
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible room-temperature phosphorescence of pure organic crystals via a radical-ion-pair mechanism†
Shinichi
Kuno
,
Hiroshi
Akeno
,
Hiroyuki
Ohtani
and
Hideya
Yuasa
*
Department of Life Science, Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, 4259 J2-10, Nagatsutacho, Midoriku, Yokohama 226-8501, Japan. E-mail: hyuasa@bio.titech.ac.jp
First published on 13th May 2015
Abstract
The afterglow of phosphorescent compounds can be distinguished from background fluorescence and scattered light by a time-resolved observation, which is a beneficial property for bioimaging. Phosphorescence emission accompanies spin-forbidden transitions from an excited singlet state through an excited triplet state to a ground singlet state. Since these intersystem crossings are facilitated usually by the heavy-atom effect, metal-free organic solids are seldom phosphorescent, although these solids have recently been refurbished as low-cost, eco-friendly phosphorescent materials. Here, we show that crystalline isophthalic acid exhibits room-temperature phosphorescence with an afterglow that lasts several seconds through a nuclear spin magnetism-assisted spin exchange of a radical ion pair. The obvious afterglow that facilitates a time-resolved detection and the unusual phosphorescence mechanism that enables emission intensification by nuclear spin managements are promising for exploiting the phosphorescence materials in novel applications such as bioimaging.
Introduction
Phosphorescent materials have been employed as the emissive layers of organic light emitting diodes (OLEDs)1–3 for enhancing the emission efficiency by taking advantage of three degenerate triplet excited states. Their susceptibility to oxygen quenching can also find applications such as ratiometric bioimaging.4 Another aspect of phosphorescent materials is their afterglow, which is applicable to a time-resolved bioimaging for the autofluorescence-free observation of cells and organisms.5 Most bioimaging-phosphorescent probes examined so far have employed Ir(III) complexes as the emitters. Ir(III) complexes are expensive and environmentally harmful and have relatively short luminescence lifetimes of micro-second order, which make the observation of time-resolved phosphorescence a costly affair. The phosphorescent materials for bioimaging should ideally have a simple structure and a long afterglow at room temperature (RT).While designing phosphorescent compounds, it is a common practice to incorporate heavy atoms to enhance spin–orbit coupling (SOC),6,7 which facilitates the spin-forbidden transitions, for example, intersystem crossings (ISCs) of the excited singlet state (S1) to the excited triplet state (T1), and T1 to the ground singlet state (S0). When it comes to organic molecules without heavy atoms, incorporation of carbonyl groups may enable ISC through 1(n–π*) → 3(π–π*) allowed transitions. However, the phosphorescence of carbonyl compounds is generally inefficient because of the poor absorption efficiency by the forbidden transition from S0 to 1(n–π*).8,9
Another approach to obtain RT phosphorescent materials is the suppression of non-radiative T1 decay by doping a host material, e.g. a filtering paper, with a compound capable of generating T110–12 or attaching a chromophore at the end of a polymer.13 The resulting structure rigidification suppresses the collisional deactivation of T1, which is responsible for the non-radiative relaxation.14 Single-molecular organic crystals, however, tend to be non-phosphorescent, owing to the enhancement of triplet–triplet annihilation (TTA) by T1 exciton migration in a crystal lattice, despite their suppressed collisional deactivation.9
In this study, we found simple organic crystals capable of emitting RT phosphorescence for several seconds. To the best of our knowledge, the visibly persistent RT phosphorescence of heavy atom-free organic single-molecular crystals has been reported only for tetraphenylmethane (Ph4C).15 Since our RT phosphorescent materials are non-phosphorescent in solution, intermolecular, aggregation-promoted T1-generation mechanisms were taken into account.16,17 We observed magnetic field effects (MFEs) on the phosphorescence of isophthalic acid (IPA) crystals, which proved the presence of singlet and triplet radical ion pairs (1RIP and 3RIP). Deuterium labeling of IPA suppressed phosphorescence intensity, suggesting a hyperfine coupling (HFC) mechanism, a nuclear spin magnetism-assisted spin conversion (1RIP–3RIP), for phosphorescence generation. We also observed IPA's charge-transfer (CT) absorption, i.e. direct transition from S0 to RIP, which was theoretically supported by time-dependent density functional theory (TD-DFT) calculations. While the presence of 1RIP and 3RIP in the phosphorescence process became evident, there is also evidence for the presence of T1, a TTA-derived delayed fluorescence. Heavy atom effects on the phosphorescence lifetime demonstrate that the phosphorescence is caused by a forbidden, radiative T1–S0 transition. Overall, photoexcitation of IPA leads to 1RIP, followed by 3RIP through an HFC-induced spin conversion, which is dissociated into T1, and then phosphorescence is emitted by T1. The phosphorescence properties of these simple aromatic compounds can be used for bioimaging even in their aggregated forms, since molecules with aggregation-induced fluorescence property has been used as bioimaging tools.5,18
Results and discussion
Afterglow
We fortuitously found that some crystalline benzoic acid derivatives showed visible afterglows persisting for more than one second at RT on irradiating with a 366 nm light from a handy UV lamp (Fig. 1). The afterglow of Ph4C on irradiation at 254 nm, the phenomenon discovered in 1939,15 is also displayed for comparison. IPA showed the strongest afterglow among the tested compounds; the afterglow was observed even with a 405 nm blue LED flashlight (Video S1, ESI†). Terephthalic acid (TPA) and pyromellitic acid (PMA) also showed afterglows. The afterglow of benzoic acid (Bz) was observable even by the naked eye, whereas its derivatives, orthophthalic acid (OPA), and a benzene hexacarboxylic acid, mellitic acid (MLA), apparently exhibited no afterglows. RT afterglows exceeding the eye's time resolution of ca. 100 ms are quite unusual for organic compounds. These compounds exhibited no afterglows in dimethyl sulfoxide solution at RT (Fig. S1, ESI†). As the emission of solid materials is usually sensitive to impurities in the commercial samples (Table S1, ESI† for elemental analyses indicating no significant impurities),19 we thoroughly purified the commercial IPA by silica gel chromatography, sublimation, and then three recrystallizations. The purified IPA showed an intensified afterglow, suggesting that impurities or crystal defects tend to suppress the afterglow (Fig. S2, ESI†). We thus hereafter disregard the impurity-induced phosphorescence as a possible emission mechanism. | ||
| Fig. 1 Afterglows of crystalline benzene derivatives on irradiation with a handy lamp at 366 nm for IPA, TPA, and PMA and at 254 nm for Ph4C. WL: white light. | ||
It is natural to assume that the afterglow with a long lifetime is attributed to a phosphorescence emission, which may be superposed by a delayed fluorescence. As discussed above, the phosphorescence of single-molecular organic crystals without heavy atoms would need to follow a special mechanism. In this context, it is intriguing that some crystalline hydrocarbons afford a very weak RT phosphorescence without SOC-assisting heavy atoms or carbonyl groups.20,21 We addressed the phosphorescence mechanism of IPA, considering all the possibilities ever studied for phosphorescence.
Delayed fluorescence
The time-resolved luminescence spectrum of IPA has three and two vibronic bands assignable to phosphorescence (2.33, 2.51, and 2.65 eV) and delayed fluorescence (3.30 and 3.46 eV), respectively, (Fig. 2). While there are several possible mechanisms for the delayed fluorescence,22,23 TTA is the most plausible mechanism in accordance with the substantial S1–T1 gap (≥0.79 eV) that eliminates a thermally activated delayed fluorescence, in which T1-to-S1 back transition can occur over the barriers of less than 0.27 eV.24 The TTA mechanism and thus the existence of T1 are corroborated by the power law25–27IDF ∝ IP2, where IDF and IP are the emission intensities of delayed fluorescence and phosphorescence, respectively. This relationship is deduced from the second-order kinetics of TTA, where a collision of two T1 molecules produces one S1 and one S0 molecules. Here, IP and IDF are proportional to phosphorescence intensity and the square of [T1] (i.e. the concentration of T1 molecules), respectively. Thus, the double-logarithmic plots for the relationship of IDF and IPvs. excitation power must show that the slope of IDF is twice that of IP. Fig. 3 shows that the corresponding slopes for IDF and IP of IPA were 1.44 and 0.74, respectively, in the excitation power range 10–104 μW, demonstrating the two-fold relationship for the TTA mechanism. We thus conclude that photoexcitation of IPA leads to T1 formation and the visible afterglow is most likely a phosphorescence emission accompanying the T1–S0 transition as discussed further in the section “Heavy atom effect”.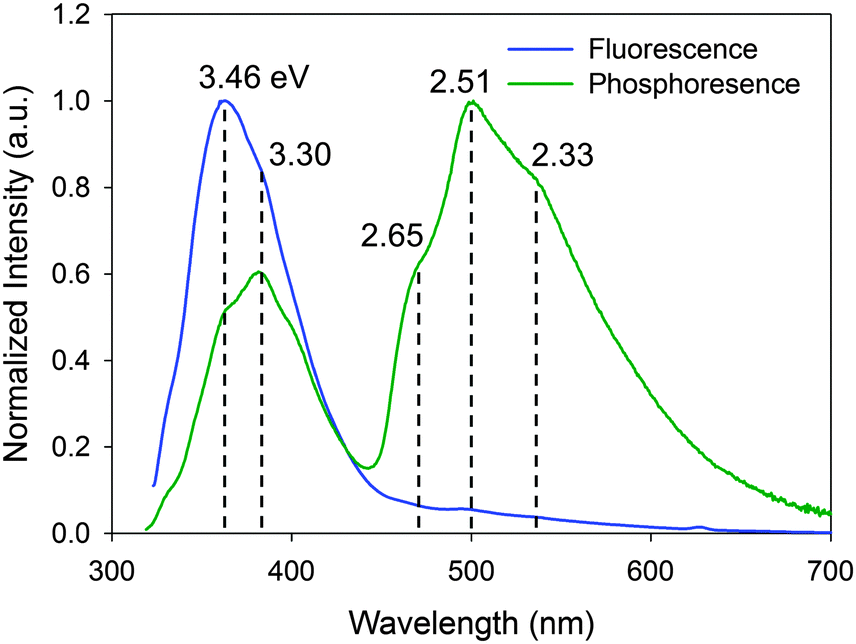 | ||
| Fig. 2 Steady-state fluorescence and time-resolved luminescence (phosphorescence) spectra of IPA at 296 K excited at 313 and 309 nm, respectively. For the time-resolved spectrum, 50–75 ms afterglows were measured. | ||
 | ||
| Fig. 3 Double-logarithmic relationship between the excitation power and the luminescence intensity. Excitation power at 350 nm was controlled by varying the bandwidth and the number of neutral density filters. The phosphorescence intensity at 500 nm and the delayed fluorescence intensity at 384 nm were measured at 0.1 s after shutting the excitation of 1 s duration. | ||
Magnetic field effect (MFE)
When TTA is observed, it is rational to consider the reverse reaction, i.e. singlet fission (SF),28 for a T1-generation mechanism. SF is an allowed S1–T1 conversion process, in which the collision of S1 with the singlet ground state (S0) leads to the formation of two T1 through electron exchanges if the energy level of S1 is more than twice that of T1, E(S1) > 2E(T1).28 The peaks at 3.46 and 2.65 eV in fluorescence and phosphorescence bands of IPA, respectively, are apparently closest to the S1 and T1 energies with the 0–0 vibronic transitions. The relationship between these S1 and T1 energies does not satisfy the SF criterion and thus SF is disregarded.Another potential T1-generation mechanism that should be taken into account for crystal phosphorescence is the HFC mechanism.29 HFC is the interaction between electron spins and nuclear spins of, in many cases, protons, and is the principal singlet-to-triplet conversion pathway for a weakly bound RIP, in which a radical cation and a radical anion are stacked together, forming CT complexes, or can be separated apart from each other in a crystal or polymer chain.30 The spin conversion between 1RIP and 3RIP (forward and backward) is faster than the S1 exciton → T1 exciton ISC, because two-electron spins in an RIP are located at different molecules, barely interacting each other, and thus the singlet–triplet energy difference is marginal. Two-electron spins in an RIP are subject to different internal magnetic fields (Bin and Bin′) generated from nuclear spins, because the electron clouds of a radical cation and a radical anion would differently shield the nuclear spins. In other words, two-electron spins precess at different angular momentum rates (Larmor frequency ω) under different internal magnetic fields ΔBin, Δω = gβΔBin, where g and β are the g-factor and Bohr magneton, respectively, (Fig. 4).31 A large difference in the precession rates means more frequent singlet-to-triplet spin exchange. Although RIPs are generally composed of two different molecules, i.e. an electron donor (D+˙) and an electron acceptor (A−˙), recent studies have established RIPs (or CT complexes) of identical molecules in the solid state.32 Though the HFC process is known for almost a half century, RT phosphorescence through these mechanisms has barely been reported. To the best of our knowledge, there is only one report on HFC-induced RT phosphorescence, in which a conjugate of Pt–porphyrin and rhodamine B in solution generated RT phosphorescence through a singlet-to-triplet conversion of RIP through an HFC mechanism.33
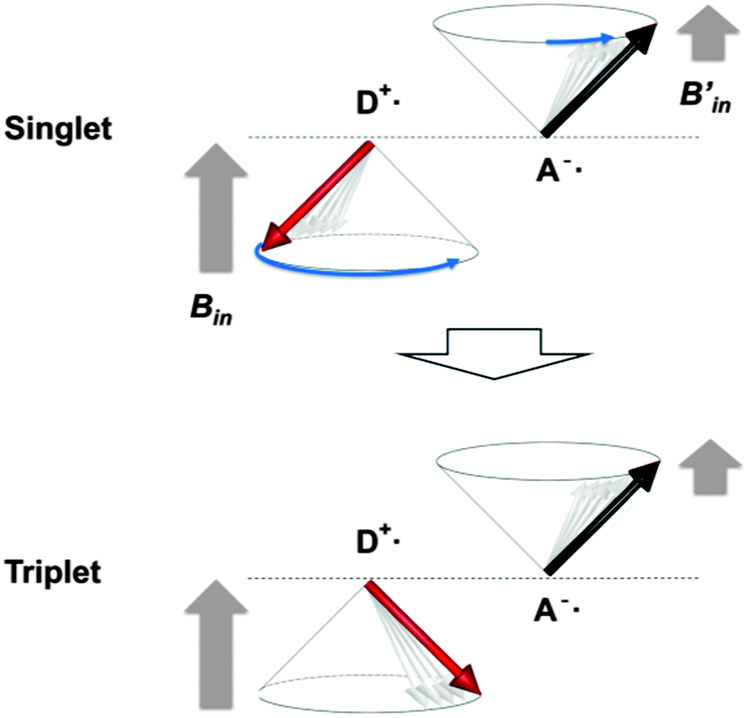 | ||
| Fig. 4 A spin exchange mechanism by electron spin precessions in an RIP consisted of an electron-donating radical cation (D+˙) and an electron-accepting radical anion (A−˙). The two spins are under different internal magnetic fields (Bin and Bin′), which causes different precession rates. The faster spin (D+˙) in the singlet state catches up the slower spin (A−˙) in due course to produce a triplet state. | ||
We thus examined the possibility of the RIP-HFC mechanism by measuring changes in the phosphorescence intensity with a magnetic field modulation.34,35 As the Zeeman energy of an electron spin at 1 T is as small as 1.2 × 10−4 eV, T1 excitons themselves are virtually indiscernible to the magnetic fields of less than 1 T, at least with regard to its luminescence emission. In such a small magnetic field, any observable changes are originated only from separated spins or RIPs that are energetically close to each other. If there is an influence of MFE on ISC in RIPs, changes in luminescence emission should be observed.
As shown in Fig. 5, the phosphorescence intensity at 500 nm markedly changed with the magnetic flux density modulation of up to 80 mT. We examined a stronger but inhomogeneous magnetic field with a neodymium permanent magnet and qualitatively observed an almost straight increase in emission strength up to 400 mT (Fig. S3, ESI†).
 | ||
| Fig. 5 Magnetic-field effects on the phosphorescence intensity at 500 nm of IPA. IPA was excited at 308 nm under an external magnetic field from an electromagnet connected to a constant voltage supply with the voltage variation between 0 and 90 V. The data represent the mean value from nine experiments and the error bars are ±s.e.m. | ||
The MFE on the phosphorescence was found to be the superposition of HFC and Δg effects.36 HFC enhances the ISC of weakly coupled radical pairs with each spin under a unique Bin generated by nuclear spins. Exposure to external magnetic fields (Bex) suppresses HFC, because three degenerate triplets, T+, T−, and T0, are split into three non-degenerate states by Zeeman splitting (Fig. 6) and thus ISCs between S0–T+ and S0–T− through HFC are weakened. The Zeeman splitting leads to a saturation of the change in the emission intensity and the saturation point appears at a relatively small Bex of about 20 mT. Under a relatively large Bex, the precession-based spin exchanges, Δω = ΔgβBex, become dominant when two electron spins are located at different molecules with unequal g-values, thus enhancing the phosphorescence emission. The fact that the phosphorescence shows considerable MFEs substantiates RIPs as the intermediates. It is intriguing to note that as one of the compounds reported to give phosphorescence on solid supports,10 sodium 2-naphthoate on a filtering paper showed an MFE similar to that of IPA crystals (Fig. S5, ESI†).
 | ||
| Fig. 6 HFC and Δg magnetic field effects on the phosphorescence of IPA. Without external magnetic fields (Bex = 0), S–T spin conversions are mediated by the internal magnetic field (Bin) generated from nuclear spins (HFC). An external magnetic field (Bex) suppresses the S–T+1 and S–T−1 spin exchanges (HFC) due to Zeeman splitting, giving a downward saturation curve. The S–T0 spin exchange is enhanced with increasing Bex by the Δg effect, where the difference in the Larmor frequency between two electron spins is amplified by Bex, giving a rising curve overriding the downward curve of HFC. [T1]rel denotes the relative concentration of T1. Here, S, T0, and T±1 denote RIPs as precursors of triplet exciton T. | ||
We also observed an MFE with delayed fluorescence that is typical for TTA37 – an increase in the emission intensity at low magnetic fields (<0.1 T), followed by a decrease at higher fields (Fig. S4, ESI†). It should be emphasized here that TTA consumes T1 and therefore the positive MFE on TTA never boosts the T1 concentration. Thus the increase of phosphorescence intensity in the Bex ranging between 20 and 100 mT is by no means caused by TTA.
Deuterium effect
Since the most abundant carbon and oxygen isotopes, 12C and 16O, have no nuclear spins, the HFC interactions in IPA must be mostly mediated by the 1H nuclear spin. To further substantiate the relevance of HFC to IPA phosphorescence, we carried out isotope substitutions.38,39 A C-deuterated derivative, IPAd4, gave a phosphorescence intensity suppressed by more than half (Fig. 7). 1H nuclear spins of a radical cation and a radical anion in an RIP can have unequal HFC constants, αH, owing to their different electron shieldings of Bin. In such a case, the differential precession torque ΔωH = gβMHΔαH, where MH is the magnetic quantum number of proton, affords a substantial turnover of ISC and thus a T1 concentration enough to emit visible phosphorescence. On the other hand, the D nuclear spin has a HFC constant (αD) 0.153 times smaller than αH, and it exhibits a relatively small spin-exchange rate given by ΔωD = gβMDΔαD = 0.153ΔωH. The observed phosphorescence suppression, therefore, is most likely due to the retarded singlet–triplet spin conversion of RIPs. If T1 were generated mainly from a usual S1–T1 ISC pathway, the phosphorescence would scarcely be affected by the nuclear spin substitution or might rather be enhanced by the diminished non-radiative relaxation via the weak C–D vibrations. | ||
| Fig. 7 Magnetic isotope effects on the phosphorescence spectra of IPA, IPA-d2, and IPA-d4. For the time-resolved spectrum, afterglows of 50–75 ms were measured. | ||
The deuterium substitution of OH groups, leading to IPAd2, resulted in a phosphorescence spectrum similar to that of the original IPA. The electron clouds containing radical spins do not spread over the oxygen atom, as illustrated by the TD-DFT calculation in the section “Charge transfer”, and hence the proton spin of the OH group is likely to be less effective toward the distant radical spin than that of the CH group. A lower vibration energy of the O–D bond relative to O–H might have suppressed the non-radiative relaxation, thus contributing to the slightly increased phosphorescence emission of IPAd2. The same tendency of deuterium effects on C–H and O–H were observed for benzoic acid (Fig. S6, ESI†). These results suggest HFC as the major pathway to T1 generation in IPA, whereas a minor contribution of the S1–T1 ISC cannot be ruled out.
Charge transfer
The above experiments demonstrate that the photoexcitation of IPA generates an RIP as an intermediate for the phosphorescence origin (T1 exciton). A question here is how the RIP is formed – by a direct excitation of S0 (CT absorption) or an electron transfer in the S1 state? The absorption spectrum of solid IPA showed a unique strong absorbance between 340 and 400 nm (Fig. 8). Absorption at this region is moderate for TPA and insignificant for OPA. Since the order of absorption strength is the same as that of phosphorescence intensity among the three compounds, the phosphorescence is likely to be related with the absorption. Interestingly, it has been reported that the phosphorescence intensity of these three compounds on silica gel showed a quite different tendency (TPA > OPA > IPA) and IPA emitted no phosphorescence.40 The absorption spectrum of IPA at this region coincides well with the corresponding phosphorescence excitation spectrum, indicating that the phosphorescence of IPA originates from photoexcitation at these wavelengths.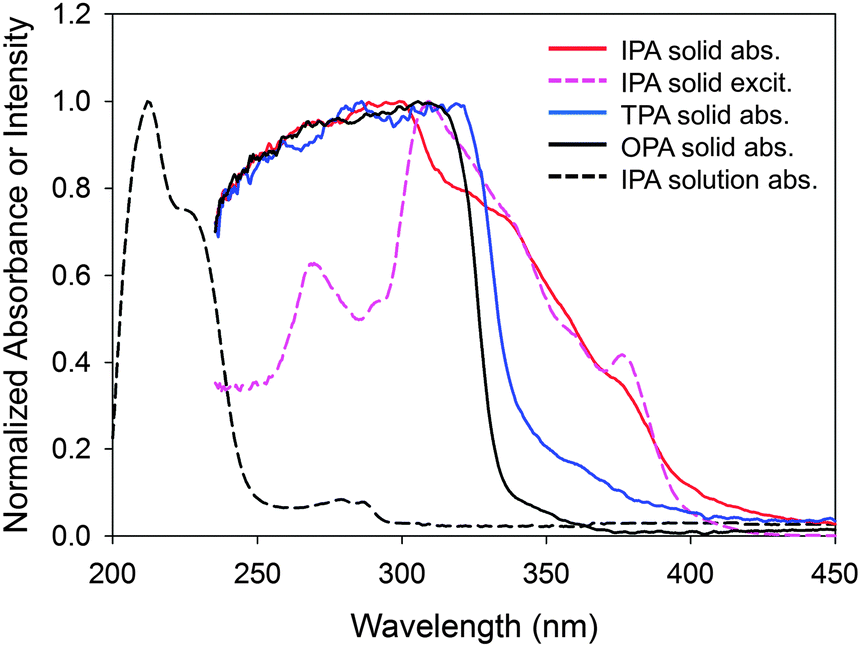 | ||
| Fig. 8 Absorption spectra of IPA, TPA, and OPA solids and IPA solution in methanol, and phosphorescence excitation spectrum with the emission at 501 nm. CT absorption is observed for IPA and TPA between 340 and 400 nm. | ||
It is most plausible that this distinctive absorption of IPA, which is absent for the non-phosphorescent compounds, is a CT absorption.41 The wavelengths of the major bands in the solid absorption spectrum are 50 nm longer than the S1–S0 bands of the methanol solution (289 and 281 nm for 0–0 and 1–0 vibronic bands, respectively). This bathochromic shift is probably due to a crystal lattice solvent effect, which may be explained as follows: the forced packing of a molecule with neighboring molecules in the crystal increases the highest occupied molecular orbital (HOMO) energy and decreases the lowest unoccupied molecular orbital (LUMO) energy, reducing the HOMO–LUMO gap and thus causing the bathochromic shift.
A TD-DFT calculation of an IPA dimer on CAM-B3LYP (631Gd),42 with a π–π distance of 3.76 Å obtained from a crystal structure of IPA (Fig. S7, ESI† for the observed and simulated XRD patterns),43 supports that there is a CT-like π–π* transition between two IPA molecules at 222.4 nm with an oscillator strength of 0.158 (Fig. 9). Similar CT-like transitions were also suggested for a benzoic acid dimer (Fig. S8, ESI†). The TD-DFT calculation for the corresponding non-phosphorescent OPA dimer produced only the local π–π* transitions in each molecule (Fig. S9, ESI†), probably because the distance between the two benzene rings (4.85 Å) is too large to permit intermolecular electron transitions.
 | ||
| Fig. 9 (A) Absorption spectrum calculated for an IPA dimer indicating a CT-like photoexcitation of isophthalic acid, πA–πB* transition at 222 nm with an oscillator strength of 0.158, using TD-DFT calculations on CAM-B3LYP at the 631Gd level using Gaussian 09. (B) Dimer structure (left) taken from the BENZDC10 file provided in Cambridge Structural DS 2013 and the molecular orbitals for the RIP dimers, πA (middle) and πB* (right). | ||
The CT complex produced directly by light absorption is considered a stacked RIP. We are uncertain if separated RIPs are formed from the CT complex via exciton migration of the radicals.
Heavy atom effect
The phosphorescence emission of IPA can occur either directly from 3RIP44,45 or from the T1 produced by 3RIP dissociation.46 In principle, the energy gap between 1RIP and 3RIP is trivial enough (<1.2 × 10−4 eV) to permit the magnetic field effect, as the two electron spins are virtually free from each other, and this is why the 1RIP–3RIP exchange through HFC and Δg effects were possible. The relatively large Stokes shift (1.37 eV) for the phosphorescence emission, therefore, suggests a T1-derived phosphorescence. In addition, the observed TTA phenomenon is consistent with the presence of T1. The emission from T1 is accompanied by a forbidden T1–S0 transition and thus the transition rate should be promoted by SOC through heavy atoms so that the phosphorescence lifetime is shortened. In practice, lifetime reduction with heavier-atom substitutions was observed for 2-halo, 3-halo, and 4-halo benzoic acids (Table 1). No such consistent relationship was seen on the phosphorescence intensity of Bz derivatives, indicating that the SOC effect does not hold for the T1-generating ISC, which also supports the 1RIP–3RIP exchange mechanism.| Halogen | λ ex (nm) | λ em (nm) | Int. (a.u.) | τ (ms) |
|---|---|---|---|---|
| a An excitation pulse cycle of 100 ms with a data acquisition delay of 50 ms and an accumulation period of 25 ms. b Weighted lifetime averages deduced from the multiexponential decay curves. c An excitation pulse cycle of 25 ms with a data acquisition delay of 11 ms and an accumulation period of 7 ms. | ||||
| o-F | 310 | 490 | 5.82 | 320 |
| o-Cl | 326 | 511 | 40.2 | 160 |
| o-Br | 348 | 557 | 5.36 | 93 |
| o-Ic | 395 | 566 | 0.171 | 16 |
| m-F | 318 | 539 | 11.8 | 940 |
| m-Clc | 324 | 525 | 2.05 | 130 |
| m-Brc | 329 | 500 | 5.06 | 48 |
| m-Ic | 388 | 553 | 10.8 | 36 |
| p-F | 294 | 498 | 0.353 | 340 |
| p-Cl | 305 | 517 | 0.160 | 230 |
| p-Br | 310 | 516 | 36.8 | 130 |
| p-Ic | 338 | 554 | 0.299 | 20 |
Conclusions
We can draw a phosphorescence mechanism of IPA as depicted in Fig. 10. IPA in solution exhibits the S1–S0 absorption bands at 289 nm, but does not show any phosphorescence because there are no sound ISC routes to the T1 state and T1, if it exists, is easily quenched by vibronic couplings. In crystals, the S0 energy is raised with the energy-increasing HOMO–HOMO interactions and the S1 energy is lowered with the energy-decreasing LUMO–LUMO interactions. The forced packing of IPA molecules enables dimer formation with the π–π distance of 3.76 Å, leading to CT absorption. As CT absorption usually has broad bands, the energy level of 1RIP can be distributed from below to above S1. Since the CT complex or a separated radical pair derived from CT has two electron spins over two molecules, the electron spins are only weakly coupled and thus the 1RIP–3RIP energy difference is marginal, facilitating the spin exchange. The spin exchange is promoted by HFC – the different internal magnetic fields around two uncoupled electron spins enable the singlet-to-triplet conversion. Thus the weakening of HFC by CH-to-CD substitution or Zeeman splitting of the degenerate three triplet states by an external magnetic field modulation results in the reduction of phosphorescence. In a stronger external magnetic field, the phosphorescence intensity becomes larger, as the singlet-to-triplet exchange is enhanced by the Δg effect. No direct emissions from these RIPs, as demonstrated by the relatively large Stokes shift (about 200 nm) for the phosphorescence emission, are allowed for an unknown reason. Dissociation of 1RIP gives S0 and S1, which emit fluorescence. From 3RIP, S0 and T1 are produced and T1 emits phosphorescence. The suppressed vibronic couplings of the crystal structure retard the non-radiative relaxation of the T1 state, which enables T1 exciton migration causing delayed S1 formation by TTA. | ||
| Fig. 10 State energy diagram for IPA phosphorescence. Green color shows the route to phosphorescence emission and blue color shows the route to fluorescence and delayed fluorescence. AbsCT: CT absorption, P: Phosphorescence, TTA: triplet–triplet annihilation, DF: delayed fluorescence, HFC: hyperfine coupling. | ||
Thus it can be concluded that the RIP-based phosphorescence mechanism presented here helps improve the development process of practical phosphorescent materials by managing nuclear spins.
Acknowledgements
We would like to thank Ms Katsumi Suda, Mr Yuichi Suzuki, and Shinpei Nakamura, Center for Advanced Material Analysis, National Corporation Tokyo Institute of Technology for XRD (K.S. and Y.S.) and ICP-AES (S.N.) measurements. This work was supported by a “Collaborative Research Based on Industrial Demand” program from Japan Science and Technology Agency (JST) and a Grant-in-Aid for Scientific Research (No. 24550148 to H.Y.) from the Japan Society for the Promotion of Science (JSPS).Notes and references
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed
.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed
- D. Chaudhuri, E. Sigmund, A. Meyer, L. Röck, P. Klemm, S. Lautenschlager, A. Schmid, S. R. Yost, T. Van Voorhis, S. Bange, S. Höger and J. M. Lupton, Angew. Chem., 2013, 125, 13691–13694 CrossRef PubMed
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed
- Y. You, Curr. Opin. Chem. Biol., 2013, 17, 699–707 CrossRef CAS PubMed
- I. V. Khudyakov, Y. A. Serebrennikov and N. J. Turro, Chem. Rev., 1993, 93, 537–570 CrossRef CAS
- C. M. Marian, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 187–203 CrossRef CAS PubMed
- S. Hoshino and H. Suzuki, Appl. Phys. Lett., 1996, 69, 224–226 CrossRef CAS PubMed
- A. Köhler and H. Bässler, Mater. Sci. Eng., R, 2009, 66, 71–109 CrossRef PubMed
- R. T. Parker, R. S. Freedlander and R. B. Dunlap, Anal. Chim. Acta, 1980, 120, 1–17 CrossRef CAS
- R. Gahlaut, H. C. Joshi, N. K. Joshi, N. Pandey, P. Arora, R. Rautela, K. Suyal and S. Pant, J. Lumin., 2013, 138, 122–128 CrossRef CAS PubMed
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS PubMed
- A. Pfister, G. Zhang, J. Zareno, A. F. Horwitz and C. L. Fraser, ACS Nano, 2008, 2, 1252–1258 CrossRef CAS PubMed
- R. T. Parker, R. S. Freedlander and R. B. Dunlap, Anal. Chim. Acta, 1980, 119, 189–205 CrossRef CAS
- D. B. Clapp, J. Am. Chem. Soc., 1939, 61, 523–524 CrossRef CAS
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CAS
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed
- X. Zhang, X. Zhang, L. Tao, Z. Chi, J. Xu and Y. Wei, J. Mater. Chem. B, 2014, 2, 4398–4414 RSC
- A. Brillante, D. P. Craig, A. W.-H. Mau and J. Rajikan, Chem. Phys. Lett., 1975, 30, 5–10 CrossRef CAS
- D. F. Williams, J. Chem. Phys., 1967, 47, 344–345 CrossRef CAS PubMed
- B. Priestley and A. Haug, J. Chem. Phys., 1968, 49, 622–629 CrossRef PubMed
- D. Hertel, H. Bässler, R. Guentner and U. Scherf, J. Chem. Phys., 2001, 115, 10007–10013 CrossRef CAS PubMed
- Z. Huang, D. Ji, A. Xia, F. Koberling, M. Patting and R. Erdmann, J. Am. Chem. Soc., 2005, 127, 8064–8066 CrossRef CAS PubMed
- M. N. Berberan-Santos and J. M. M. Garcia, J. Am. Chem. Soc., 1996, 118, 9391–9394 CrossRef CAS
- A. Gerhard and H. Bässler, J. Chem. Phys., 2002, 117, 7350–7356 CrossRef CAS PubMed
- A. Hayer, H. Bässler, B. Falk and S. Schrader, J. Phys. Chem. A, 2002, 106, 11045–11053 CrossRef CAS
- S. A. Bagnich, H. Bässler and D. Neher, J. Chem. Phys., 2004, 121, 9178–9183 CrossRef CAS PubMed
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed
- C. B. Grissom, Chem. Rev., 1995, 95, 3–24 CrossRef CAS
- H.-J. Werner, H. Staerk and A. Weller, J. Chem. Phys., 1978, 68, 2419–2426 CrossRef CAS PubMed
- C. R. Timmel and K. B. Henbest, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 2573–2589 CrossRef CAS PubMed
- S. Sharifzadeh, P. Darancet, L. Kronik and J. B. Neaton, J. Phys. Chem. Lett., 2013, 4, 2197–2201 CrossRef CAS
- T. Mani, D. M. Niedzwiedzki and S. A. Vinogradov, J. Phys. Chem. A, 2012, 116, 3598–3610 CrossRef CAS PubMed
- U. E. Steiner and T. Ulrich, Chem. Rev., 1989, 89, 51–147 CrossRef CAS
- B. Hu, L. Yan and M. Shao, Adv. Mater., 2009, 21, 1500–1516 CrossRef CAS PubMed
- Y. Sakaguchi, H. Hayashi and S. Nagakura, Bull. Chem. Soc. Jpn., 1980, 53, 39–42 CrossRef CAS
- R. E. Merrifield, J. Chem. Phys., 1968, 48, 4318–4319 CrossRef CAS PubMed
- N. J. Turro and B. Kraeutler, J. Am. Chem. Soc., 1978, 100, 7432–7434 CrossRef CAS
- C. Baleizão and M. N. Berberan-Santos, ChemPhysChem, 2011, 12, 1247–1250 CrossRef PubMed
- C. D. Ford and R. J. Hurtubise, Anal. Chem., 1978, 50, 610–612 CrossRef CAS
- J. N. Murrell, Quart. Rev., 1961, 15, 191–205 RSC
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS PubMed
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed
- S. Iwata, J. Tanaka and S. Nagakura, J. Chem. Phys., 1967, 47, 2203–2209 CrossRef CAS PubMed
- J. Herbich, A. Kapturkiewicz and J. Nowacki, Chem. Phys. Lett., 1996, 262, 633–642 CrossRef CAS
- T. Mani, M. Tanabe, S. Yamauchi, N. V. Tkachenko and S. A. Vinogradov, J. Phys. Chem. Lett., 2012, 3, 3115–3119 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, four tables, and thirteen figures. A video file is separately given. See DOI: 10.1039/c5cp01203a |
| This journal is © the Owner Societies 2015 |