
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTime-resolved IR spectroscopy of a trinuclear palladium complex in solution†
M.
Zimmer
a,
F.
Rupp
b,
P.
Singer
b,
F.
Walz
c,
F.
Breher
*c,
W.
Klopper
*d,
R.
Diller
*b and
M.
Gerhards
*a
aChemistry Department and Research Center Optimas, TU Kaiserslautern, Erwin-Schrödinger-Straße 52, 67663 Kaiserslautern, Germany. E-mail: gerhards@chemie.uni-kl.de; Fax: +49 631 2052750; Tel: +49 631 2052537
bPhysics Department, TU Kaiserslautern, Erwin-Schrödinger-Straße 46, 67663 Kaiserslautern, Germany. E-mail: diller@physik.uni-kl.de; Fax: +49 631 2053902; Tel: +49 631 2052323
cInstitute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: breher@kit.edu; Fax: +49 721 60847021; Tel: +49 721 60844855
dInstitute of Physical Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 2, 76131 Karlsruhe, Germany. E-mail: klopper@kit.edu; Fax: +49 721 60847225; Tel: +49 721 60847263
First published on 1st May 2015
Abstract
This paper presents a combined spectroscopic and theoretical analysis of a trinuclear [Pd3{Si(mtMe)3}2] complex (mtMe = methimazole) which has been demonstrated to be a potential catalyst for coupling reactions. It is a highly symmetric model system (D3 in the electronic ground state) for the investigation of electronic states and the structure of polynuclear transition metal complexes. Different time-resolved IR spectroscopic methods covering the femtosecond up to the microsecond range as well as density functional computations are performed to unravel the structure and character of this complex in the electronically excited state. These are the first time-resolved IR studies on a trinuclear Pd complex. Based on the interplay between the computational results and those from the IR studies a 3A state is identified as the lowest lying triplet state which has C2 symmetry.
1 Introduction
Polynuclear transition metal complexes are known to have chemically interesting properties and applications, e.g. in catalysis1–4 or light harvesting processes,5,6 which can be explored in photochemical and photophysical investigations.7–9 The afore-mentioned properties can be influenced by cooperative effects between the metal centers,10–13i.e. the interactions (couplings) between the metal centers and the ligand(s) have to be investigated. These investigations require versatile spectroscopic techniques and detailed theoretical analyses. In recent years structure-sensitive X-ray14–18 and vibrational spectroscopy19–28 with sub-ps up to μs time resolution have been applied to explore structure and dynamics of electronically excited transition metal containing complexes.In this context a very interesting trinuclear palladium complex29 (1, Fig. 1) has been investigated by our research groups with a variety of state-of-the-art photophysical and computational methods.30 These analyses gave a detailed insight into the photophysical properties and into the electronically excited states. Based on these investigations we could characterize the lowest lying triplet state from which a long-lived (τAr = 1.3–1.7 μs, τAir = 222 ± 3 ns)30 luminescence occurs. Extensive theoretical work was involved to obtain conclusions on the electronically excited states of 1. However, no conclusive investigations on the structures of the electronic states have been performed up to now, which would yield more precise information on these states and thus a better and more reliable assignment. For the investigations presented here we use structure sensitive time-resolved IR spectroscopy from the femto- to the microsecond time range in combination with further theoretical analyses that go beyond our previous work;30 this is the first application of such a strategy to a trinuclear Pd complex. Our chosen combination of methods is especially suited to analyse the structure of the complex in its ground and excited states.
 | ||
| Fig. 1 Structural arrangement of [Pd3{Si(mtMe)3}2] (1). | ||
To cover the nano- and microsecond range, time-resolved step-scan FTIR spectroscopy (TR-FTIR) is applied. Publications of TR-FTIR measurements on transition metal complexes exist since the early 1990s.22–25 Most of those investigations are about electronically excited states, especially with respect to metal-to-ligand charge transfer (MLCT) transitions in complexes including one or two metal centers. In these early investigations so called “reporter” ligands (carbonyl and cyanide) have been introduced which bear the advantage of a huge oscillator strength for the ν(CO) and ν(CN) stretching vibration(s) and a high sensitivity for structural changes.22 Investigated systems without a “reporter” ligand are for example Ru(bpy) complexes (bpy = 2,2′-bipyridine) including the well analyzed [Ru(bpy)3]2+.26,27 Regarding the number of metal atoms very few examples for tri- and tetrametallic investigations with TR-FTIR can be found in the literature, some of them focus on the energy transfer in rhenium complexes, e.g. [(bpy)(CO)3Re(trans-dppene)Re(phen)(CO)2(trans-dppene)Re(bpy)(CO)3]3+ (phen = phenanthroline, dppene = 1,2-bis(diphenylphosphino)ethene) and Re4(CO)12(4,4′-bpy)4Cl4.24,28
Having none of the above mentioned “reporter” groups in our system (1) our investigations are even more demanding. The transient (step-scan) FTIR spectroscopy still benefits from the advantages of the FTIR technique among them the possibility to record a complete spectral region simultaneously (multiplex advantage). Thus it is possible to capture the whole mid-IR region (and in principle also NIR and FIR). In our case the mid-IR region, especially from 1600 to 1100 cm−1, is of interest.
After a detector rise time of about 30 ns our step-scan set up enables us to perform measurements with a resolution of about 10 ns (see Section 2). Beyond this, ultrafast transient IR spectroscopy is applied to the trinuclear palladium complex to investigate the primary photoinduced vibrational dynamics in the femto and picosecond time domain, thereby covering the entire excited state lifetime from its very beginning. Femtosecond transient IR spectroscopy has been applied successfully to the investigation of divers photoinduced phenomena in metal complexes such as CO release,31–33 excited state dynamics in Cr(acac)334 and a Ru(II) catalyst35 as well as high-spin state formation in Fe(II) spin-crossover systems.21 Thereby ultrafast vibrational dynamics can be related to electronic dynamics as intersystem crossing and internal conversion.36,37
2 Experimental methods
The Pd3 complex (1) was prepared following the synthesis described in ref. 29. DMSO-d6 (<0.02% water, 99.8% D) was purchased from VWR Chemicals and purged with argon prior to usage. All experiments described in Sections 2.1 and 2.2 were performed at room temperature.2.1 Time-resolved step-scan FTIR spectroscopy
For the time-resolved FTIR experiments a Bruker VERTEX 80v spectrometer equipped with a step-scan interferometer module was used (for a detailed description of the experimental set-up, cf. Fig. S1 of the ESI†). The spectrometer, using a liquid-nitrogen cooled mercury–cadmium–telluride (MCT) detector connected to a fast preamplifier, was coupled with a Q-switched Nd:YAG laser (Lumonics HY750, frequency tripled to 355 nm) that generates excitation pulses with a half-width of about 10 ns and a repetition rate of 10 Hz. The timing of the laser pulse and the step-scan triggering was performed by a Stanford Research Systems DG535 delay generator. The UV pump beam was adjusted to have an overlap with the spectrometer's IR probe beam inside the flow cell. To avoid backscattering of the UV pulse into the detector or interferometer compartment, AR-coated germanium filters were placed inside the sample compartment.A mM solution of 1 in argon purged DMSO-d6 was pumped by a peristaltic pump (Masterflex Easy-Load II, variable flow rate) through an optical cell that is part of a closed-cycle continuous flow tubing system including a sample reservoir. The cell was composed of housing, CaF2 windows, separated by two Mylar® spacers with a variable thickness (0.006 to 0.5 mm). In our case 0.26 mm spacers and a flow rate of about 40 mL min−1 are chosen. The sample cell as well as the sample reservoir were purged by argon to assure an excited state lifetime of τAr = 1.3–1.7 μs, since it is quenched by oxygen to τAir = 222 ns.30 The 355 nm excitation laser beam was attenuated to 0.6–0.8 mJ per shot at a diameter of about 9 mm. The temporal resolution was set to 20 ns for these measurements. Five adjacent 20 ns intervals were joined and averaged, so a temporal resolution of 100 ns was obtained. The step-scan experiment was triggered 400 ns before the Q-Switch of the laser was triggered which means that the laser beam reached the sample 600 ns after the experiment starts. Hence this time was set as zero point in all spectra. A total number of 465 coadditions at each interferogram point was recorded. The spectral region was limited by undersampling to 0–2633 cm−1 with a spectral resolution of 2 cm−1 (resulting in 2665 interferogram points). For the prevention of problems by performing a Fourier transformation an IR longpass filter (transmission 4.73–9.00 μm) was used (i.e. no IR intensity outside the measured region should be observed).
Both amplified AC and DC signals of the MCT detector were fed to a 14-bit transient recorder board (Spectrum Germany, M3I4142, 400 MS/s), which in turn was connected to an external computer running Bruker OPUS software for spectrometer control and data manipulation/analysis. For data analysis the recorded DC and AC spectra were used to calculate the ΔA (change in absorbance) spectra with:
 | (1) |
2.2 Ultrafast transient IR absorption
For femtosecond mid-IR experiments, the complex was dissolved at concentration equivalent to an OD = 2 at 330 nm in dimethylsulfoxide (DMSO) for probe around 1166 cm−1 and in deuterated DMSO (DMSO-d6) for probe around 1453, 1295, 1378 cm−1 (see Section 4). The solution was placed between two UV-grade CaF2 windows (Crystal GmbH) of 2.54 cm in diameter with 250 μm path length. Before and after each femtosecond transient experiment steady state UV/Vis spectra (JASCO, V-670) were recorded to ensure sample integrity.The femtosecond pump–probe experiments were based on a Ti:sapphire regenerative amplifier system to generate ultra-short pump and probe pulses (CPA-2001, Clark MXR: rep. rate 1 kHz, pulse energy 1 mJ, central wavelength 775 nm, pulse duration 150 fs (FWHM)).
A home-build two stage non-collinear optical parametric amplifier was used to generate femtosecond pump pulses at 330 nm with energy of 250–300 nJ at the sample position. Another portion of the fundamental beam was fed in a two stage optical parametric amplifier with consecutive difference frequency generation, yielding the spectrally tunable probe pulse between 6–12 μm with a spectral width of 50–60 cm−1 as described before.38 Pump and probe pulses were superimposed in the sample with focal width of about 200 and 100 μm, respectively. The sample was rotated to exchange the excited volume between two consecutive laser shots. The transmitted mid-IR probe light was spectrally dispersed and detected by a polychromator with a 32-element MCT (mercury–cadmium–telluride) detector array (Infrared-Systems-Development) with ≈2 cm−1 spectral width per element. In all experiments the pump beam was chopped at half the repetition rate (500 Hz), allowing the calculation of pump-induced difference absorbance spectra ΔOD(λ,t) on a shot-to-shot basis as a function of probe wavelength and pump–probe delay. Time zero of the experiment was determined via a transient absorption experiment on a thin silicon wafer. The pump/probe cross correlation time was typically ≈350 fs (FWHM, Gauss).
The transient data was analyzed by a global fit routine, describing the time dependent absorbance change ΔOD(λ,t) as a sum of exponential decay functions
 | (2) |
3 Theory
Quantum-chemical calculations were performed with the TURBOMOLE program package39 at the level of density-functional theory (DFT) using the hybrid functional B3LYP. The calculations were performed in exactly the same basis as used in ref. 30, that is, dhf-TZVP-2c for Pd, S, and Si, and def2-SV(P) for H, C, and N. This basis is referred to as ‘‘TZVP’’ for short. In ref. 30, the geometries of the 3A1 and 3A2 excited states as well as the 1A1 ground state were optimized in the framework of self-consistent Kohn–Sham calculations (ΔSCF) at the B3LYP/TZVP level in D3 symmetry. Subsequently, in the course of the present work, we computed the harmonic vibrational frequencies at these geometries using a parallelized version40 of the module AOFORCE,41 which allows for the analytical calculation of force constants. For the 1A1 ground state, we have found that all computed harmonic vibrational frequencies are real, meaning that the D3 equilibrium structure is a true minimum on the potential energy hypersurface. For each of the two triplet excited states, however, we obtained one (two-fold degenerate) imaginary frequency of e symmetry (26i cm−1 for the 3A1 state and 122i cm−1 for 3A2). Therefore, these states were re-optimized in C2 symmetry, now yielding true minima with real harmonic vibrational frequencies. In C2 symmetry, the 3A1 (D3) state becomes a 3A state, whose adiabatic ΔSCF energy is found 1.79 eV above the well depth of the ground state (this value was 1.81 eV for its D3-symmetric counterpart) while the 3A2 (D3) state becomes a 3B state, whose adiabatic energy is found 2.01 eV above the ground state (this value was 2.32 eV in D3 symmetry). Hence, the energy lowering due to the geometry relaxation in C2 symmetry amounts to 0.02 and 0.31 eV for the 3A and 3B states, respectively. In the 3A state, the three Pd atoms form an isosceles triangle with two equal sides of 270.2 pm and one longer side of 275.4 pm while in the 3B state, they form an isosceles triangle with two equal sides of 285.9 pm and one shorter side of 281.3 pm (in D3 symmetry, the Pd–Pd distances were 271.5 pm and 288.2 pm, respectively, in the 3A1 and 3A2 states).For gaining insight into the observed energy lowering due to geometry relaxation, we computed a correlation diagram (cf. ESI,† Fig. S2) of the frontier orbitals as a function of a distortion coordinate x, which is defined as follows: x = 0 corresponds to an unbiased D3-symmetric geometry that is obtained by averaging the 3A1 and 3A2 geometries optimized in ref. 30; x = −1 corresponds to the C2-symmetric geometry of the 3B state and x = +1 corresponds to the C2-symmetric geometry of the 3A state. Separate linear interpolations of the structures are performed between the x = −1 and x = 0 geometries on the one hand and between the x = 0 and x = +1 geometries on the other. The LUMO is stabilized for both positive (x > 0) and negative (x < 0) distortions. This can be understood as pseudo-Jahn–Teller effect42 due to interactions with a close-lying two-fold degenerate e level (LUMO+1, 72e). At the same time, the HOMO is destabilized towards x = +1 while the HOMO−1 is destabilized towards x = −1. The latter even becomes the HOMO in the 3B geometry. The interplay between destabilization of the relevant ground-state occupied orbitals and stabilization of the LUMO appears to be responsible for the energy lowering upon distortion. Consistent with the correlation diagram, Fig. S3 (ESI†) shows the energies of the excited states of interest as a function of the distortion coordinate, as obtained from TDDFT computations at the B3LYP/TZVP level using the lowest triplet state as reference.
The Cartesian coordinates of the ground state and the two triplet states are given in the ESI† (Tables S1–S3).
4 Spectroscopic results and discussion
Static IR spectra
In the spectral region from 1150 to 1550 cm−1 four IR absorption bands belonging to 1, at 1172, 1301, 1383, 1457 cm−1 are identified (see Fig. S4, ESI† and lower trace of Fig. 2 in the region down to 1250 cm−1); furthermore a broad, not well structured absorption region from 1390–1450 cm−1 is observed. In comparison to the DFT calculations the four defined peaks can be assigned to δ(C–H)aliphatic 1457 cm−1 (1454 cm−1), ν(C–N) 1301 cm−1 (1300 cm−1) and skeletal vibrations 1383 and 1172 cm−1 (1379 and 1159 cm−1), calculated values in parentheses (scaling factor 0.98, see below). | ||
| Fig. 2 Bottom: steady state FTIR, solvent DMSO-d6 subtracted; top: TR-FTIR spectra, 0.5, 1.5, 2.0 and 3.5 μs after excitation at 355 nm; inset: TR-FTIR spectra, 0.1, 0.2 and 0.3 μs after excitation at 355 nm. | ||
Nano- and microsecond time regime
After laser excitation five bleach bands appear at 1174, 1303, 1385, 1402 and 1458 cm−1 (Fig. 2 and 3) in the step-scan FTIR spectrum, which can definitely be assigned to the electronic ground state vibrations. (Remark: the values observed in depletion differ by a few wavenumbers from the values obtained in the static FTIR spectrum due to a significant spectral overlap of the ground- and electronically excited state vibrational bands. This is illustrated in Fig. S5, ESI†). In addition, four new bands appear that are 9–14 cm−1 red shifted at 1162, 1289, 1373 and 1449 cm−1. These bands originate from the electronically excited species. Furthermore the relative depletion intensities of the ΔA peaks match that of the steady state spectrum (see Fig. 2 and Fig. S6, ESI†).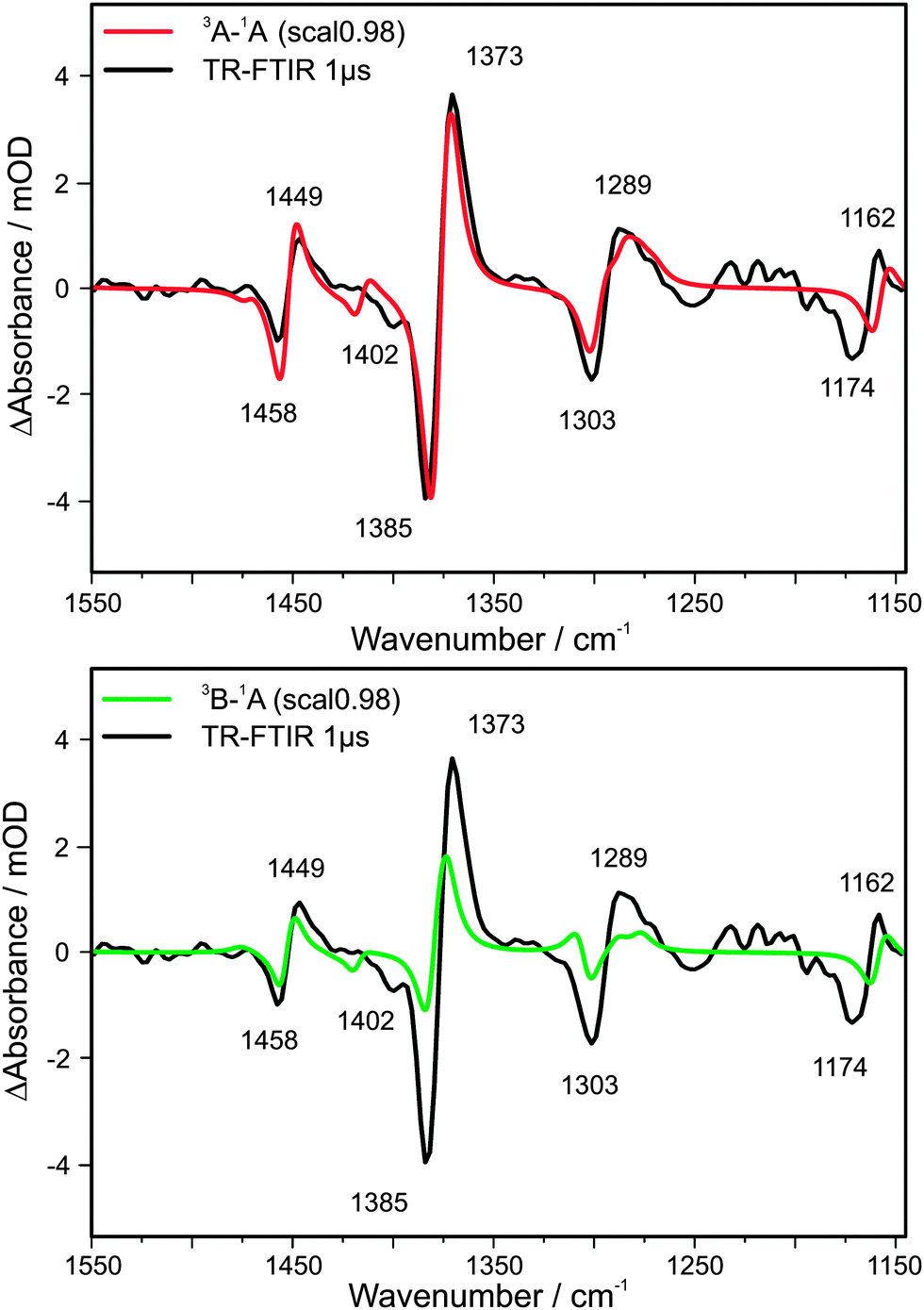 | ||
| Fig. 3 TR-FTIR spectrum 1.0 μs after excitation; top: in comparison with calculated excitation into a 3A state; bottom: in comparison with calculated excitation into a 3B state. | ||
In the first 100 ns interval after excitation the bands at 1303/1289, 1385/1373 and 1458/1449 cm−1 are already observable (see inset of Fig. 2). To obtain a better S/N ratio and since no dynamics in this temporal range are visible spectra are also averaged to 500 or 1000 ns steps. The signal disappears completely after about 3.5 μs, confirming the TCSPC lifetime measurements30 and giving a hint that the argon purged sample chamber and argon purged housing of the sample reservoir prevent the sample from oxygen. (For a better illustration of the decay within microseconds, the transient IR spectra up to 3.5 μs are shown in steps of 500 ns in the ESI,†cf. Fig. S7).
In order to compare calculated harmonic frequencies with experimental values the computed vibrational frequencies have to be scaled. A typical empirical factor for the chosen theoretical ansatz (cf. Section 3) is 0.98 (cf. e.g.ref. 43). The reliability of this factor can be tested by fitting the calculated IR spectrum of the electronic ground state (1A) as well as the spectra of the electronically excited states (3A and 3B) to the experimental values. Indeed the best agreement is achieved for a factor of 0.98. (In case of the 3A state the scaling factors vary between 0.979 and 0.983 whereas for the 3B state the variation is larger, between 0.970 and 0.983, indicating a better description of the experimental frequencies for the 3A state.)
In the DFT calculations a red shift of the 3A state vibrations compared to the ground state 1A (modes) in the order of about 10 cm−1 is observable, for example 16 cm−1 for the 1285/1301 cm−1 peaks or 7 cm−1 for the 1373/1380 cm−1 peaks (Fig. S8). In contrast, the vibrational modes of the 3B state are closer to the values of the 1A state. It should be mentioned that the investigated spectral region shows small but significant differences for the calculated 3A and 3B spectra. Other parts of the spectrum are either less significant or cannot be observed with our experimental set-ups (cf. Fig. S9 of the ESI†).
In order to quantify the comparison between calculated and experimentally observed frequencies the calculated spectrum of the electronic ground state (1A) has to be subtracted from the calculated spectra obtained for the 3A and 3B states. In order to simulate the vibrational spectra, Lorentzian functions are used. By fixing the band positions (scaling factor 0.98), different full widths at half maximum (FWHM) are chosen. By testing a constant width or variable widths (between 10 and 20 cm−1) within one fit it turns out that 14 cm−1 is the best average value. The goodness of the fit can be described by the R2/χ2 parameter which is about a factor of three to four better for the 3A state compared to 3B (by using a scaling factor of 0.98 and a constant band width of 14 cm−1 or a variable band width between 10 and 20 cm−1 optimized for each transition). Fig. 3 shows the comparison of experiment and theory: Fig. 3 (top) ground state (1A) subtracted from 3A and Fig. 3 (bottom) 1A subtracted from 3B. It can be seen that the calculated band positions (relative to the experimental values) are better predicted for the 3A state, especially in the case of the transitions at 1289, 1373 and 1449 cm−1. In case of the 3B state also an additional band around 1310 cm−1 would be expected which is not observed in the experimental spectrum. Furthermore, the calculated difference spectra reveal also that only the 3A state gives an appropriate description of the experimentally observed band intensity ratios, especially for the transitions at 1373/1385 cm−1. In case of this most prominent pair of transitions both increase and depletion in the ΔA spectrum are of similar intensity for the 3A state (in agreement with the experiment) whereas for the 3B state the increase should be by a factor of 3 larger than the depletion. Taking the qualitative and quantitative aspects of calculated frequencies and intensities into account, it can be concluded that the excited state probed on the nano- and microsecond time scale is a 3A state.
Another experiment was performed with a frequency doubled Nd:YLF laser (edgewave INNOSLAB IS8 II – ET, output of about 0.4 mJ per shot at 261.5 nm) that was operated at 100 Hz. According to our previous experimental study30 it is known that the UV/Vis spectrum shows two strong and broad absorptions: the first electronic transition has its maximum around 333 nm and a second one is around 262 nm. By exciting the second electronic transition with a wavelength of 261.5 nm, the same IR transitions as for the excitation of the first electronic transitions (with 355 nm) are observed in the step-scan FTIR spectrum. These finding clearly show that after electronic excitation the same long-lived electronic state is reached.
Femto- and picosecond time regime
In our earlier work,30 femtosecond transient absorption in the UV/Vis already showed the ultrafast formation of an electronically excited state within much less than 1 ps with no subsequent spectral changes up to 700 ps. In order to investigate the formation of this long lived state via its IR spectrum transient IR spectroscopy is performed. Fig. 4 (top) displays IR difference absorption signals at selected delay times of 0.5 and 340 ps after photoexcitation at 330 nm between 1145 and 1480 cm−1. Fig. 4 (bottom) depicts two kinetic traces at 1366 and 1381 cm−1. They show that after instantaneous absorption rise and bleach, respectively, the signal decreases within a few picoseconds and merges into stationary amplitude. | ||
| Fig. 4 Top: mid-IR difference absorption spectra at selected delay times (0.5 and 340 ps) after photoexcitation at 330 nm. Wavenumbers indicate the (apparent) peak positions at 340 ps. Bottom: kinetic trace at 1366 cm−1 (blue) and 1381 cm−1 (black) and global monoexponential fit with τ1 = 7.8 ± 0.4 ps (red). | ||
The stationary difference spectrum at 340 ps exhibits negative peaks at 1176, 1303, 1386, 1402 and 1460 cm−1 and positive peaks at 1164, 1291, 1375 and 1450 cm−1. As in the TR-FTIR spectra the bleach peaks (neg.) are assigned to the electronic ground state and the positive vibrational bands are assigned to the long living excited state, the latter showing a rather uniform bathochromic shift of about 10 cm−1 with respect to the former. The difference spectrum at early times (0.5 ps) is very similar to that at 340 ps, however, the respective difference bands are of larger amplitude with increased positive absorbance on the low-energy side. This feature is easily explained by vibrational cooling, i.e. the dissipation of the excess energy to the solvent: Anharmonic coupling between thermally populated low-energy vibrational modes and high-energy modes (fingerprint region) causes asymmetrically broadened vibrational bands. Thus, the observed apparent blue-shift on the picosecond time scale indicates the cooling process of the vibrationally hot (unrelaxed) molecule in the excited electronic state. It should be noted that the spectral shapes (widths) of the positive bands in the transient spectra are not uniform since they are determined by band-specific parameters such as (1) spectral width (dephasing time) of the particular band in the excited electronic state and (2) the anharmonic coupling constants of the particular band to all other vibrational modes in the excited electronic state, especially to the low frequency modes.
Although the relation between vibrational temperature and time-dependency of the observed spectral changes is intrinsically non-exponential, the global fit according to eqn (2) is used in order to derive a characteristic time constant. This yields τVR = 7.8 ± 0.4 ps with the decay associated spectrum A1(λ) given in Fig. 5. It now describes the observed blue-shift of each product vibrational band (Fig. 4) in terms of monoexponentially decaying positive (low energy lobe) and negative (high energy lobe) absorbance strength as an approximation for the cooling process. A0(λ) is basically identical with the 340 ps difference spectrum, confirming the quality of the fit. Note the excellent match of A0(λ) and the microsecond-FTIR spectra (Fig. 3). No further, sub-ps time constant (as found in the transient UV/Vis data reported earlier (380 fs)30) could be derived from the data. This might be due to the highly transitory character of the Franck–Condon state, leading to spectrally extremely broad infrared bands with possibly low absorption cross section. In addition the given (limited) time resolution and S/N-ratio add to this effect. Thus the IR data together with the transient UV/Vis data reported earlier (ref. 30) indicate that ultrafast intersystem-crossing (ISC), internal conversion (IC) and thermalization via intramolecular vibrational relaxation (IVR) lead to the vibrationally hot 3A excited state within less than 400 fs. Subsequently the excess energy of about 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1 (according to λexc = 330 nm and λFluor = 675 nm30) is dissipated into the solvent.
500 cm−1 (according to λexc = 330 nm and λFluor = 675 nm30) is dissipated into the solvent.
 | ||
| Fig. 5 Decay associated mid-IR spectra (DAS, according to eqn (2)), describing the cooling process (A1(λ)) and the “stationary” difference absorption spectrum (A0(λ)) (cf. text for details). | ||
The ultrafast dynamics, i.e. ISC within a few hundred femtoseconds followed by vibrational cooling on the picosecond time scale is typical for photoexcited transition-metal complexes36 and has been frequently reported.21,35 In addition, it turns out that the femtosecond transient IR spectra, by their strongly structured spectra, allow for a much clearer identification of the cooling/relaxation process as compared to broad and unstructured UV/Vis spectra, where corresponding spectral broadening and shifts are often much weaker or not even detectable as in our earlier work30 on this complex.
5 Conclusions
This paper reports on the first characterization of electronically excited states in a trinuclear Pd complex investigated by a combination of time-resolved IR spectroscopy and quantum chemical calculations. Step-scan FTIR and femtosecond IR spectroscopy are chosen to cover a time range from femtoseconds to microseconds. The same vibrational spectrum is obtained for the whole time range (from some ps to about 2 μs) indicating that within less than 400 fs a long-lived electronic state is populated. From the TR-FTIR spectra the decay time of this long-lived excited state obtained via luminescence spectroscopy is confirmed. Quantum chemical calculations performed for the excited electronic states show that the lowest energy state is of 3A symmetry. Calculated IR spectra for the optimized 3A state match very well with the experimentally observed ones in contrast to the spectra calculated for the energetically higher 3B state. Thus an unambiguous assignment of the experimentally observed long-lived triplet state to a 3A state can be given in addition to the assignment via excited state electronic absorption.30Furthermore the femtosecond transient IR data reveal vibrational cooling characterized by a monoexponential time constant of 7.8 ps and no further changes up to 1 ns. Thus the vibrational excess energy dissipates on a picosecond time scale.
The presented extensive spectroscopic and theoretical studies on a highly symmetric trinuclear Pd complex allow an assignment of electronically excited states and its dynamical behaviour. A long-lived electronically excited state is formed by interaction of the three Pd atoms. The geometry of the complex changes from D3 symmetry in the electronic ground state to C2 symmetry in the 3A excited state, i.e. a pseudo-Jahn–Teller effect is observed. The trinuclear Pd complex has proved to be a rewarding object to study photophysics of a oligonuclear metal complex by the interplay of experiment and theory. The application of a variety of spectroscopic methods (UV/Vis, luminescence, molecular beam)30 including now the structure sensitive IR techniques in combination with high level DFT and TDDFT calculations provide a detailed analysis on the structural changes by going from the electronic ground to an electronically excited state. These investigations are a starting point for further examples of interacting metal centers which can be of photophysical, magnetic or catalytical relevance.
Acknowledgements
Financial support by the German Science Foundation (DFG) and Collaborative Research Center CRC/Transregio 88, “Cooperative effects in homo- and heterometallic complexes (3MET)” is gratefully acknowledged (Projects B4, C1, C2, C4).Notes and references
- H. B. Gray and A. W. Maverick, Science, 1981, 214, 1201 CAS.
- F. Y. Pétillon, P. Schollhammer, J. Talarmin and K. W. Muir, Coord. Chem. Rev., 1998, 178–180, 203–247 CrossRef.
- D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840–850 CrossRef CAS PubMed.
- M. H. Pérez-Temprano, J. A. Casares and P. Espinet, Chem. – Eur. J., 2012, 18, 1864–1884 CrossRef PubMed.
- S. Serroni, S. Campagna, F. Puntoriero, F. Loiseau, V. Ricevuto, R. Passalacqua and M. Galletta, C. R. Chim., 2003, 6, 883–893 CrossRef CAS PubMed.
- J. A. Faiz, R. M. Williams, M. J. J. Pereira Silva, L. De Cola and Z. Pikramenou, J. Am. Chem. Soc., 2006, 128, 4520–4521 CrossRef CAS PubMed.
- T. Suzuki, T. Nakagawa, K. Ohkubo, S. Fukuzumi and Y. Matsuo, Chem. Sci., 2014, 5, 4888–4894 RSC.
- S. Tschierlei, M. Presselt, C. Kuhnt, A. Yartsev, T. Pascher, V. Sundström, M. Karnahl, M. Schwalbe, B. Schäfer, S. Rau, M. Schmitt, B. Dietzek and J. Popp, Chem. – Eur. J., 2009, 15, 7678–7688 CrossRef CAS PubMed.
- B. Dietzek, W. Kiefer, J. Blumhoff, L. Böttcher, S. Rau, D. Walther, U. Uhlemann, M. Schmitt and J. Popp, Chem. – Eur. J., 2006, 12, 5105–5115 CrossRef CAS PubMed.
- I. Bratko and M. Gómez, Dalton Trans., 2013, 42, 10664–10681 RSC.
- J. Park and S. Hong, Chem. Soc. Rev., 2012, 41, 6931–6943 RSC.
- R. M. Haak, S. J. Wezenberg and A. W. Kleij, Chem. Commun., 2010, 46, 2713–2723 RSC.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- N. Huse, H. Cho, K. Hong, L. Jamula, F. M. F. de Groot, T. K. Kim, J. K. McCusker and R. W. Schoenlein, J. Phys. Chem. Lett., 2011, 2, 880–884 CrossRef CAS.
- S. E. Canton, K. S. Kjær, G. Vankó, T. B. van Driel, S.-I. Adachi, A. Bordage, C. Bressler, P. Chabera, M. Christensen, A. O. Dohn, A. Galler, W. Gawelda, D. Gosztola, K. Haldrup, T. Harlang, Y. Liu, K. B. Møller, Z. Németh, S. Nozawa, M. Pápai, T. Sato, T. Sato, K. Suarez-Alcantara, T. Togashi, K. Tono, J. Uhlig, D. A. Vithanage, K. Wärnmark, M. Yabashi, J. Zhang, V. Sundström and M. M. Nielsen, Nat. Commun., 2015, 6 DOI:10.1038/ncomms7359.
- C. Bressler, C. Milne, V. T. Pham, A. ElNahhas, R. M. van der Veen, W. Gawelda, S. Johnson, P. Beaud, D. Grolimund, M. Kaiser, C. N. Borca, G. Ingold, R. Abela and M. Chergui, Science, 2009, 323, 489–492 CrossRef CAS PubMed.
- G. Vankó, A. Bordage, M. Pápai, K. Haldrup, P. Glatzel, A. M. March, G. Doumy, A. Britz, A. Galler, T. Assefa, D. Cabaret, A. Juhin, T. B. van Driel, K. S. Kjær, A. Dohn, K. B. Møller, H. T. Lemke, E. Gallo, M. Rovezzi, Z. Németh, E. Rozsályi, T. Rozgonyi, J. Uhlig, V. Sundström, M. M. Nielsen, L. Young, S. H. Southworth, C. Bressler and W. Gawelda, J. Phys. Chem. C, 2015, 119, 5888–5902 Search PubMed.
- S. E. Canton, X. Zhang, L. M. Lawson Daku, A. L. Smeigh, J. Zhang, Y. Liu, C.-J. Wallentin, K. Attenkofer, G. Jennings, C. A. Kurtz, D. Gosztola, K. Wärnmark, A. Hauser and V. Sundström, J. Phys. Chem. C, 2014, 118, 4536–4545 CAS.
- A. L. Smeigh, M. Creelman, R. A. Mathies and J. K. McCusker, J. Am. Chem. Soc., 2008, 130, 14105–14107 CrossRef CAS PubMed.
- P. Tourón Touceda, S. Mosquera Vázquez, M. Lima, A. Lapini, P. Foggi, A. Dei and R. Righini, Phys. Chem. Chem. Phys., 2012, 14, 1038–1047 RSC.
- M. M. N. Wolf, R. Gross, C. Schumann, J. A. Wolny, V. Schünemann, A. Døssing, H. Paulsen, J. J. McGarvey and R. Diller, Phys. Chem. Chem. Phys., 2008, 10, 4264–4273 RSC.
- J. R. Schoonover, G. F. Strouse, K. M. Omberg and R. B. Dyer, Comments Inorg. Chem., 1996, 18, 165 CrossRef CAS PubMed.
- M. W. George and J. J. Turner, Coord. Chem. Rev., 1998, 177, 201 CrossRef CAS.
- J. M. Butler, M. W. George, J. R. Schoonover, D. M. Dattelbaum and T. J. Meyer, Coord. Chem. Rev., 2007, 251, 492–514 CrossRef CAS PubMed.
- B. D. Moore, M. Poliakoff, M. B. Simpson and J. J. Turner, J. Phys. Chem., 1985, 89, 850 CrossRef CAS.
- P. Chen, K. M. Omberg, D. A. Kavaliunas, J. A. Treadway, R. A. Palmer and T. J. Meyer, Inorg. Chem., 1997, 36, 954 CrossRef CAS.
- K. M. Omberg, J. R. Schoonover, J. A. Treadway, R. M. Leasure, R. B. Dyer and T. J. Meyer, J. Am. Chem. Soc., 1997, 119, 7013–7018 CrossRef CAS.
- A. Gabrielsson, F. Hartl, H. Zhang, J. R. L. Smith, M. Towrie, A. Vlček and R. Perutz, J. Am. Chem. Soc., 2006, 128, 4253 CrossRef CAS PubMed.
- F. Armbruster, J. Meyer, A. Baldes, P. Ona-Burgos, I. Fernández and F. Breher, Chem. Commun., 2011, 47, 221 RSC.
- Y. Schmitt, K. Chevalier, F. Rupp, M. Becherer, A. Grün, A. M. Rijs, F. Walz, F. Breher, R. Diller, M. Gerhards and W. Klopper, Phys. Chem. Chem. Phys., 2014, 16, 8332–8338 RSC.
- T. P. Dougherty and E. J. Heilweil, Chem. Phys. Lett., 1994, 227, 19–25 CrossRef CAS.
- P. Rudolf, F. Kanal, J. Knorr, C. Nagel, J. Niesel, T. Brixner, U. Schatzschneider and P. Nuernberger, J. Phys. Chem. Lett., 2013, 4, 596–602 CrossRef CAS.
- J. B. Asbury, K. Hang, J. S. Yeston, J. G. Cordaro, R. G. Bergman and T. Q. Lian, J. Am. Chem. Soc., 2000, 122, 12870–12871 CrossRef CAS.
- E. M. S. Maôas, R. Kananavicius, P. Myllyperkio, M. Pettersson and H. Kunttu, J. Am. Chem. Soc., 2007, 129, 8934–8935 CrossRef PubMed.
- D. Imanbaew, Y. Nosenko, C. Kerner, K. Chevalier, F. Rupp, C. Riehn, W. R. Thiel and R. Diller, Chem. Phys., 2014, 442, 53–61 CrossRef CAS PubMed.
- A. Vlček, Coord. Chem. Rev., 2000, 200, 933–977 CrossRef.
- P. S. Wagenknecht and P. C. Ford, Coord. Chem. Rev., 2011, 255, 591–616 CrossRef CAS PubMed.
- F. Peters, J. Herbst, J. Tittor, D. Oesterhelt and R. Diller, Chem. Phys., 2006, 323, 109–116 CrossRef CAS PubMed.
- TURBOMOLE V6.6 2014, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, 2007, available from http://www.turbomole.com Search PubMed.
- C. van Wüllen, J. Comput. Chem., 2011, 32, 1195–1201 CrossRef PubMed.
- P. Deglmann, F. Furche and R. Ahlrichs, Chem. Phys. Lett., 2002, 362, 511–518 CrossRef CAS.
- I. B. Bersuker, Chem. Rev., 2013, 113, 1351–1390 CrossRef CAS PubMed.
- M. K. Kesharwani, B. Brauer and J. M. L. Martin, J. Phys. Chem. A, 2015, 119, 1701–1714 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp00959f |
| This journal is © the Owner Societies 2015 |