adding 0.8 g of the precursor to 0.56 g of Pluronic 127, 1.2 ml of ethanol and 62 mg of mesitylene. The mixture was stirred under gentle heating until homogenization had occurred. Then 3.17 g of HCl–KCl buffer with pH = 1.9 was added. After a few minutes of stirring the mixture was heated up to 65 °C for 3 h. Afterwards the sol was aged in an open glass container for one week and the template was removed by twofold extraction for 2 d in 30 g of ethanol and 30 g of concentrated hydrochloric acid.
UKON2a was synthesized by adding 0.26 g of Pluronic F127 and 1.20 g of ethanol to 0.46 g of the precursor. The solution was stirred under gentle heating until homogenization occurred. Then 0.26 g of 1 M HCl was added and the solution was stirred for additional 30 min. Afterwards the sol was aged in an open glass container for one week and the template was removed by extraction for 4 d in first 30 g of concentrated H2SO4 and 30 g of distilled water and then in 30 g of ethanol and 30 g of concentrated hydrochloric acid.
2.2. Materials characterization
Solid-state NMR spectra were recorded using a Bruker AVANCE III spectrometer operating at 400 MHz equipped with a 4 mm PH MAS DVT 400W1 BL4 N-P/H CGR probe head with a magic angle gradient. TEM images were obtained on a Zeiss Libra 120 at 120 kV acceleration voltage. The TEM-samples were prepared by shortly dipping a carrier covered with a holey carbon foil (Plano company, S147) into a dispersion of the grinded materials in THF. Small-angle X-ray scattering (SAXS) measurements were conducted using a Bruker AXS Nanostar. N2-physisorption measurements were recorded on a Micromeritics Tristar. SEM images were obtained by a Zeiss 249 CrossBeam 1540XB. Isothermal titration calorimetry (ITC) measurements were performed on an iTC200 micro calorimeter from Malvern. Analytical ultracentrifugation sedimentation experiments have been carried out on an Optima XL-I analytical ultracentrifuge of Beckman Coulter.The NMR samples were prepared by degassing 70 mg of the desired material and then introducing 170 μl of an oxygen free 1 mol l−1 TEMPONE-OH solution. The solution was infiltrated overnight and the supernatant solution was removed prior to the introduction of the material into the zirconia rotor.
Cw-EPR measurements were performed on continuous wave (cw)-X-band EPR Miniscope spectrometer MS400 from magnettech equipped with a variable temperature unit (Temperature Controller TC-H03, magnettech GmbH). The Helmholtz coils were connected to a heat exchanger (Haake SC100 from Thermo Scientific) to reduce signal shifting during the scan averaging. All solutions and materials were degassed at least 12 times by pump–freeze–thaw cycles prior to use. The samples were prepared by adding 2.5 ml of a 1-oxyl-2,2,6,6-tetramethyl-4-piperidone (TEMPONE) solution of various concentrations (c = 5 × 10−4 mol l−1, 5 × 10−2 mol l−1, 7.5 × 10−2 mol l−1 or 1 × 10−1 mol l−1) to 60 mg of the mesoporous material and infiltrated under argon overnight. Afterwards the supernatant was removed and the materials were washed three times with pure degassed ethanol to remove adsorbed spin probes from the outer particle surface. At every temperature, the samples were allowed to stabilize for at least 15 minutes prior to the measurement. All spectra were analysed by simulating the spectra using the free MATLAB toolbox Easyspin.52
CW-EPR imaging experiments were performed in the X-band at room temperature on a Bruker E 580 spectrometer in an ER 4180 TMHS resonator. Spatial resolution was provided by an E540 GCX2 gradient coil system. The samples were placed inside a shrinking tube connected to sample tubes with an inner diameter of 2 mm on the top and bottom. The sample tubes and the sample were filled with ethanol and TEMPONE was added to the sample tube at the top. Applying a magnetic field gradient of 146 G cm−1 in the y-direction (sample access axis) spectra were recorded every 390 seconds. From each spectrum a 1d projection of the spin density ρ1d(y) was calculated by deconvolving the spectrum using the spectrum in the absence of a magnetic field gradient as the deconvolution kernel and taking the resonator profile into account. The resulting time evolution ρ1d(y,t) of ρ1d(y) was simulated numerically by solving the diffusion equation
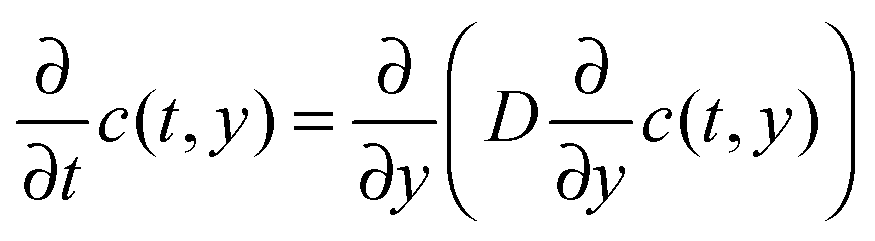

The macroscopic translational diffusion coefficient D was then determined using least-squares minimization of the difference between the simulated and experimental ρ1d(y,t).
3. Results and discussion
3.1. Mesoporous host materials
Two types of mesoporous silica materials were used for the current study (see also the Experimental part). For a start, unmodified, pure silica materials varying in pore-size (DP = 4.1 to 12.2 nm) were prepared.50 A summary of some textural data is given in Table 1 and a set of typical analytical data for porous solids (including N2 physisorption, SAXS, TEM, 13C-NMR and 29Si-NMR) is given for one exemplary material in the ESI-1.†| Material | Pore diameter [nm] | Pore volume [cm3 g−1] | BET surface area [m2 g−1] |
|---|---|---|---|
| a MPS ≅ mesoporous silica; MPOSA ≅ mesoporous organosilica containing terminal trimethylsiloxane groups; UKON1 ≅ PMO containing bridging bromobenzene groups;51 UKON2A ≅ PMO containing bridging benzoic acid groups.51 | |||
| MPS-41 | 4.1 | 2.64 | 523 |
| MPS-46 | 4.6 | 2.83 | 528 |
| MPS-54 | 5.4 | 3.1 | 464 |
| MPS-80 | 8.0 | 3.89 | 488 |
| MPS-122 | 12.2 | 3.09 | 401 |
| MPOSA-48 | 4.8 | 1.46 | 342 |
| UKON1-53 | 5.3 | 1.00 | 392 |
| UKON2A-56 | 5.6 | 1.45 | 623 |
In addition, mesoporous materials with different surface properties of a preferably similar pore-size were generated using organosilica chemistry.43 A hydrophobic environment was generated by attaching trimethylsilyl groups via post-modification of mesoporous silica.53 Mesoporous hosts with surfaces characterized by functional groups (R–Br, R–COOH) were selected from the PMO class. Here, the so-called UKON-materials26,27,51,53–56 established in our group were prepared, which are characterized by a bridging phenyl ring substituted in the 3-position. Whenever possible, materials with the size of the porous particles extending the 1 mm scale (see ESI-2†) were used for the experiments. The utilization of large particles of the mesoporous host has the advantage that the diffusion path probed by NMR and EPR is smaller than the particle extension and this means that external, non-confined diffusion in the spaces between particles becomes negligible.25
3.2. Diffusion studies for the non-confined reference state
 | ||
| Chart 1 TEMPONE (1) used as an EPR spin probe and TEMPONE-OH (2) used for NMR studies. | ||
MAS PFG NMR experiments were performed using TEMPONE-OH (2) as a probe molecule. A double stimulated echo pulse sequence with bipolar gradient pulses and an Eddy current delay were chosen to compensate for possible convection and Eddy currents, which might occur inside mesoporous materials (vide infra).58 Thus, the signal intensity I in the experiment is described using eqn (1).
 | (1) |
Fig. 1 shows the result of the MAS PFG NMR measurement. The logarithmic intensity is plotted against the squared gradient strength. A linear decay indicating a single diffusing component is found. The diffusion coefficient was calculated using eqn (1).
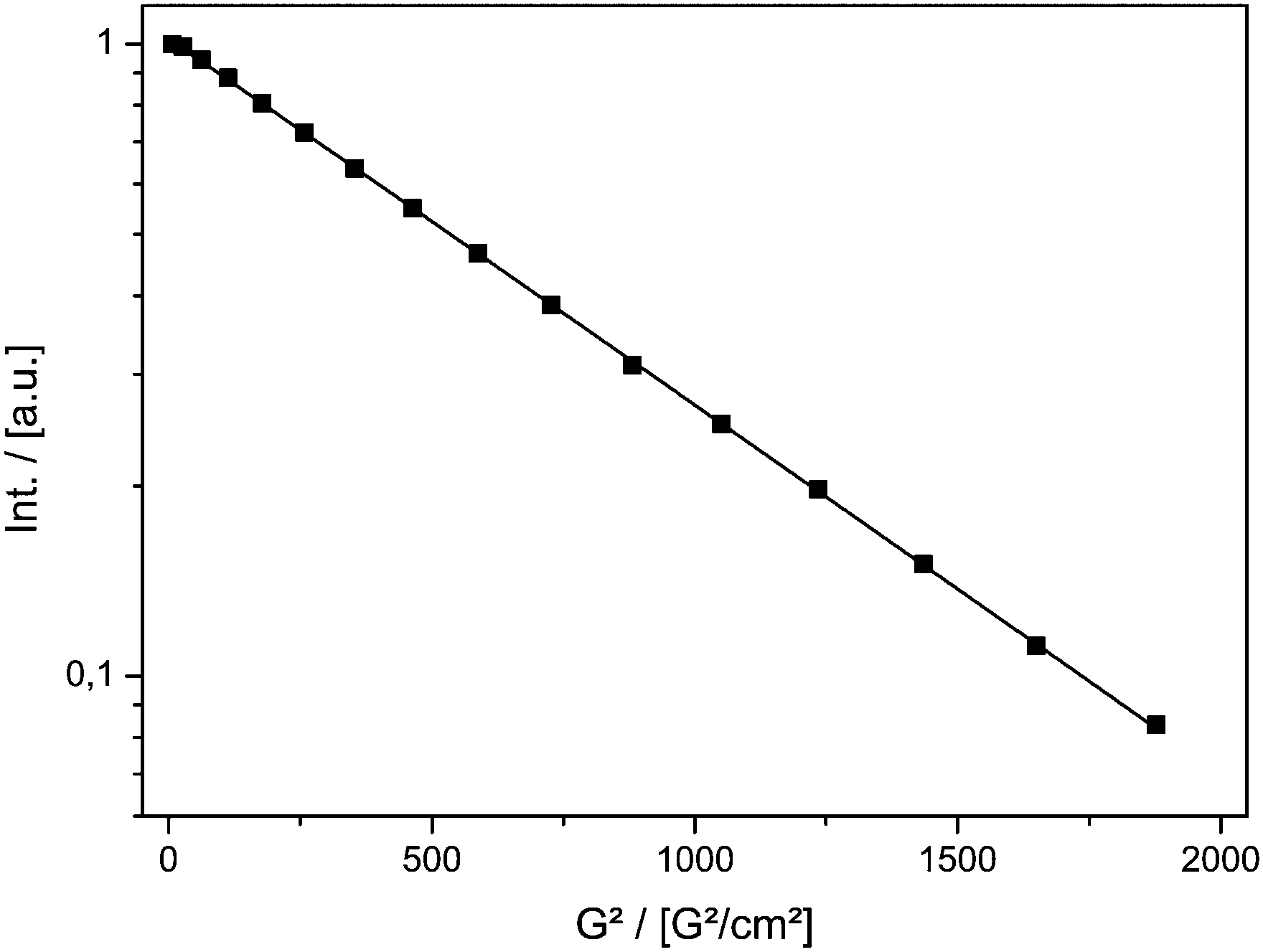 | ||
| Fig. 1 Signal decay of TEMPONE-OH in ethanol measured by PFG MAS NMR as a function of the squared gradient strength of the double stimulated echo experiment. | ||
The value at T = 298 K Dtr(NMR) of 5.56(±0.08) × 10−10 m2 s−1 can be compared to the value reported in the literature obtained using the Taylor dispersion method (Dtr = 8.4 × 10−10 m2 s−1).57Dtr is in the correct range, but is underestimated by ≈30%.
Information about the molecular diffusion of TEMPONE can be obtained from cw-EPR data by measuring the concentration broadening of the EPR lines. Temperature dependent EPR studies of TEMPONE in ethanolic solution were performed for two different concentrations c (see Fig. 2a). For c = 0.5 × 10−3 mol l−1 a well-resolved spectrum composed of three lines is observed. This spectral appearance is expected because of the coupling of the unpaired electron of the nitroxide group with the nuclear spin of the nitrogen atom.59,60 By increasing the temperature from 223 K to 303 K, the intensity of the high field line increases, because of an increase of molecular tumbling of the radicals.61 The line width, measured as the peak to peak distance ΔBpp from maximum to minimum of a single spectral line, does not change significantly within the observed temperature range for the low concentrated solution.
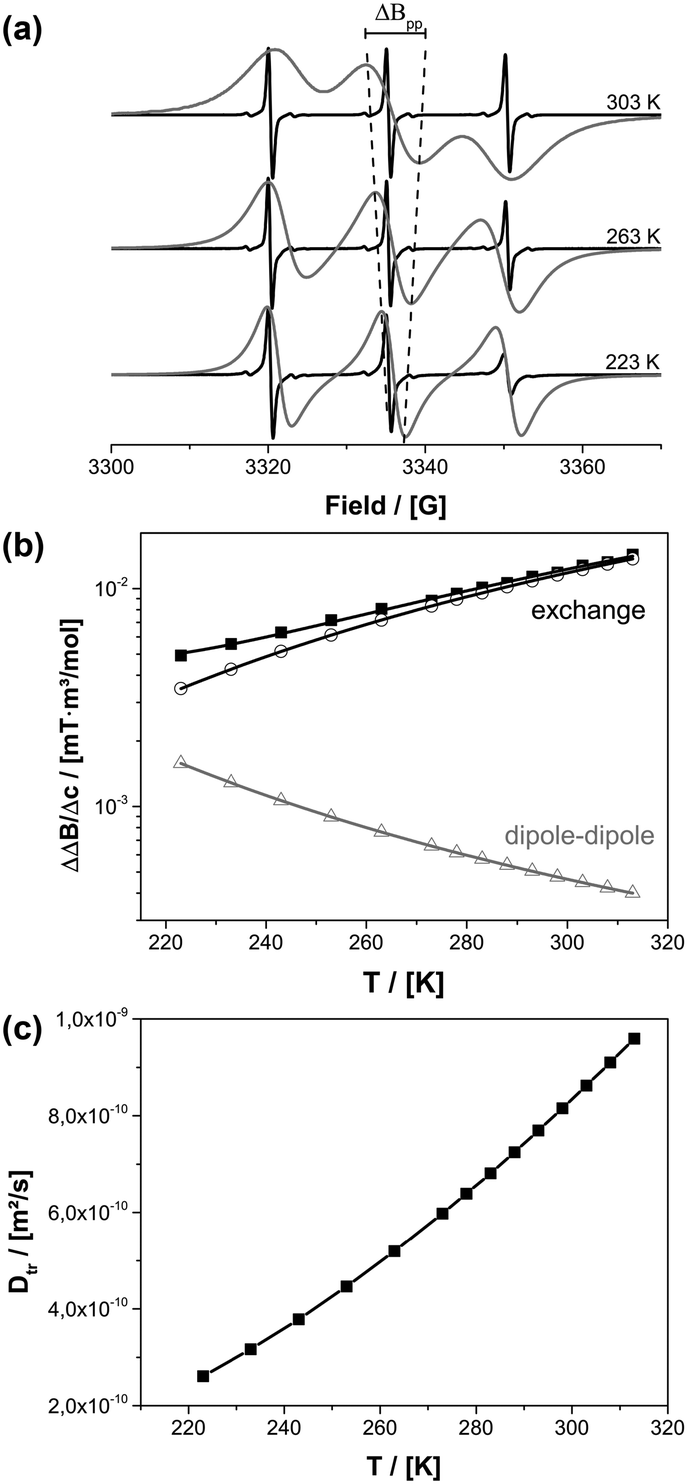 | ||
| Fig. 2 (a) EPR spectra for TEMPONE in ethanol at different temperatures and concentrations of 0.5 mM (black) and 50 mM (grey). The line width change of the middle field transition is shown as a dotted line for clarity. (b) Difference between the intrinsic line width at 0.5 mM and the broadened line width at 50 mM (divided by the concentration difference) ΔΔB/Δc of the low field transition for different temperatures. (c) Dtr calculated for all temperatures from cw-EPR data of (b). | ||
In contrast to this, an increase in peak to peak line broadening is observed for the spectra obtained at a concentration of c = 0.5 × 10−2 mol l−1 as indicated by the dotted line in Fig. 2a. The concentration broadening increases continually from 223 K to 303 K until the different transitions of the spectrum interfere with each other at 303 K. There are two reasons for the concentration dependence: dipole–dipole interaction and Heisenberg spin exchange. The dipole–dipole interaction depends on rotational correlation time, which is proportional to η/T. At low temperature the line width is large due to an increased viscosity of the solvent and line broadening of paramagnetic species is determined mainly by dipole–dipole interactions with nearby radicals. By increasing the temperature, viscosity decreases and the dipole–dipole interactions average out leading to a smaller line width. At the same time diffusion increases and therefore Heisenberg spin exchange increases due to an enhanced collision frequency of the radicals. This again results in EPR line broadening at a higher temperature.
To date, there is no exact analytical solution to determine the fraction of dipole–dipole and exchange interactions from the line width.62 A procedure was developed by Freed and coworkers.63 The line broadening for a given concentration is described according to eqn (2).
| ΔΔBc = ΔBpp(Δc + c0) − ΔBpp(c0) | (2) |
 | (3) |
ΔΔB was calculated for the peak to peak distance of the low field line. Fig. 2b shows the contribution of Heisenberg spin exchange and dipole–dipole interaction calculated according to eqn (3) by varying A, B and Eatr. It is seen that over the entire temperature range spin exchange dominates the line width. Lower temperatures could not be considered because, in agreement to earlier studies, it was found that in this temperature region an analysis according to eqn (3) is questionable.40,64 For temperatures below 223 K it is not possible to differentiate the contributions of spin exchange and dipole–dipole interaction because of a strong change of the intrinsic line width originating from an increase in viscosity (see ESI-3†). One can now extract the spin exchange rate constant ke (eqn (4)).
 | (4) |
The spin exchange rate constant is connected to the diffusion coefficient by using the Einstein–Smoluchowski equation (eqn (5)).
| ke = 16πfrDtr | (5) |
The diffusion of TEMPONE (in ethanol) as a function of temperature was calculated and is shown in Fig. 2c. The value at T = 298 K Dtr(EPR) = 8.16(±0.90) × 10−10 m2 s−1 is in good agreement with the value reported in the literature using the Taylor dispersion method (Dtr = 8.8 × 10−10 m2 s−1).57 However, it has to be mentioned that the calculated diffusion coefficient from cw-EPR depends on the encounter distance of the radicals for spin exchange. In agreement with Nayeem et al., we used an encounter distance of 6.4 Å instead of the crystallographic molecule radius of 3.2 Å.63 They also discussed an encounter distance of 4.6 Å derived from other literature data, which would lead to an excellent agreement with our NMR experiment (Dtr = 5.87 × 10−10 m2 s−1). It is obvious that the used encounter distance can lead to a certain failure and therefore we calculated the diffusion coefficient independently by the Stokes–Einstein approach. In this case the rotational correlation time of TEMPONE was determined from the spectra by simulation and used to first calculate the hydrodynamic radius from the Stokes–Einstein–Debye equation (6a)65 and afterwards to calculate Dtr using again Stokes–Einstein for translational diffusion (6b):
 | (6a) |
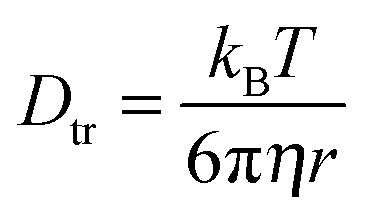 | (6b) |
It is seen that the diffusion coefficients obtained from EPR yield reliable results, and EPR seems to be suitable as a complementary technique for PFG NMR. This is the first important result of our study.
Thus, the adsorption properties of TEMPONE and TEMPONE-OH on a silica surface, avoiding any confinement situation, have been characterized by isothermal titration calorimetry (ITC). For realizing a comparably large surface area, compared to the nanoporous solids, the adsorption has been studied in the presence of compact silica nanoparticles with a diameter of 8 nm (see ESI-4†). ITC is able to measure binding affinities of any type of molecular interaction and thermodynamic parameters in solution. It is possible to determine weak and non-specific surface interactions between the used probe molecules and a silica surface.66 An adsorption enthalpy of (7.8 ± 1.1) kJ mol−1 and (1340 ± 117) binding sites per nanoparticle for TEMPONE and (7.4 ± 0.4) kJ mol−1 and (1240 ± 25) binding sites for TEMPONE-OH, respectively, were observed. Furthermore, the adsorption enthalpy is low, which is why one should not expect any significant effects due to non-specific interaction with silica surfaces. The binding sites fit well to a bimolecular adsorption shell for both probe molecules. Approximately 624 possible adsorption sites for one monolayer have been calculated from geometric considerations. The equal adsorption enthalpies of TEMPONE and TEMPONE-OH confirm identical surface interactions. It will thus be possible to compare the diffusion results for the confined solution in the porous materials.
The different electronic structures of the probe molecules may also lead to different molecule–solvent interactions especially hydrogen bonds in alcohols. A possibility to reveal this is the temperature dependency of the diffusion coefficient. Therefore, temperature-dependent studies have not only been performed for TEMPONE (EPR; Fig. 2c) but also for TEMPONE-OH (NMR; see ESI-5†). As expected, Dtr of TEMPONE-OH increases with temperature. Comparison with Dtr of TEMPONE from cw-EPR shows that the temperature dependency of both probe molecules is comparable within the investigated interval (see ESI-5†). This confirms that there is no major difference observable in the solvent interaction within our study. This seems to be irritating since the changed electronic structure should influence the hydrogen bonding behaviour, but the observation was in accordance with the literature. Terazima reported that the differences in diffusion between stable radicals like nitroxides and their closed shell derivatives are very small in many solvents and therefore we think they are ideal probe molecules to study diffusion on different length scales by EPR and NMR.57
3.3. Diffusion studies on mesoporous materials
It was demonstrated above that MAS PFG NMR and cw-EPR deliver comparable results for diffusion studies on spin probes performed under non-confinement conditions. It is important to note that the situation changes for confinement in mesoporous hosts. Because NMR is sensitive to the diffusion path length on the order of up to hundreds of μm, and via EPR one probes the sub 10 nm scale, there should be differences as soon as the mean free path becomes smaller than the mentioned, characteristic dimensions (see Scheme 1). The latter scenario is obviously given in nanoporous materials with pore sizes below 10 nm (see Table 1). | ||
| Fig. 3 Signal decay of TEMPONE-OH in ethanol confined to MPS-54 measured by MAS PFG NMR as a function of the squared gradient strength of the double stimulated echo experiment. The result of a two component alignment is shown as a black line. | ||
Next, the diffusion coefficient of the slow, confined component D2tr was determined for different pore-sizes (see Fig. 4a). There is a significant influence of pore-sizes below 8 nm. In this region D2tr decreases with decreasing pore size. Above 8 nm the dependency is weaker, but D2tr still remains smaller compared to the unconfined situation. The latter results show that the confinement conditions, as expected, strongly influence diffusion on the mesoscale.
 | ||
| Fig. 4 (a) D2tr coefficients provided by NMR data using TEMPONE-OH (black squares) and by EPR data using TEMPONE with c = 0.1 mol l−1 (Δc = 0.0995 mol l−1; grey circles) and c = 0.05 mol l−1 (Δc = 0.0495 mol l−1; grey triangles) within mesoporous silica materials of various pore sizes. Dependence of the D2tr(EPR) parameter on Δc for TEMPONE in free solution (b) and confined in a mesoporous host (MPS-54) (c). The dotted line in (b) represents D2tr calculated from the Stokes–Einstein equation. | ||
Analogous investigations were performed using cw-EPR and TEMPONE. As was found for the NMR experiment, two components were identified. The results are discussed for MPS-54 as a representative case (see Fig. 5). The spectra recorded at different temperatures are dominated by a set of sharp, three-line signals characteristic of mobile TEMPONE species. In contrast to the behaviour in the free solution (see above; Fig. 2), the rotational correlation time is reduced compared to the reference state and remains almost constant by increasing the temperature in the mesoporous material.
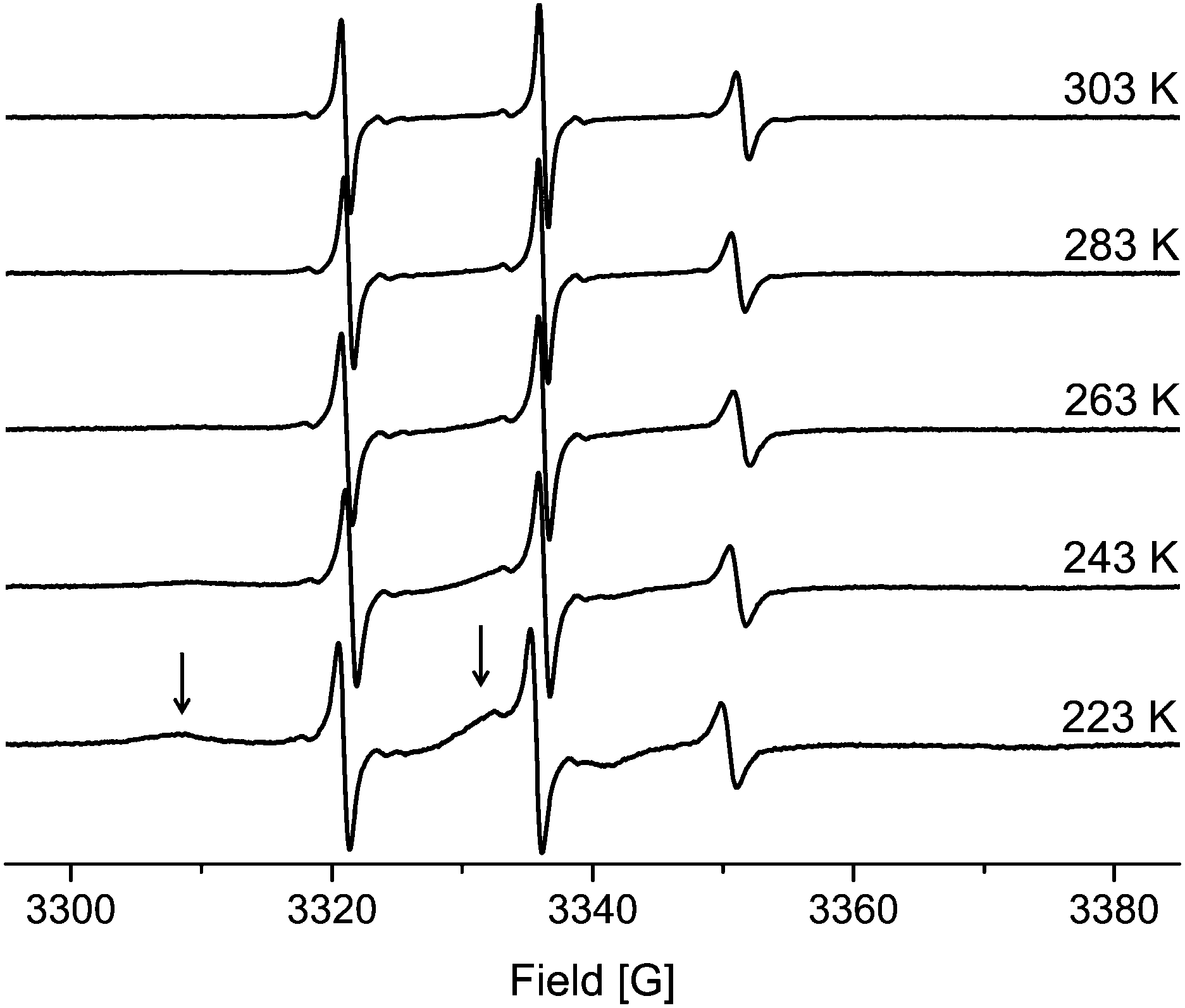 | ||
| Fig. 5 EPR spectra of TEMPONE in ethanol confined to MPS-54. The arrows indicate the adsorbed molecules within the material for the 223 K spectra. | ||
On closer examination, an additional, broad signal is observed at lower temperature. Such spectral features agree with spin probes in a state of slow tumbling. In agreement with the literature this component can be identified as a surface adsorbed species.22 The latter is supported by analysing the temperature dependency. The fraction of the broad, slow rotating component decreases with increasing temperature while the intensity of the sharp signals corresponding to the fast rotating component increases. Higher temperature leads to less adsorption and to more freely diffusing molecules. Thus, the origin of the two components mentioned is different compared to the species identified by NMR spectroscopy.
Only diffusing molecules can show spin exchange, and only these molecules should be considered for the calculation of Dtr(EPR) under confinement. Thus, for a quantitative evaluation of the data using eqn (2) and (3), one has to account for the fraction of surface-bound TEMPONE. A corrected concentration c(T) is defined in eqn (7).
| c(T) = ffree(T)·c0 | (7) |
All spectra have been simulated to determine the exact fraction of free and adsorbed TEMPONE radicals within the materials for each temperature. Considering this correction term, D2tr values have been obtained from cw-EPR measurements using the same methodology introduced for the non-confined situation. We found that in MPS-41 the line broadening is dominated by dipole–dipole interaction and spin exchange is negligible (see ESI-7†). Therefore, D2tr was determined only for porous materials with pore-sizes larger than 4.1 nm (Fig. 4). Because EPR is sensitive towards short-range interactions and/or collision with nearby radicals on a molecular scale (sub 10 nm), it is expected that TEMPONE will experience confinement effects much more strongly compared to the NMR studies as NMR probes molecular movement over mesoscopic length scales (up to a few micrometres). The immense impact of the spatial constraints resulting from the nanoporous environment can be demonstrated by performing concentration dependent experiments (see Fig. 4c). In comparison to the free solution (Fig. 4b), where we find (as expected) no correlation of D2tr with Δc, inside mesopores the parameter varies with the TEMPONE concentration (Fig. 4c). The latter was a surprising result and requires further explanation (see Scheme 2).
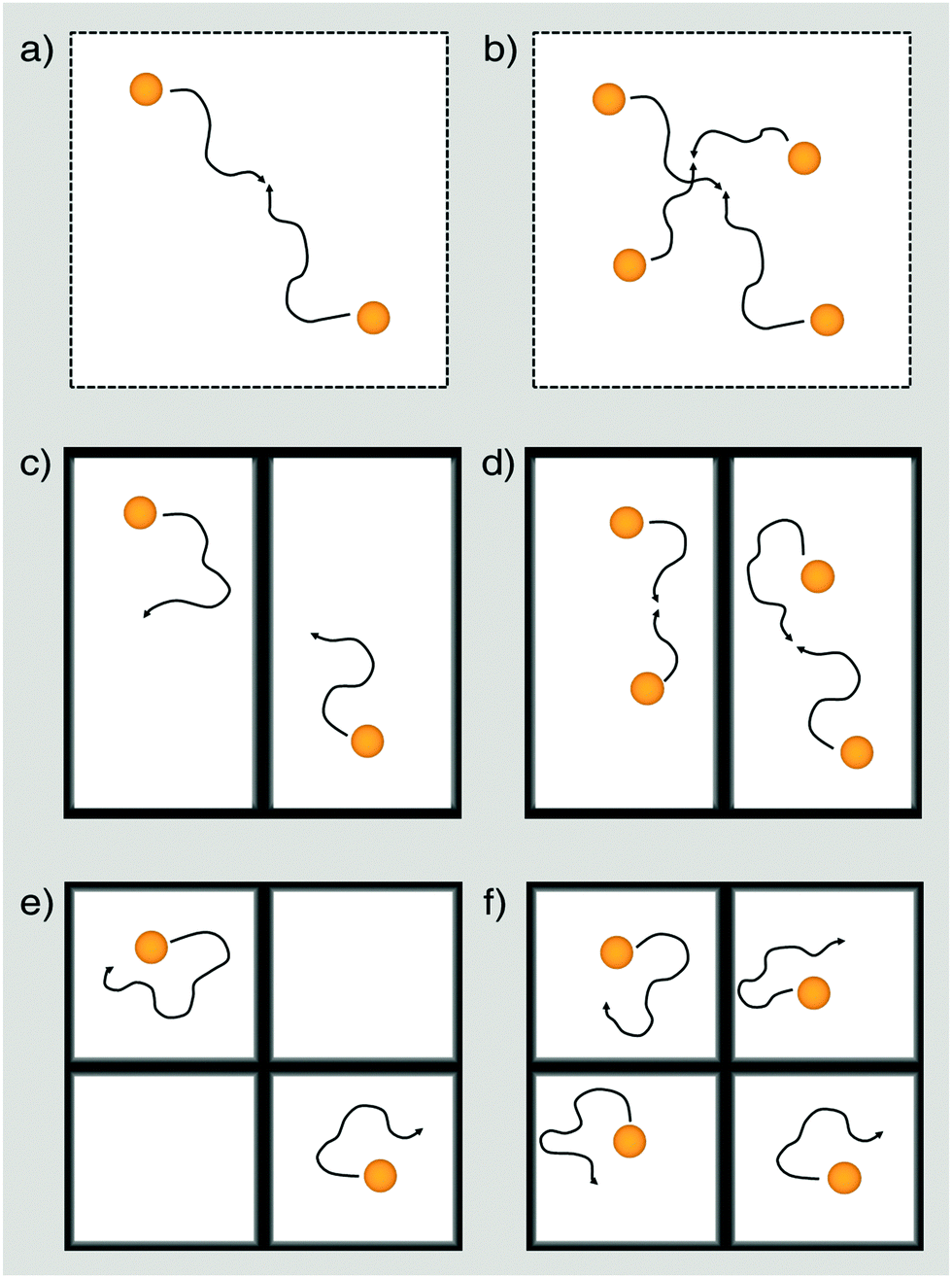 | ||
| Scheme 2 Diffusion and collision of TEMPONE as seen by cw-EPR under non-confined (a, b), weakly confined (c, d) and strongly confined conditions for a low concentrated (a, c, e) and a high concentrated solution (b, d, f). The weak confining conditions represent materials of a large pore size (e.g. MPS-122 or MPS-80), while the strong confining conditions correspond to materials of a small pore size (e.g. MPS-54). At a low concentration (in our case 0.05 mol l−1) cw-EPR is only sensitive for collision between TEMPONE radicals in a non-confined solution (a) but not under weak (c) or strong (e) confinement. For high concentrated solutions, cw-EPR is capable of non-confining conditions (b) and materials showing weak confining conditions. | ||
In agreement with previous studies performed on confinement effects on excimer formation,67 it is seen that compartmentalization leads to a reduction of the probability for intermolecular pathways. Because spin exchange is an intermolecular process, it will also be influenced by confinement. When the concentration of the spin probe is too low, there is only insufficient spin exchange and the evaluation of D2tr is hampered. This can be seen in Fig. 4a, where D2tr(EPR) and its pore-size dependence is shown for c = 0.05 mol l−1 (Δc = 0.0495 mol l−1). Although confinement is enhanced for smaller pore-sizes, and this expectedly affects D2tr(NMR), D2tr(EPR) remains unaltered (Scheme 2c and e). In contrast to this, for a TEMPONE concentration of c = 0.1 mol l−1 (Δc = 0.0995 mol l−1) D2tr(EPR) is sensitive to diffusion and reproduces the tendencies found by MAS PFG NMR. Higher concentrations of the spin-probe were not tested, because the solubility of TEMPONE is restricted, and based on our experiments (see also below) we are sure that we are above the threshold, when the spin probes show spin exchange. In addition, at higher concentration spin exchange leads to line narrowing, making it impossible to evaluate the data and to calculate diffusion coefficients.
The confinement influence on spin exchange is very interesting and is examined further in detail in Fig. 6. As discussed for the free solution, from the ΔΔB/Δc plots one can gather information about how the spin probe is influenced by dipole–dipole interaction versus spin exchange (see Fig. 2b). One clearly sees (Fig. 6a) that for c = 0.075 mol l−1 (Δc = 0.0745 mol l−1) and inside 5.4 nm pores the behaviour is dominated by dipole–dipole interaction. The existence of dipole–dipole interactions shows that the spin probes are in proximity to each other, but they cannot get close enough for spin exchange. Exchange interaction can only be observed at higher temperature. A good criterion for this is the temperature of minimal line width Tm, which is observed at 303 K (Fig. 6a). At this point dipole–dipole interaction and spin exchange equally contribute to the observed line width (compare eqn (3)). Above this temperature the line width mainly depends on exchange interaction while, at lower temperature, the line width is mainly determined by the dipole–dipole interaction. The observation of strong dipole–dipole interaction indicates that the inter-spin vector cannot strongly reorient on the timescale of the cw-EPR experiment. This is an expected behaviour for two molecules moving in relatively small pores which are not left on that time scale since the inter-spin vector between them cannot reorient significantly on that time scale (compare Scheme 2e). Consequently, an explanation for the reduced diffusion coefficient might be that at a low concentration we observe diffusion between different pores or rather collisions between radicals from different pores. The situation changes for MPS-80, which is identical to MPS-54 except for the greater pore diameter (8 nm). The temperature of the minimal line width is already observed at 269 K (Fig. 6a). Fig. 6b shows the influence of the TEMPONE concentration. Increasing the concentration of TEMPONE does not change the fraction of exchange and dipole–dipole interaction for a given material as seen for MPS-80, also indicated by only small shifts in Tm, but the overall interactions gain in intensity (see Scheme 2). At a high concentration cw-EPR delivers a similar diffusion coefficient like MAS PFG NMR (see Fig. 4a). From this we conclude that cw-EPR is now sensitive to diffusion within a pore.
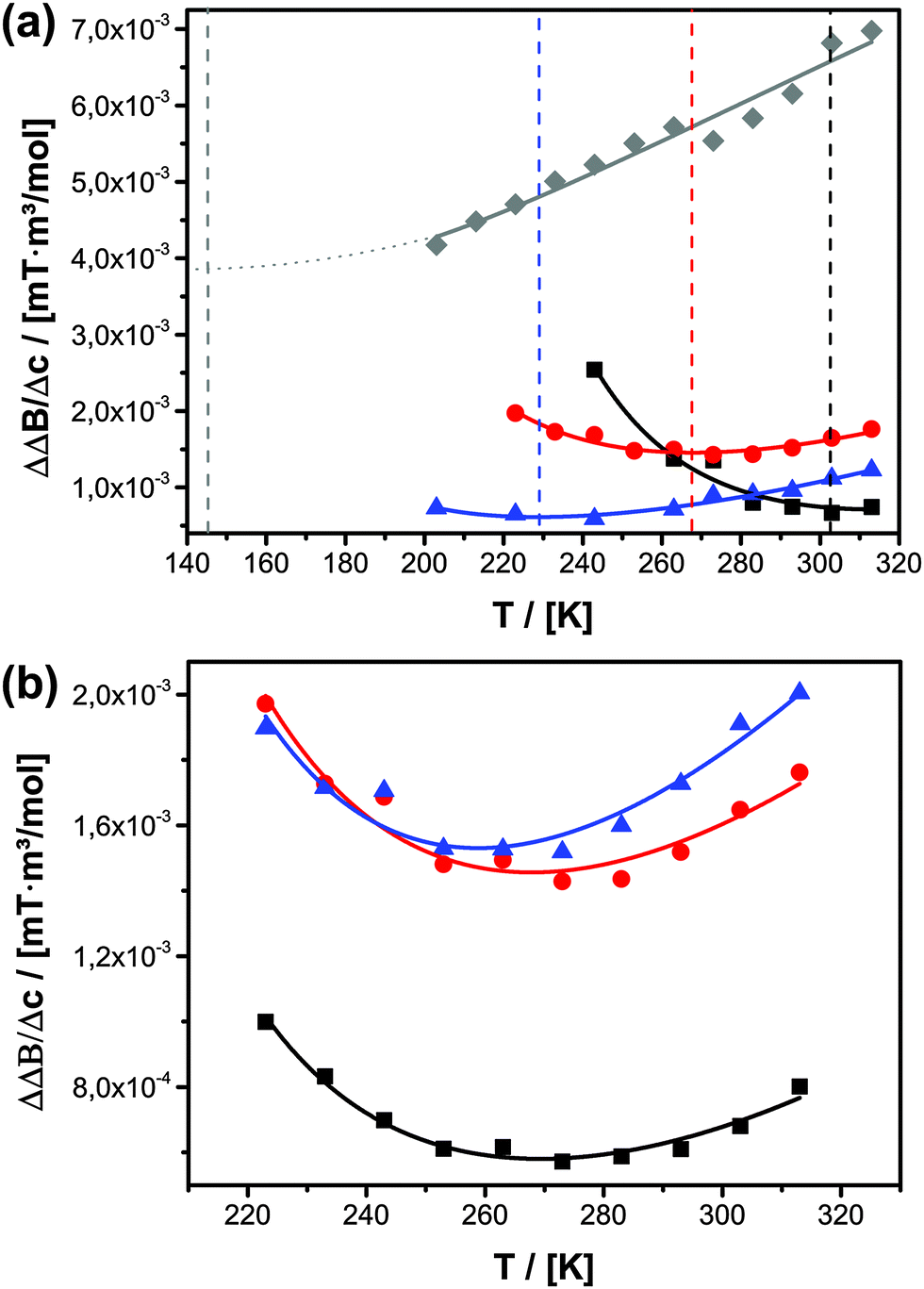 | ||
| Fig. 6 (a) Temperature-dependent ΔΔB/Δc plots for Δc = 0.0745 mol l−1 for TEMPONE confined in MPS-54 (black squares), MPS-80 (red circles), UKON-2a56 (grey diamonds) and MPOSA-48 (blue triangles). The vertical lines indicate the temperature of the minimal line width. (b) Temperature-dependent ΔΔB/Δc plots for three different concentrations. Δc = 0.0495 mol l−1 (black squares); Δc = 0.0745 mol l−1 (red circles); Δc = 0.0995 mol l−1 (blue triangles) for TEMPONE confined in MPS-80. | ||
Thus, by using the EPR spin probe it is possible to precisely adjust the (diffusion) length scale when the probe molecules start to “feel” each other with respect to spin exchange (Scheme 2). As a consequence, we expect that very sensitive studies focusing on the molecular scale (Scheme 1) can be conducted also addressing molecular factors influencing mass transport inside mesoporous materials, most importantly surface effects. The disadvantage is that D2tr(EPR) depends on several parameters and is not anymore a constant as it is in the bulk phase. In context of cw-EPR as a complementary technique to PFG NMR this is no disadvantage, since it enables to get further information about molecular processes, which are not available by PFG NMR (Scheme 1). In conclusion, it is important for the EPR studies to employ considerably higher concentrations of TEMPONE as in the free solution (Fig. 4b and c). Only when Δc is 0.0995 mol l−1 or higher, one is able to see similar confinement effects on D2tr(EPR) compared to D2tr(NMR). A higher concentration of the spin probes was not investigated due to several reasons: the solubility of TEMPONE is restricted and it is not sure how the solubility within mesoporous materials changes compared to bulk solution. Our data already show that adsorption slightly increases at 100 mM compared to the lower concentrated solutions. As shown in Fig. 6b the difference between Δc = 74.5 mM and 99.5 mM is small compared to the difference between Δc = 49.5 mM and 74.5 mM. Therefore we are sure that we observe Dtr which is very close to the true value. In addition, at a higher concentration spin exchange leads to line narrowing, making it impossible to evaluate the data and to calculate diffusion coefficients. In conclusion the scaling effect is unlikely for diffusion between molecular diffusion and mesoscale diffusion. This is probably a consequence of small adsorption on the silica materials at room temperature. In contrast to this, we observed a stronger surface influence for the functional materials and an expected scale effect between molecular and mesoscale diffusion (see Section 3.3.2).
Finally, an important question is if the observed tendencies for molecular-scale (EPR) and mesoscale (NMR) diffusion are in agreement with observations on macroscale diffusion. Therefore, we resort to an EPR imaging approach combined with numerical simulations. In comparison to other techniques, EPR imaging is still in its infancy.39,68–70 However, it has for example been used for measuring macroscopic diffusion through two different types of porous Al2O3.69 In the context of biological membranes, cw-EPR and EPR imaging has also been combined to reveal differences between molecular and macroscopic diffusion.39Fig. 7 shows the time evolution of the 1d projection of the spin density ρ1d(y,t) during the diffusion process of TEMPONE through a monolithic MPS-54 particle filled with ethanol (initial concentration of 10 mM TEMPONE above the monolith).
 | ||
| Fig. 7 Macroscale diffusion of TEMPONE through a monolithic MPS-54 particle from top to bottom (see also ESI-8†). Left: experimentally obtained time evolution of the 1d projection of the spin density during the diffusion process, right: corresponding numerical simulation. | ||
The diffusion process was monitored by EPR imaging (left) and was numerically simulated (right). Each vertical slice corresponds to the concentration profile of TEMPONE within the particle at a given time, e.g. at the beginning of the experiment the spin label is concentrated at the top of the sample. Fitting with the numerical simulation revealed the macroscopic translational diffusion coefficient within the sample (Dtr,MPS-54 = 7.07(±2.2) × 10−11 m2 s−1). First, it is seen, that the diffusion coefficient is in the order for DIPH, proving that the method yields reliable values. For MPS-54 diffusion is reduced on the macroscale compared to mesoscale. This is a reasonable result because at a larger scale confined diffusion is expected to differ, because now the number of parameters impeding it has become larger. Therefore, EPR mapping can give valuable information about how a DIPH is affected by other factors beyond the molecular scale, like pore-connectivity, grain size of the porous particles reducing the mass transport rate. Instead, significant parts of the open volume inside a porous material would lead to an increase in D compared to the lower scales (see also ESI-8 and ESI-9†). Using the combination of complementary methods cw-EPR (for the molecular scale DIPH studies <10 nm), MAS PFG NMR (for the mesoscale diffusion >10 nm) and EPR-mapping (for the macroscopic scale >μm) it might be possible in the future to separate the different contributions to DIPH from each other, and to achieve a more rational design of porous materials. Furthermore, a potential we have not explored in the current paper is that EPR mapping can be used to obtain information about the local diffusive behaviour in macroscopic porous bodies like chromatography columns.
| Material | D 2tr(NMR) [10−11 m2 s−1] | D 2tr(EPR) [10−11 m2 s−1] | σ r(EPR) [10−11 s] |
|---|---|---|---|
| No confinement | 55.6 ± 0.1 | 81.6 ± 9.0 | 1.74 |
| MPOSA-48 | 5.25 ± 0.06 | 14.06 ± 4.40 | 4.66 |
| UKON1-53 | 10.10 ± 4.54 | 15.82 ± 0.66 | 4.45 |
| UKON2A-56 | 17.60 ± 1.10 | 31.60 ± 2.18 | 3.55 |
It is seen that the surface groups on the pore-walls have a significant effect on diffusion inside the porous host. The changes in D2tr are now in the range ΔD2tr ≈ 17 × 10−11 m2 s−1, which is higher compared to the changes provoked by confinement alone within our study (ΔD2tr < 6 × 10−11 m2 s−1). A correlation of D2tr with the surface functional groups of the organosilica materials is difficult to derive due to the amphiphilic character of TEMPONE. From MPOSA-48 (trimethylsilyl surface) over UKON1-53 (bromobenzene surface) to UKON2a-56 (benzoic-acid surface) the surface polarity increases but for MPOSA-48 and UKON1-53 similar diffusion coefficients are observed. Whereas short-range van der Waals interactions are always present, in the case of polar surfaces additional dipole–dipole interactions with a longer range and eventually hydrogen bonding (for UKON-2a) will also influence the spin probes. Furthermore, one has to consider a potential amphiphilic character of TEMPONE, which was discussed by us in a previous paper.22 It was derived that the mobility of the spin probe is high if the polarity difference between the spin probe, the solvent and the surface is small. The fastest D2tr is found for UKON2A-56 which is only by 60% smaller than the value determined for TEMPONE in free solution. This is in agreement with our earlier study, in which the highest mobility for 3-carboxy-PROXYL (carboxy-2,2,5,5-tetramethyl-1-pyrrolidinyloxy), a spin probe with a carboxy functional group, in ethanol and UKON2A was observed. In all cases molecular scale diffusion is faster than mesoscale diffusion indicating that the surface influences diffusion stronger on a longer scale.
Again, we can now obtain additional information about the system from the EPR-data, respectively from the ΔΔB/Δc plots (Fig. 6a). It is seen that the influence of different surface groups is much bigger than an increased pore size and the intermolecular interactions are dominated by spin exchange. In MPOSA-48 Tm is 228 K and in UKON2A-56 Tm is even lower than 200 K. Unfortunately, no reliable EPR data could be obtained at lower temperatures due to the same reasons mentioned before.
There are two possibilities how the surface functional groups of the mesoporous material could influence the diffusivity of confined guests. Most likely the guest–wall interaction changes the overall mobility of the spin-probe within the pore system. Strong interaction should lead to low mobility, because there is a higher chance for the guest species for being adsorbed at the surface. It should be noted again that regarding D2tr(EPR) we are only considering the mobile fraction of TEMPONE. Furthermore, we only systematically varied the pore surface for a fixed pore size. Therefore, an alternative explanation is required. Because the voids in mesoporous materials are so small, there is an intimate contact between solvent molecules and surface groups as well (Scheme 1). This contact could change the properties of the solvent itself and this in conclusion affects the mobility of the spin probe. The information contained in EPR spectra about the rotation of the spin probe is important in this context. On the time scale of the cw-EPR experiment it is possible to clearly distinguish between surface adsorbed and free rotating spin probes, which are surrounded by ethanol. Under non-confined conditions TEMPONE shows σr = 1.74 × 10−11 s. Within the series of the organosilica materials σr is smallest (TEMPONE rotation is fastest) also in UKON2a (see Table 2). Thus, there are several pieces of evidence that the functional groups present at the surfaces of porous materials can indeed influence the rotational and translational mobility of TEMPONE in the confined liquid. This in reverse explains the differing influence of spin exchange on the line width. Because the rotation correlation time σr depends on solvent viscosity (eqn (6a)), one possible explanation is that the surface bound groups of the mesoporous host could also have an effect on the solvent viscosity, at least close to the surface, and this might alter the diffusional behaviour. This interesting hypothesis will be investigated in further detail in future studies.
4. Summary and conclusions
The macroscopic diffusion of dissolved molecular species inside porous materials is undoubtedly a consequence of mass transport processes at the mesoscale (10 nm to 1 μm). However, it is also clear that mesoscale diffusion itself is a direct consequence of processes at the molecular level (0–10 nm) (see Scheme 1). We have seen that it is indeed extremely difficult to accomplish a detailed picture of those molecular processes in very small pores (5–15 nm), because of a complex interplay between various factors. These factors involve temperature, confinement effects, probe–solvent interactions, solvent–surface interactions and probe–surface interactions. Because EPR spectroscopy fits ideally with regard to time and length scale, we wanted to establish it as an experimental method for unravelling some of the effects molecular probes experience under strong confinement conditions, in particular, when the effects of nearby surfaces cannot be excluded. Our idea was to extract information from the dipole–dipole and spin-exchange interaction between dissolved, molecular spin probes (persistent nitroxide radicals), and to verify the findings using MAS PFG NMR spectroscopy as a complementary method, which is sensitive to mesoscale diffusion but not to the above-mentioned molecular interactions. In addition, a novel method (EPR imaging) was applied to gather information about macroscale diffusion.First, we succeeded in demonstrating that the results of cw-EPR and MAS PFG NMR experiments for a conventional, free solution of the spin-probes as a reference agree well with each other. Then, investigations on different porous materials followed. The pore size and the type of surface bound organic functionalities have been varied systematically. As expected we observed a significant influence of pore size. Diffusion decreases all the more the smaller the pores get. Roughly, the diffusion coefficient is reduced by a factor of ten in the pore size regime of 5–15 nm compared to the free solution. Surface bound groups located at the pore walls exert an even larger influence on the diffusive behaviour of the guest species. These strong effects could be explained by a change of the solute–solvent properties under confinement. It was seen that not only guest–surface interactions play an important role, but the solvent–surface interaction seems to affect the mobility of the spin probe decisively.
We can conclude that there are considerable advantages of the EPR methodology presented in the current paper compared to other methods used for the investigation of diffusion inside porous hosts like MAS PFG NMR. Cw-EPR spectroscopy is more sensitive towards porous hosts, with functionalized surfaces. Whereas for pure silica materials, there were barely any differences in mass transport observed using cw-EPR and PFG-MAS-NMR, differences were observed for organically functionalized materials. This is because cw-EPR is more suitable for the characterization of the microenvironment, for example intermolecular interactions or adsorption. Furthermore, we could access in-depth information about the intermolecular processes (spin exchange, dipole–dipole interaction) and the physical behaviour (rotation, adsorption) of the spin probes under confinement, which is not accessible by PFG NMR alone. It is also worth mentioning that MAS PFG NMR requires rather sophisticated, non-standard equipment, while cw-EPR can be performed using any standard spectrometer, which significantly unburdens DIPH studies.
Acknowledgements
MW and SP acknowledge the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) for funding within the SPP 1570 (PO 780/14-1). Further financial support from the DFG (DR 743/7-1) is gratefully acknowledged.Notes and references
- F. Schuth, K. S. W. Sing and J. Weitkamp, Handbook of Porous Solids, Wiley-VCH Verlag GmbH & Co. KGaA, 2002 Search PubMed.
- T. K. Perkins and O. C. Johnston, Transactions of the Society of Petroleum Engineers of Aime, 1963, 228, 70–84 CrossRef PubMed.
- F. J. Keil, Catal. Today, 1999, 53, 245–258 CrossRef CAS.
- R. Krishna, J. Phys. Chem. C, 2009, 113, 19756–19781 CAS.
- I. Medved and R. Cerny, Microporous Mesoporous Mater., 2011, 142, 405–422 CrossRef CAS PubMed.
- R. Krishna, Chem. Soc. Rev., 2012, 41, 3099–3118 RSC.
- D. Ruthven, S. Brandani and M. Eic, in Adsorption and Diffusion, ed. H. Karge and J. Weitkamp, Springer, Berlin, Heidelberg, 2008, vol. 7, pp. 45–84 Search PubMed.
- J. Kärger, T. Binder, C. Chmelik, F. Hibbe, H. Krautscheid, R. Krishna and J. Weitkamp, Nat. Mater., 2014, 13, 333–343 CrossRef PubMed.
- J. Kärger and R. Valiullin, Chem. Soc. Rev., 2013, 42, 4172–4197 RSC.
- C. Chmelik and J. Kärger, Chem. Soc. Rev., 2010, 39, 4864–4884 RSC.
- J. Kärger and R. Valiullin, eMagRes, 2011 Search PubMed.
- A. Pampel, F. Engelke, P. Galvosas, C. Krause, F. Stallmach, D. Michel and J. Kärger, Microporous Mesoporous Mater., 2006, 90, 271–277 CrossRef PubMed.
- M. Fernandez, A. Pampel, R. Takahashi, S. Sato, D. Freude and J. Kärger, Phys. Chem. Chem. Phys., 2008, 10, 4165–4171 RSC.
- M. Fernandez, J. Kärger, D. Freude, A. Pampel, J. M. van Baten and R. Krishna, Microporous Mesoporous Mater., 2007, 105, 124–131 CrossRef CAS PubMed.
- C. Carrara, G. Pages, C. Delaurent, S. Viel and S. Caldarelli, J. Phys. Chem. C, 2011, 115, 18776–18781 CAS.
- F. Stallmach, A. Graser, J. Kärger, C. Krause, M. Jeschke, U. Oberhagemann and S. Spange, Microporous Mesoporous Mater., 2001, 44, 745–753 CrossRef.
- F. Guenneau, K. Panesar, A. Nossov, M.-A. Springuel-Huet, T. Azais, F. Babonneau, C. Tourne-Peteilh, J.-M. Devoisselle and A. Gedeon, Phys. Chem. Chem. Phys., 2013, 15, 18805–18808 RSC.
- C. Carrara and S. Caldarelli, J. Phys. Chem. C, 2012, 116, 20030–20034 CAS.
- J. L. Coffman, E. N. Lightfoot and T. W. Root, J. Phys. Chem. B, 1997, 101, 2218–2223 CrossRef CAS.
- M. Sharifi, M. Wark, D. Freude and J. Haase, Microporous Mesoporous Mater., 2012, 156, 80–89 CrossRef CAS PubMed.
- F. Lin, M. Mertens, P. Cool and S. Van Doorslaer, J. Phys. Chem. C, 2013, 117, 22723–22731 CAS.
- M. Wessig, M. Drescher and S. Polarz, J. Phys. Chem. C, 2013, 117, 2805–2816 CAS.
- M. Okazaki and K. Toriyama, J. Phys. Chem. B, 2005, 109, 13180–13185 CrossRef CAS PubMed.
- V. Chechik and A. Caragheorgheopol, Electron Paramagn. Reson., 2006, 20, 96–130 Search PubMed.
- S. Polarz and A. Kuschel, Chem. – Eur. J., 2008, 14, 9816–9829 CrossRef CAS PubMed.
- A. Kuschel, M. Luka, M. Wessig, M. Drescher, M. Fonin, G. Kiliani and S. Polarz, Adv. Funct. Mater., 2010, 20, 1133–1143 CrossRef CAS PubMed.
- A. Kuschel, M. Drescher, T. Kuschel and S. Polarz, Chem. Mater., 2010, 22, 1472–1482 CrossRef CAS.
- M. F. Ottaviani, X. G. Lei, Z. Q. Liu and N. J. Turro, J. Phys. Chem. B, 2001, 105, 7954–7962 CrossRef CAS.
- S. Anandan and M. Okazaki, Microporous Mesoporous Mater., 2005, 87, 77–92 CrossRef CAS PubMed.
- M. Okazaki, S. Seelan and K. Toriyama, Appl. Magn. Reson., 2009, 35, 363–378 CrossRef CAS.
- A. M. Sheveleva, D. I. Kolokolov, A. A. Gabrienko, A. G. Stepanov, S. A. Gromilov, I. K. Shundrina, R. Z. Sagdeev, M. V. Fedin and E. G. Bagryanskaya, J. Phys. Chem. Lett., 2014, 5, 20–24 CrossRef CAS.
- F. Lin, X. Meng, M. Mertens, P. Cool and S. Van Doorslaer, Phys. Chem. Chem. Phys., 2014, 16, 22623–22631 RSC.
- J. Y. Zhang, Z. Luz, H. Zimmermann and D. Goldfarb, J. Phys. Chem. B, 2000, 104, 279–285 CrossRef CAS.
- S. Ruthstein, V. Frydman, S. Kababya, M. Landau and D. Goldfarb, J. Phys. Chem. B, 2003, 107, 1739–1748 CrossRef CAS.
- A. Moscatelli, A. Galarneau, F. Di Renzo and M. F. Ottaviani, J. Phys. Chem. B, 2004, 108, 18580–18589 CrossRef CAS.
- M. Okazaki, P. Jin, K. Ohta and K. Toriyama, J. Phys. Chem. C, 2009, 113, 11086–11094 CAS.
- J. H. Freed, Annu. Rev. Biophys. Biomol. Struct., 1994, 23, 1–25 CrossRef CAS PubMed.
- J.-H. Sachse, M. D. King and D. Marsh, J. Magn. Reson., 1987, 71, 385–404 CAS.
- Y. K. Shin, U. Ewert, D. E. Budil and J. H. Freed, Biophys. J., 1991, 59, 950–957 CrossRef CAS.
- M. Okazaki and K. Toriyama, J. Phys. Chem. C, 2007, 111, 9122–9129 CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. Mccullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- F. Hoffmann, M. Cornelius, J. Morell and M. Froba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS PubMed.
- C. Sanchez, C. Boissiere, D. Grosso, C. Laberty and L. Nicole, Chem. Mater., 2008, 20, 682–737 CrossRef CAS.
- C. Sanchez, P. Belleville, M. Popall and L. Nicole, Chem. Soc. Rev., 2011, 40, 696–753 RSC.
- W. Wang, J. E. Lofgreen and G. A. Ozin, Small, 2010, 6, 2634–2642 CrossRef CAS PubMed.
- N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC.
- P. Van der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC.
- I. Sierra, D. Perez-Quintanilla, S. Morante and J. Ganan, J. Chromatogr. A, 2014, 1363, 27–40 CrossRef CAS PubMed.
- B. Smarsly, S. Polarz and M. Antonietti, J. Phys. Chem. B, 2001, 105, 10473–10483 CrossRef CAS.
- A. Kuschel and S. Polarz, Adv. Funct. Mater., 2008, 18, 1272–1280 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- A. Kuschel and S. Polarz, J. Am. Chem. Soc., 2010, 132, 6558–6565 CrossRef CAS PubMed.
- M. Luka and S. Polarz, Microporous Mesoporous Mater., 2013, 171, 35–43 CrossRef CAS PubMed.
- J. Gehring, D. Schleheck, M. Luka and S. Polarz, Adv. Funct. Mater., 2014, 24, 1140–1150 CrossRef CAS PubMed.
- A. Kuschel, H. Sievers and S. Polarz, Angew. Chem., Int. Ed., 2008, 47, 9513–9517 CrossRef CAS PubMed.
- M. Terazima, Acc. Chem. Res., 2000, 33, 687–694 CrossRef CAS PubMed.
- A. Jerschow and N. Müller, J. Magn. Reson., 1997, 125, 372–375 CrossRef CAS.
- M. Pannier, S. Veit, A. Godt, G. Jeschke and H. W. Spiess, J. Magn. Reson., 2000, 142, 331–340 CrossRef CAS PubMed.
- S. Schreier, C. F. Polnaszek and I. C. P. Smith, Biochim. Biophys. Acta, 1978, 515, 375–436 Search PubMed.
- J. S. Hwang, R. P. Mason, L. P. Hwang and J. H. Freed, J. Phys. Chem., 1975, 79, 489–511 CrossRef CAS.
- K. M. Salikhov, Appl. Magn. Reson., 2010, 38, 237–256 CrossRef CAS.
- A. Nayeem, S. B. Rananavare, V. S. S. Sastry and J. H. Freed, J. Chem. Phys., 1989, 91, 6887 CrossRef CAS PubMed.
- B. Berner and D. Kivelson, J. Phys. Chem., 1979, 83, 1406–1412 CrossRef CAS.
- B. L. Bales, F. L. Harris, M. Peric and M. Peric, J. Phys. Chem. A, 2009, 113, 9295–9303 CrossRef CAS PubMed.
- A. Thibaut, A. M. Misselyn-Bauduin, J. Grandjean, G. Broze and R. Jerome, Langmuir, 2000, 16, 9192–9198 CrossRef CAS.
- A. Thomas, S. Polarz and M. Antonietti, J. Phys. Chem. B, 2003, 107, 5081–5087 CrossRef CAS.
- D. A. Cleary, Y. K. Shin, D. J. Schneider and J. H. Freed, J. Magn. Reson., 1988, 79, 474–492 CAS.
- O. E. Yakimchenko, E. N. Degtyarev, V. N. Parmon and Y. S. Lebedev, J. Phys. Chem., 1995, 99, 2038–2041 CrossRef CAS.
- A. Marek, J. Labsky, C. Konak, J. Pilar and S. Schlick, Macromolecules, 2002, 35, 5517–5528 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ESI-1: a representative data set for one porous material (UKON2a). ESI-2: SEM micrograph of mesoporous materials with monolithic character. ESI-3: peak-to-peak line width of a 0.5 mM solution of TEMPONE in ethanol. ESI-4: adsorption studies of TEMPONE and TEMPONE-OH on silica nanoparticles. ESI-5: temperature dependency of the cw-EPR and PFG MAS NMR experiments. ESI-6: diffusion of molecules between and inside the porous particles. ESI-7: line width difference between 50 mM and 0.5 mM TEMPONE solution in MPS-41M. ESI-8: diffusion data of MPS-122 and MPOSA-48 from EPR imaging. ESI-9: MPS-122 particle from the EPR imaging experiment after the removal of the shrinking tube. See DOI: 10.1039/c5cp01369k |
| This journal is © the Owner Societies 2015 |