
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A study of the relationship between water and anions of the Hofmeister series using pressure perturbation calorimetry†
Jordan W.
Bye
and
Robert J.
Falconer
*
Department of Chemical & Biological Engineering, ChELSI Institute, University of Sheffield, Sheffield, S1 3JD, UK. E-mail: r.j.falconer@sheffield.ac.uk; Fax: +44 (0)114 2227501; Tel: +44 (0)114 2228253
First published on 30th April 2015
Abstract
Pressure perturbation calorimetry (PPC) was used to study the relationship between water and sodium salts with a range of different anions. At temperatures around 25 °C the heat on pressurisation (ΔQ) from 1 to 5 bar was negative for all solutions relative to pure water. The raw data showed that as the temperature rose, the gradient was positive relative to pure water and the transition temperature where ΔQ was zero was related to anion surface charge density and was more pronounced for the low-charge density anions. A three component model was developed comprising bulk water, the hydration layer and the solute to calculate the molar expansivity of the hydration layer around the ions in solution. The calculated molar expansivities of water in the hydration layer around the ions were consistently less than pure water. ΔQ at different disodium hydrogen phosphate concentrations showed that the change in molar enthalpy relative to pure water was not linear even as it approached infinite dilution suggesting that while hydration layers can be allocated to the water around ions this does not rule out interactions between water and ions extending beyond the immediate hydration layer.
Introduction
Water is a unique solvent that plays a critical role in supporting life on earth. Despite this there is still ongoing discussion between scientists on the nature of water and its association with inorganic and organic molecules. A unique property of water is its propensity to rapidly swap protons between water molecules at a picosecond timescale, referred to as hydrogen bonding. When a salt is added to water it dissociates into anions and cations as described by Arrhenius in 1887.1 These ions have electrical fields which interact with the dipolar water molecules that arrange themselves around the ions. These arranged water molecules are often referred to as the hydration layer. There is discussion around whether the ion's effect extends just to the water molecules at the interface with the ion, into a second hydration layer or beyond into more distant water.In 1888 Franz Hofmeister published two papers on the effect salts had on protein solubility in water.2,3 An English translation of these papers is available in Kunz et al. 2004.4 The observation was made that ions could be ordered according their ability to precipitate or solubilise protein ensembles. Hofmeister explained his observations in terms of the hydration strength of the ions (their ability to absorb water). Ions with strong hydration compete with the protein for the water and it is this that causes the protein's precipitation. A similar ordering of the ions was observed in surface tension measurements by Adolf Heydweiller in 1910.5 An alternative explanation for the Hofmeister effect was published in 1930 by Cox and Wolfenden which explained the observed effect of ions on viscosity in terms of the degree of polymerisation of the solvent in the presence of the ions.6 Over a period of years the explanation for the Hofmeister effect evolved as the ability of an ion to alter the hydrogen bond population of the solvent7 often referred to as the structure making and structure breaking theory. This theory relied on the ions having the ability to interact with water molecules beyond their first hydration layer. In 2003, the structure making and structure breaking theory was directly challenged using data from femtosecond pump–probe spectroscopy that suggested there was no measurable interaction with water beyond the first hydration layer.8 Since then evidence using neutron diffraction,9 molecular dynamic simulation10 and calorimetry techniques11 have suggested long-range interactions beyond the first hydration layer are likely.
In this paper we add pressure perturbation calorimetry (PPC) at low pressure changes to the existing palate of techniques for studying electrolyte solutions and discuss the results with reference to the Hofmeister effect. The current PPC technology was preceded by instrumentation that subjects the samples to high pressure changes (up to 4000 bar), which were suitable for studying phase transitions in organic solvents and polymers, and had been around since the 1970s.12–15 More recently a modification to differential scanning microcalorimetry (DSC) instrumentation enabled this type of analysis to be carried out at lower pressure changes (4 bar) with a high degree of sensitivity, which enabled the analysis of the heat change on pressurisation for diverse solutes in water.16,17 These devices measure the heat change in the solution as the pressure above the solution is altered. From this the thermal expansion coefficient (α) of the solute has been calculated and the hydrogen-bond population of the water at the solute–water interface studied.18,19 This approach has been applied to a diverse range of solutes including polymers,16 amino acids,17 small inorganic and organic molecules,18 proteins,16–28 lipid micelles and bilayers,29–31 and nucleic acids.32,33
The calculation of the thermal expansion coefficient from PPC data assumed a two-state model for water with a relatively low density and a denser liquid species. PPC relies on the Le Chatelier's principle, that on pressurisation the water will try to equilibrate by moving towards the higher density form. The derivation of the equation that has been used to calculate the thermal expansion coefficient uses a two-component system for small molecules in solution where Vtot is the total volume, V0 is the molar volume of solvent and ![[V with combining overline]](https://www.rsc.org/images/entities/i_char_0056_0305.gif) is the partial specific volume of the solute in the solution which includes any volume changes induced in the solvent; x0 is molar fraction of the solvent and xs is molar fraction of the solute.17,18
is the partial specific volume of the solute in the solution which includes any volume changes induced in the solvent; x0 is molar fraction of the solvent and xs is molar fraction of the solute.17,18
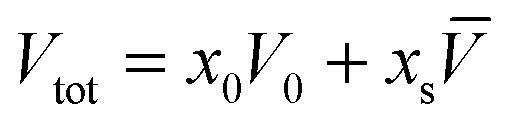 | (1) |
Data analysis
The authors initially calculated values for the coefficient of thermal expansion using the equation derived in Lin et al. 200217 using published apparent partial volume () values34 and the experimental PPC results for 100 mM salt solutions (see Fig. S1 in the ESI†). While the coefficient of thermal expansion for sodium chloride, sodium bromide, sodium iodide, sodium thiocyanate and sodium perchlorate are plausible, those for sodium fluoride and disodium sulphate were unusual. Sodium flouride and disodium sulphate both have high charge density and their conventional apparent partial volume () values34 are markedly lower than their intrinsic volumes (Vint)35 (see Fig. S2 in the ESI†). It was concluded that the two-component model was problematic for high charge density ions. It was this observation that led the authors to suggest a three-component model for salt solutions where Vb is the molar volume of bulk solvent, Vh is the average molar volume of the solvent within the hydration layer and Vs is the molar volume of solute; xb is the molar fraction of the bulk solvent, xh is the molar fraction of the solvent within the hydration layer and xs is the molar fraction of the solute.| Vtot = xbVb + xhVh + xsVs | (2) |
| Q = −TΔPαV | (3) |
| Q = −TΔP[xbVbαb + xhVhαh + xsVsαs] | (4) |
The difference in heat between the sample and reference cells is
| ΔQ = TΔP[x0V0α0 − xbVbαb − xhVhαh − xsVsαs] | (5) |
As V0 = Vb, α0 = αb and x0 is 1 the equation can be simplified to
| ΔQ = TΔP[(1 − xb)(Vbαb) − xhVhαh − xsVsαs] | (6) |
| ΔQ = TΔP[(xs + xh)(Vbαb) − xhVhαh − xsVsαs] | (7) |
| ΔQ = TΔP[(n + 1)(xsVbαb) − nxsVhαh − xsVsαs] | (8) |
| ΔQ = TΔPxs[(n + 1)(Vbαb) − nVhαh − Vsαs] | (9) |
| ΔQ = TΔPxs[(n + 1)(Vbαb) − nVhαh − Vsαs] + A | (10) |
Determining the thermal expansion coefficient (αh) is more difficult as a value for Vh is needed. The difference between experimentally determined apparent partial molal volumes34 and the intrinsic molal volume35 could be used to estimate the change in molar volume of the water within the hydration layer. An alternative approach would be to calculate the effect of electrostriction on the molar volume of the water within the hydration layer from molecular dynamic simulation data.38 Both these method would be estimates at best so the calculations used by the authors were confined to calculating the molar expansivity of the hydration layer around the ions.
Methods and materials
Sample preparation
Ultra-pure water and all salts were sourced from Sigma Aldrich, Gillingham, UK with purities >99.9%. Sodium was used as the counter-ion for all anions. 2000 mM stock solutions at pH 7 were made for all salts (sodium fluoride stock solutions were made to 100 mM at pH 8.4 due to solubility limits); stock solution dilutions were performed by adding the required amount of pure water.Pressure perturbation calorimetry (PPC) measurements
PPC measurements were obtained using a capillary Nano-DSC (TA Instruments, New Castle, DE, USA). Samples were degassed for 1 hour at 30 °C to remove dissolved gas from samples and eliminate bubble formation during the scan. Heat effects (ΔQ) were measured during alternating pressure pulses of ±4 bar from 1 bar to 5 bar at 1 °C intervals from 7–92 °C, giving a usable data range of 10–90 °C. A heating rate of 0.1 °C min−1 was used to satisfy isothermal conditions required during pressure pulses; this scanning rate is slower than the instrument feedback.33 The instrument was held at a constant temperature for 30 minutes before each scan to ensure that any asymmetry between the reference and sample cells was minimal. Heat changes during pressurisation steps were used for data analysis and were calculated using the NanoAnalyze software (TA Instruments, New Castle, DE, USA) provided by the manufacturer. Water baseline scans were performed with pure water in reference and sample cell, scans with salt present were performed with pure water in the reference cell and salt solution in the sample cell. The area under each thermal power spike was calculated by integration using NanoAnalyze software (TA Instruments, New Castle, DE, USA) and was used as the heat change during pressurisation for that temperature, shown in Fig. 1a–d. | ||
Fig. 1 (a–c) Show the raw data from a pressure perturbation scan of 1000 mM disodium hydrogen phosphate, sodium chloride and sodium thiocyanate in pure water at pH 7 respectively. Alternating pressure pulses from 1–5 bar and then 5–1 bar were applied to the sample at 1 °C intervals from 7–92 °C, with a heating rate of 0.1 °C min−1. (d) Shows a close up of (a) from 70 °C to 75 °C, with data for pressure increases and decreases shown (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ). Here the area under the power spike for the pressure pulses was integrated by the software provided to calculate the energy absorbed and released by the sample relative to the reference cell containing pure water. For the pressurisation steps shown in (d) the change in heat is positive (endothermic) with reference to the pure water baseline. During the depressurisation steps the heat change is negative (exothermic) with reference to the pure water baseline. ). Here the area under the power spike for the pressure pulses was integrated by the software provided to calculate the energy absorbed and released by the sample relative to the reference cell containing pure water. For the pressurisation steps shown in (d) the change in heat is positive (endothermic) with reference to the pure water baseline. During the depressurisation steps the heat change is negative (exothermic) with reference to the pure water baseline. | ||
Calculation of molar expansivity
Application of eqn (10) is simple in practice for solutes where the specific volume (Vs), thermal expansivity (αs) are known and n can be estimated. The specific volume (Vb) and thermal expansivity (αb) of pure water are known.36A was determined by adjusting it until the calculated ΔQ fitted experimental ΔQ data.Eqn (10) was rearranged as follows so molar expansivity (Vhαh) can be calculated.
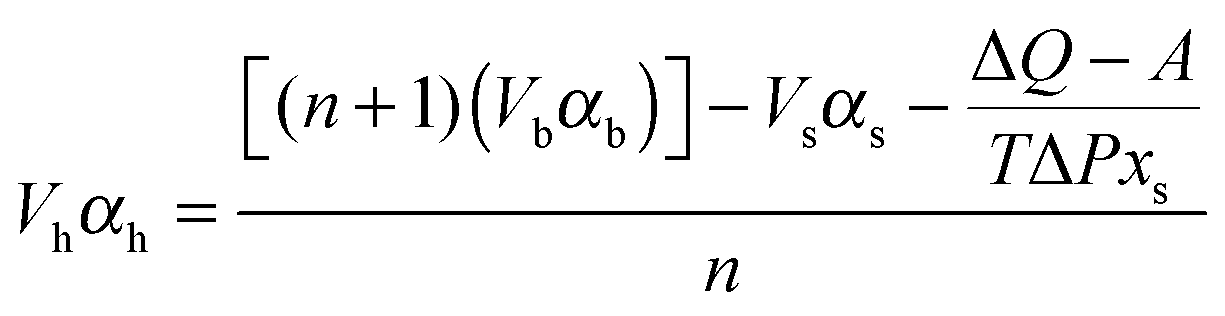 | (11) |
Results
2 M stock solutions were made for disodium hydrogen phosphate (at pH 7.0 this is a mixture of sodium dihydrogen phosphate and disodium hydrogen phosphate), disodium sulphate, sodium chloride, sodium bromide, sodium iodide, sodium thiocyanate and sodium perchlorate at pH 7.0 (sodium fluoride at pH 8.4 was only made to 100 mM due to solubility limits). These salts were chosen so that a range of anions from the Hofmeister series could be studied, including the simple monoatomic halogen anions and more complex oxoanions.Thermal data was collected as microwatts (μW) of power required to keep the reference and sample cell temperatures identical upon pressurisation and depressurisation steps, the raw data for 1000 mM disodium hydrogen phosphate, sodium chloride and sodium thiocyanate, is shown in Fig. 1a–c, respectively. Fig. 1d shows a small temperature range in which the area under the thermal spikes (which was integrated) can be more clearly seen, along with the pressure change which caused the thermal spike. The power function was converted into heat absorbed or released (ΔQ) in micro joules (μJ) by the sample during pressure changes between 1–5 atm.
The ΔQ associated with pressurisation of the salt solutions at 100 mM and 1000 mM, respectively, is shown in Fig. 2a and b. The plotted heat energies show the difference in energy added to the sample cell compared to the reference cell which contained pure water. Sodium fluoride was not tested at 1000 mM due to its low solubility. For all salts tested a negative ΔQ is observed at lower temperatures upon sample pressurisation, which indicates that an exothermic process is taking place. The temperature where ΔQ upon sample pressurisation is zero (Ti) was found to be salt dependent and independent of salt concentration. The Ti were found to be 60.5 °C for disodium hydrogen phosphate, 59.5 °C for disodium sulphate, 54 °C for sodium fluoride, 59 °C for sodium chloride, 65.5 °C for sodium bromide, 76.5 °C for sodium iodide, 90.5 °C for sodium thiocyanate and 104 °C for sodium perchlorate at all salt concentrations. The temperature where ΔQ upon pressurisation became endothermic was not reached for sodium perchlorate concentrations. The temperature where its ΔQ became endothermic was calculated by extrapolation for sodium perchlorate. The surface charge density of the ions tested plotted against the temperature where the heat changes upon pressurisation of the salt solution becomes endothermic is shown in Fig. 3. Charge, ionic and apparent ionic radii for sulphate, fluoride, chloride, bromide, iodide, thiocyanate and perchlorate were used to calculate the surface charge density of each ion.29,30 It should be noted that phosphate was omitted from this graph due to phosphate being composed of a mixture of H2PO4− and HPO42− at pH 7.0. There is a trend of increasing Ti values with a decrease in surface charge density. Thiocyanate does not fit the trend as well as the other ions which may be due to it being treated as a sphere to obtain its ionic radius, when it is not spherical. It should be noted that the ordering of these ions by the Ti value closely agrees with the order seen in the Hofmeister series.2–4
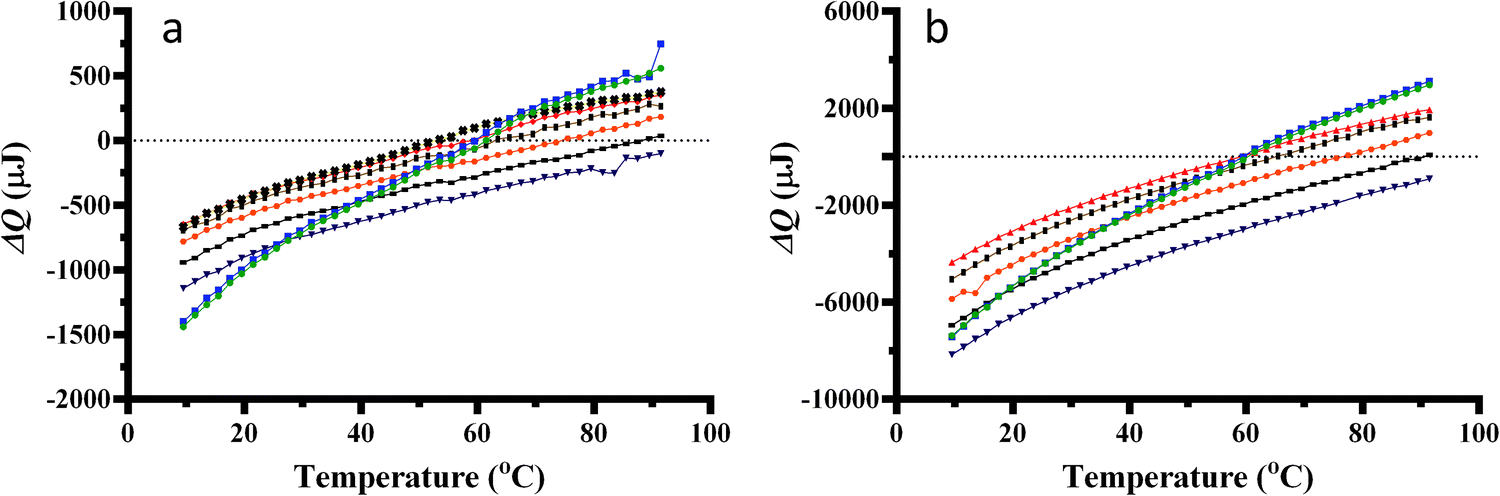 | ||
Fig. 2 (a) Heat changes for pressure increase (1 to 5 bar) from 7–92 °C for (a) 100 mM and (b) 1000 mM disodium hydrogen phosphate ( ), disodium sulphate ( ), disodium sulphate ( ), sodium fluoride ( ), sodium fluoride ( ), sodium chloride ( ), sodium chloride ( ), sodium bromide ( ), sodium bromide ( ), sodium iodide ( ), sodium iodide ( ), sodium thiocyanate (–) and sodium perchlorate ( ), sodium thiocyanate (–) and sodium perchlorate ( ). The sample size was 300 μL. Lines between data points do not represent experimental data and are only shown for guidance. ). The sample size was 300 μL. Lines between data points do not represent experimental data and are only shown for guidance. | ||
 | ||
Fig. 3 Ion surface charge density against the temperature (Ti) where the ΔQ equals zero for 100 mM disodium sulphate ( ), sodium fluoride ( ), sodium fluoride ( ), sodium chloride ( ), sodium chloride ( ), sodium bromide ( ), sodium bromide ( ), sodium iodide ( ), sodium iodide ( ), and sodium perchlorate ( ), and sodium perchlorate ( ). Note thiocyanate was omitted as it is not spherical. ). Note thiocyanate was omitted as it is not spherical. | ||
The average gradient on the ΔQ versus temperature plot for the eight salts was tested at 100 mM from 9.5–91.5 °C. The average gradient for each salt (100 mM) was calculated by dividing the difference in ΔQ upon pressurisation at 9.5 °C and 91.5 °C. From Fig. 2 it can clearly be seen that ions with a −2 charge release more energy at lower temperatures and require more energy at higher temperatures upon pressurisation than ions with a −1 charge. It is worth remembering that the divalent anion, sulphate has two sodium cations so the gradient cannot be ascribed to the anion alone. The gradients of ions with a −1 charge were similar to each other, so are not dependent on ion charge density, size or shape.
The effect of different phosphate concentrations on ΔQ versus temperature is shown in Fig. 4. The change in molar enthalpy relative to pure water of the ΔQ versus temperature was dependent on phosphate concentration. The ΔQ versus phosphate concentration at different temperatures is shown in Fig. 5a and b. It is worth noting that change in molar enthalpy relative to pure water is not linear even at concentrations around 100 mM where there are over 500 water molecules per anion and cation.
 | ||
Fig. 4 Heat changes for pressure increase (1–5 bar) from 7–92 °C at different concentrations of disodium hydrogen phosphate; 50 ( ), 100 ( ), 100 ( ), 250 ( ), 250 ( ), 500 ( ), 500 ( ), 750 ( ), 750 ( ) and 1000 mM (●). The sample size was 300 μL. ) and 1000 mM (●). The sample size was 300 μL. | ||
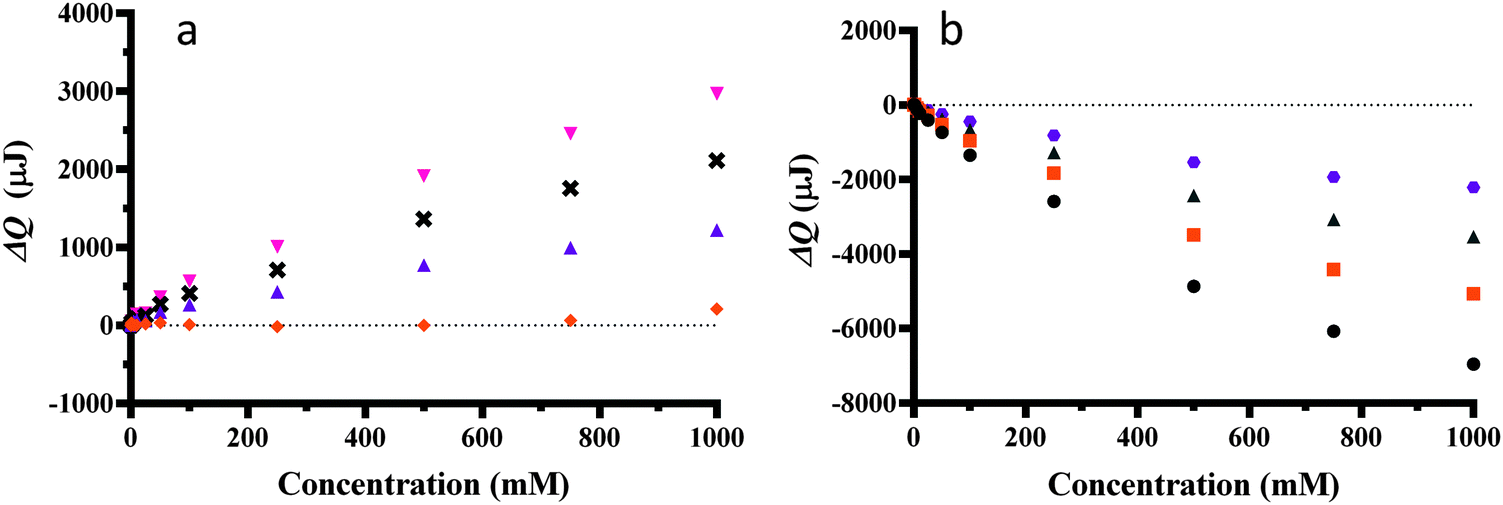 | ||
Fig. 5 Heat changes for pressure increase (1–5 bar) at different concentrations of disodium hydrogen phosphate and different temperatures; (a) 11.5 (●), 21.5 ( ), 31.5 ( ), 31.5 ( ), (b) 41.5 ( ), (b) 41.5 ( ), 61.5 ( ), 61.5 ( ), 71.5 ( ), 71.5 ( ), 81.5 (x) and 91.5 °C ( ), 81.5 (x) and 91.5 °C ( ). The sample size was 300 μL. ). The sample size was 300 μL. | ||
The values for the molal volume (Vb) and thermal expansion coefficient of pure water (αb) are known from the literature,36 and T, ΔP and xs values are controlled during each experiment. The variables are n and molar expansivity (Vhαh). Vh and αh cannot be separated by this approach. Vsαs is known for crystalline salts at a set temperature.38 In this paper we assume the Vsαs value for salt and the n does not vary with temperature calculations. The ΔQ values calculated using eqn (10) for 100 mM NaCl (Fig. 6) are consistent with an n value of 13.3 and a Vhαh value 0.55 times that of the known Vbαb for bulk water at temperatures between 5 and 95 °C. The calculated values for all the sodium halide salts using both the single hydration layer and double hydration layer models are shown in Table 1. The Vhαh values approach that of pure water as the charge density reduces but are all lower than pure water.
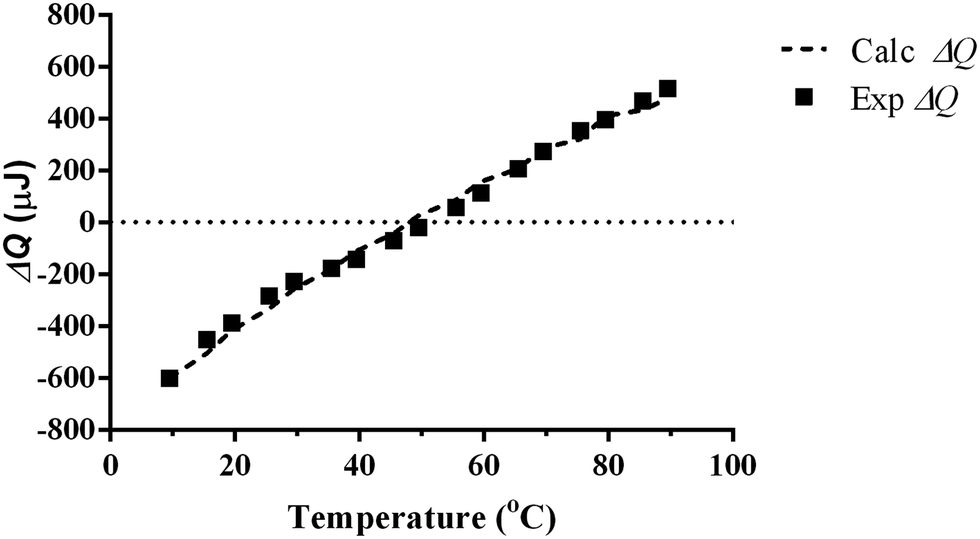 | ||
| Fig. 6 Experimental values for the ΔQ for 100 mM NaCl fitted against values for ΔQ calculated using eqn (10) using the assumptions that n is 13.3 and the molar expansivity of the hydration layer is 0.55 that of pure water. | ||
| n | V s α s (m3 mol−1 K−1) | V h α h/Vbαbc | A (J) | |
|---|---|---|---|---|
| a The number of water molecules per sodium and halide ions was determined by MD simulation.40 b The Vsαs is for crystalline sodium halides.41 c The Vbαb is derived from pure water density measurements published in Kell, 1967.38 The Vhαh/Vbαb and A values were calculated using eqn (10). | ||||
| 1 Layer model | ||||
| NaF | 12.3 | 1.48 × 10−9 | 0.54 | 7.35 × 10−7 |
| NaCl | 13.3 | 3.16 × 10−9 | 0.55 | 6.60 × 10−7 |
| NaBr | 13.1 | 4.46 × 10−9 | 0.62 | 7.05 × 10−7 |
| NaI | 14.0 | 5.51 × 10−9 | 0.65 | 7.80 × 10−7 |
| 2 Layer model | ||||
| NaF | 34.3 | 1.48 × 10−9 | 0.83 | 7.50 × 10−7 |
| NaCl | 40.8 | 3.16 × 10−9 | 0.85 | 6.75 × 10−7 |
| NaBr | 40.5 | 4.46 × 10−9 | 0.87 | 7.50 × 10−7 |
| NaI | 46.1 | 5.51 × 10−9 | 0.89 | 8.10 × 10−7 |
Discussion
Interpretation of the PPC results for the different salt solutions has to take into account that the measured ΔQ is a result of both the sodium and the anion and cannot be separated. The monovalent anions all have the same concentration of sodium so differences between the ΔQ values will be due to the anion alone. The second factor that has to be considered is that the measured ΔQ is relative to the pure water in the reference cell. By applying eqn (10) the Vhαh values for the hydration layer were calculated.Heat release has been interpreted as bond formation between water molecules40 so the initial exothermic ΔQ can be interpreted as the hydration layer strengthening or forming additional hydrogen bonds relative to pure water. At temperatures over the Ti value this relationship reverses and ΔQ became endothermic can be interpreted as the hydration layer weakening or breaking more hydrogen bonds relative to pure water. The temperature where this transition between bond formation and bond breaking is observed is dependent on charge density of the anion. This is clearly seen for the halides which are simple spheres but the general trend is also evident with the more complex structure of the oxoanions and thiocyanate (which is more linear than spherical). It is possible that PPC is providing evidence about the hydrogen bond population of water around the anions. Electrostatic interaction impact on the hydration layer is also evident as valency of the anion has an obvious effect on the average gradient of the ΔQ versus temperature plot though this work does not differentiate between this being driven by the valency of the anion or the number of sodium ions present.
The experimental data where the concentration of phosphate was varied (Fig. 4, 5a and b) demonstrated that change in molar enthalpy relative to pure water at a temperature below the Ti value was reduced as the xs increased. The non-linearity that occurs below 100 mM phosphate suggests that there are longer-range interactions between water and ions that extends beyond the first two layers of water and are probably electrostatic interactions.10 This does call into question where the n value should be set so the use of eqn (10) should have the assumptions used to select the n value clearly explained.
The equation for the analysis of PPC data presented in this paper (eqn (10)) is based on a three component model comprising bulk water (unperturbed water); hydration water (water that is measurably perturbed by the ions) and the ions. Eqn (10) accepts that the hydration water has its own molar volume and thermal expansion coefficient that may differ from the bulk water and this should not be ignored. The n value is dependent on where the boundary between the hydration layer and bulk water is set. Molecular dynamic simulation has been used to estimate the n value around the halides, see Table 1.38 For example, if the boundary is set at a single layer of water molecules per ion, a sodium and chloride would have an n value of 13.3; but if a double layer is chosen the n value will be 34.3. The researcher has a choice of a single thickness or double thickness model when using eqn (10) and should remember that the calculated Vhαh will be an average for the water molecules within the hydration layer.
The apparent partial molar volumes of water around salts at infinite dilution32 and the ionic volume43 have been observed to be quite different for over 50 years,6 see Fig. S1 in the ESI.† This has been explained as electrostriction, a reduction in the volume of the water interacting with the ion's electrical field and is particularly noticeable for high charge density ions like sodium and fluoride.34 The Vhαh values calculated are consistently lower than bulk water suggesting that any decrease in the Vh value is due to electrostriction, the αh value or a combination of the two.
Partial molal adiabatic compressibility of sodium salts in water at 25 °C determined from the velocity of sound through the sample41 like the molar expansivity of the hydration layer (Vhαh) calculated here also follows a series that mirrors the Hofmeister series. It is attractive to think the three phenomenon are related and have to do with the interaction of the salt with water.
Franz Hofmeister's original observation was that certain salts were consistent in their ability to precipitate and dissolve proteins.2–4 Hofmeister put this observation down to the salt absorbing the water. In the 1930s the idea that ions have a long-range effect on the hydrogen bond structure of water was used to explain the viscosity of electrolyte solutions.5 This lead to the theory of some ions being “structure makers” and others being “structure breakers”. Since the 1930s there has been extensive research undertaken to better study the interaction between water and ions using an incredibly diverse collection of analytical techniques including nuclear magnetic resonance, optical Kerr effect spectroscopy, dielectric relaxation spectroscopy, transient vibrational adsorption spectroscopy, terahertz spectroscopy, X-ray scattering and molecular dynamic simulation. The femtosecond pump–probe spectroscopy results first used to challenge the long-range effect of ions on water came to the conclusion that ions had little effect on water beyond it's immediate single molecule thick hydration layer.8 A hydration layer composed of a single water layer has been used to explain experimental data for a range of techniques including nuclear magnetic resonance,42 optical Kerr effect spectroscopy,43 transient vibrational adsorption spectroscopy,42 and terahertz spectroscopy.44 The problem with the model of water being limited to a single layer around the ion is that it failed to explain the Hofmeister effect while destroying the established structure maker structure breaker theory. The new orthodoxy of a single water molecule thick hydration layer has been criticised using data from neutron scattering9 and femtosecond infrared spectroscopy45 which detected a second layer of water around ions. When the potential long-range effects of the ion's electrical field is taken into consideration along with the dynamic nature of water the selection of a hydration layer's boundary is probably a measure of each analytical method's sensitivity. PPC can be used to justify either a single or double water layer around ions. It can also be used to challenge the idea of the interaction between water and ions being solely short range. The slope of the ΔQ versus phosphate ion concentration plot shown in Fig. 5 and 6 did not show linearity as it approached zero concentration. This phenomenon was also observed when determining apparent partial molar volumes of water around salts where an infinite dilution method was needed to estimate the partial molar volume.34 Both observations could be used to argue for long-range interactions between ions and water beyond a one or two molecule thick hydration layer. While the authors would not argue that structure making structure breaking theory is an adequate explanation for the Hofmeister effect the femtosecond pump–probe spectroscopy evidence that was used to criticise the theory8 was not as conclusive as it looked at the time.
The majority of the papers that used PPC to study amino acids,17 small inorganic and organic molecules,18 proteins17–28 and DNA32,33 used the equation derived in Lin et al. 200217 to calculate the thermal coefficient of expansion of the solvent volume. The authors of this paper suggest that eqn (10) is worth applying to PPC studies of diverse solutes in aqueous solutions. Small organic and inorganic molecules as well as macromolecules have hydration layers. There is an increasing body of work using techniques such as terahertz spectroscopy that have been used to detect hydration layers around diverse organic molecules including sugars,46 peptides47 and proteins48–50 suggesting hydration in many cases is not limited to a single layer at the solutes surface, but can be extensive. Whether the boundary of the hydration layer around a solute is a single layer, a double layer or more complex this population of water has to be defined in terms of its own average molar volume and thermal expansion coefficient like the water around salts in this paper. In studies using macromolecules such as globular proteins as the solute, it is also worth noting that the molar expansivity (Vsαs) will have to be estimated from the known partial volumes of the protein's constituent groups51 and that estimating a value for n in eqn (10) will be problematic with the data available at the time of writing.
Acknowledgements
The authors would like to thank the Department of Chemical and Biological Engineering at the University of Sheffield for financing Jordan Bye's studentship, and the Engineering and Physical Sciences Research Council (EPSRC) Knowledge Transfer Account for part funding the purchase of the DSC. The authors would also like to thank Dr James McGregor of the University of Sheffield for his advice into the interpretation of the PPC results.References
- S. Arrhenius, Z. Phys. Chem., 1887, 1, 631–648 Search PubMed.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 24, 247–260 Search PubMed.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 25, 1–30 Search PubMed.
- W. Kunz, J. Henle and B. W. Ninham, Curr. Opin. Colloid Interface Sci., 2004, 9, 19–37 CrossRef CAS PubMed.
- A. Heydweiller, Ann. Phys., 1910, 33, 145–185 CrossRef CAS PubMed.
- W. M. Cox and J. H. Wolfenden, Proc. R. Soc. London, Ser. A, 1934, 92, 475–488 CrossRef.
- R. W. Gurney, Ionic Processes in Solution, McGraw-Hill, New York, 1953 Search PubMed.
- A. W. Omta, M. F. Kropman, S. Woutersen and H. J. Bakker, Science, 2003, 301, 347–349 CrossRef CAS PubMed.
- R. Mancinelli, A. Botti, F. Bruni, M. A. Ricci and A. K. Soper, Phys. Chem. Chem. Phys., 2007, 9, 2959–2967 RSC.
- S. J. Irudayam and R. H. Henchman, J. Chem. Phys., 2013, 137, 034508 CrossRef PubMed.
- J. W. Bye and R. J. Falconer, J. Phys. Chem. B, 2014, 118, 4282–4286 CrossRef CAS PubMed.
- L. Ter-Minassian, J. C. Petit, N. Vankiet and C. Brunaud, J. Chim. Phys., 1970, 67, 265–269 Search PubMed.
- L. Ter-Minassian and F. Milliou, J. Phys. E: Sci. Instrum., 1983, 16, 450–455 CrossRef CAS.
- S. L. Ranzio, J. Phys. E: Sci. Instrum., 1984, 11, 1058–1061 CrossRef.
- S. L. Ranzio, Thermochim. Acta, 1997, 300, 29–41 CrossRef.
- P. Kujawa and F. M. Winnik, Macromolecules, 2001, 34, 4130–4135 CrossRef CAS.
- L. N. Lin, J. F. Brandts, J. M. Brandts and V. Plotnikov, Anal. Biochem., 2002, 302, 144–160 CrossRef CAS PubMed.
- J. D. Batchelor, A. Olteanu, A. Tripathy and G. J. Pielak, J. Am. Chem. Soc., 2004, 126, 1958–1961 CrossRef CAS PubMed.
- R. Ravindra and R. Winter, Z. Phys. Chem., 2003, 217, 1221–1243 CrossRef CAS.
- W. Dzwolak, R. Ravindra, J. Lendermann and R. Winter, Biochemistry, 2003, 42, 11347–11355 CrossRef CAS PubMed.
- C. Nicolini, R. Ravindra, B. Ludolph and R. Winter, Biophys. J., 2004, 86, 1385–1392 CrossRef CAS.
- R. Ravindra and R. Winter, ChemPhysChem, 2004, 5, 566–571 CrossRef CAS PubMed.
- R. Ravindra, C. Royer and R. Winter, Phys. Chem. Chem. Phys., 2004, 8, 1938–1943 Search PubMed.
- L. Mitra, N. Smolin, R. Ravindra and R. Winter, Phys. Chem. Chem. Phys., 2006, 8, 1249–1265 RSC.
- R. Winter, D. Lopes, S. Grundzielanek and K. Vogtt, J. Non-Equilib. Thermodyn., 2007, 32, 41–97 CAS.
- A. Cooper, D. L. Cameron, J. Jakus and G. W. Pettigrew, Biochem. Soc. Trans., 2007, 35, 1547–1550 CrossRef CAS PubMed.
- D. L. Cameron, J. Jakus, S. R. Pauleta, G. W. Pettigrew and A. Cooper, J. Phys. Chem. B, 2010, 114, 16228–16235 CrossRef CAS PubMed.
- A. D. Tsamaloukas, N. K. Pyzocha and G. I. Makhatadze, J. Phys. Chem. B, 2010, 114, 16166–16170 CrossRef CAS PubMed.
- M. Nazari, H. Y. Fan and H. Heerklotz, Langmuir, 2012, 28, 14129–14136 CrossRef CAS PubMed.
- N. Tamai, Y. Nambu, S. Tanaka, M. Goto, H. Matsuki and S. Kaneshina, Colloids Surf., B, 2012, 92, 232–239 CrossRef CAS PubMed.
- H. Y. Fan, M. Nazari, S. Chowdury and H. Heerklotz, Langmuir, 2011, 27, 1693–1699 CrossRef CAS PubMed.
- G. Rayan, A. D. Tsamaloukas, R. B. Macgregor and H. Heerklotz, J. Phys. Chem. B, 2009, 113, 1738–1742 CrossRef CAS PubMed.
- A. I. Dragan, D. J. Russel and P. L. Privalov, Biopolymers, 2009, 91, 95–101 CrossRef CAS PubMed.
- F. J. Millero, Chem. Rev., 1971, 71, 147–176 CrossRef CAS.
- Y. Marcus, Ions Properties, CRC Press, Boca Raton, 1997 Search PubMed.
- G. S. Kell, J. Chem. Eng. Data, 1967, 12, 66–69 CrossRef CAS.
- H. Ohtaki and T. Radnai, Chem. Rev., 1993, 93, 1157–1204 CrossRef CAS.
- S. H. Lee and J. C. Rasaiah, J. Phys. Chem., 1996, 100, 1420–1425 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, 88th edn, 2008 Search PubMed.
- B. Hribar, N. T. Southall, V. Vlachy and K. A. Dill, J. Am. Chem. Soc., 2002, 124, 12302–12311 CrossRef CAS PubMed.
- F. J. Millero and J. D. Sharp, J. Chem. Eng. Data, 2013, 58, 3458–3463 CrossRef CAS.
- H. J. Bakker, Chem. Rev., 2008, 108, 1456–1473 CrossRef CAS PubMed.
- I. A. Heisler, K. Mazur and S. R. Meech, J. Phys. Chem. B, 2011, 115, 1863–1973 CrossRef CAS PubMed.
- M. Heyden, E. Bründermann, U. Heugen, G. Niehues, D. M. Leitner and M. Havenith, J. Am. Chem. Soc., 2008, 130, 5773–5779 CrossRef CAS PubMed.
- K. J. Tielrooij, N. Garcia-Araez, M. Bonn and H. J. Bakker, Science, 2010, 328, 1006–1009 CrossRef CAS PubMed.
- M. Heyden, E. Bründermann, U. Heugen, G. Niehues, D. M. Leitner and M. Havenith, J. Am. Chem. Soc., 2008, 130, 5773–5779 CrossRef CAS PubMed.
- T. Ding, R. Li, J. A. Zeitler, T. L. Huber, L. F. Gladden, A. P. J. Middelberg and R. J. Falconer, Opt. Express, 2010, 18, 27431–27444 CrossRef CAS PubMed.
- S. Ebbinghaus, S. J. Kim, M. Heyden, X. Yu, U. Heugen, M. Gruebele, D. M. Leitner and M. Havenith, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20749–20752 CrossRef CAS PubMed.
- B. Born, H. Weingärtner, E. Bründermann and M. Havenith, J. Am. Chem. Soc., 2009, 131, 3752–3755 CrossRef CAS PubMed.
- J. W. Bye, S. C. Meliga, D. Ferachou, J. A. Zeitler and R. J. Falconer, J. Phys. Chem. A, 2014, 118, 83–88 CrossRef CAS PubMed.
- G. I. Makhatadze, V. N. Medvedkin and P. L. Privalov, Biopolymers, 1990, 30, 1001–1010 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp00571j |
| This journal is © the Owner Societies 2015 |