
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Qualifying label components for effective biosensing using advanced high-throughput SEIRA methodology†
Andrea
Hornemann
*a,
Diane
Eichert
b,
Sabine
Flemig
c,
Gerhard
Ulm
a and
Burkhard
Beckhoff
a
aPhysikalisch-Technische Bundesanstalt, Abbestr. 2-12, 10587 Berlin, Germany. E-mail: andrea.hornemann@ptb.de; Fax: +49 30 3481 7103; Tel: +49 30 3481 7183
bElettra-Sincrotrone Trieste S.C.p.A. di interesse nazionale, Strada Statale 14, 34149 Trieste, Italy
cBAM Bundesanstalt für Materialforschung und-prüfung, Richard-Willstätter-Str.10, 12489 Berlin, Germany
First published on 3rd March 2015
Abstract
The need for technological progress in bio-diagnostic assays of high complexity requires both fundamental research and constructing efforts on nano-scaled assay recognition elements that can provide unique selectivity and design-enhanced sensitivity features. Nanoparticle induced sensitivity enhancement and its application related to multiplexed capability Surface-Enhanced InfraRed Absorption (SEIRA) assay formats are well suitable for these purposes. The potential of diverse fluorophore–antibody conjugates, being chemisorbed onto low-cost gold nanoparticulate SEIRA substrates, has been explored with respect to their spectral discriminability. These novel biolabels deliver molecular SEIRA fingerprints that have been successfully analyzed by both uni- and multivariate analyzing tools, to discriminate their multiplexing capabilities. We show that this robust spectral encoding via SEIRA fingerprints opens up new opportunities for a fast, reliable and multiplexed high-end screening in biodiagnostics.
1. Introduction
Nanotechnology has an increasing impact on both the design and functionality of biomedical and bio-diagnostic devices for reliable high-performance analysis. High-throughput capabilities require the simultaneous detection of various analytes combined with appropriate bioassay components. Additionally, reduction of the biosensors' cost by using, for instance, low cost nanoparticles (NPs) instead of artificial nanostructures is another important aspect. Various bioassay configurations may be used, such as the immobilization of targeting elements such as antibodies on glass slides coated with various targeting ligands, as microarrays. To design assays for antibodies that function as molecular probes, a selective binding event has to be optimized with respect to the overall physicochemical properties which may influence the successful readout of signals. This constitutes the core of the first design steps to elaborate multi-purpose bio-assay platforms. Bio-assays are standardized tests usually implemented to support multiple intended uses such as the detection of pharmaceuticals, antigens, hormones, or toxins in a complex sample. However, there is usually a long way to go for the establishment of a bio-assay setup, which necessitates preliminary steps such as (i) the development of performance-based methods, (ii) the analysis of biological assays and (iii) their validation. This calls for advanced methodologies and readout procedures. Surface-Enhanced InfraRed Absorption (SEIRA) in combination with statistical tools addresses the challenges of accuracy, specificity, precision, and robustness by providing highly resolved vibrational information of complex molecular compounds. Surface-Enhanced InfraRed Absorption (SEIRA) constitutes an ideal platform to isolate vibrational spectroscopic signatures of targeted and active molecules (e.g. antibodies) of the bio-assay, and challenges the Surface-Enhanced Raman Spectroscopy (SERS) readout.1,2 Dissimilarly to SERS providing sensitive access to Raman-active modes in molecules, SEIRA allows for efficient detection of IR-active modes, thus providing complementary speciation information. Enhancement processes (see comparison in the ESI†) of chemisorption to physisorption, for SERS and SEIRA, can modify the similarity of the effective speciation sensitivities of both methods.3–5The present work addresses one of the design steps of an assay platform – the targeted biolabels consisting, in our case, of fluorophore-labeled antibodies chemisorbed onto gold nano-aggregates (Au NA), acting as nano-antennas for biocompatible, low-cost, amplified SEIRA detection6 and efficient fast discrimination for appropriate read-out in tunable and pre-selectable spectral ranges.3
Systematic analyses were performed on the model antibody Goat Anti-Mouse IgG and Goat Anti-Mouse IgG conjugated with fluorophores such as fluorescein isothiocyanate (FITC), rhodamine B isothiocyanate (RITC) and sulphorhodamine B dye (SULFO) fluorophores. These biolabels adsorbed at/nearby NP-functionalised (silanized) surfaces present additional and enhanced spectral signatures due to plasmonic modifications that were compared with those delivered from non-chemisorbed gold NA films simply incubated with the label components.
Extensive areas of dried sample films were analyzed by IR/SEIRA spectroscopy and the resulting complex hyperspectral datasets were submitted to automated statistical analysis, namely, principal component analysis (PCA). The relationship and dependency between chemical functional groups of various antibody–fluorophore conjugation systems were determined. Consequently, this paper presents profound insights into multivariate discrimination capabilities by direct visualization of derived structural clusters among various biolabel configurations.
2. Material and methods
For further details, please confer to the ESI.†2.1 Synthesis of Au NP SEIRA substrates and immobilization of Au NPs on a MirrIR low-e-substrate
Au NPs were synthesized according to a modified protocol of Lee and Meisel,7 yielding an estimated concentration of 1 × 10−9 M (ca. 7 × 1014 NP/L) and characterized by UV/Vis-NIR spectroscopy (see Fig. S-1, ESI†). For the preparation of SEIRA substrates, the immobilization of Au NPs was performed according to the protocol illustrated in ref. 8, using mid-infrared (MIR) low-e-slides.2.2 Synthesis of fluorophore-labeled antibody conjugates
Different fluorescent dyes that are widely used for biomedical applications were studied. The conjugation of fluorophore–antibody was performed by a covalent linkage between the free residual amino groups of the antibody9 and the thiocyanate and sulfonyl groups of the fluorophores,10 and further characterized by MALDI-TOF-MS (Fig. S-2, ESI†).2.3 Atomic force microscopy
Au NP modified low-e-slides were studied using a NeaSNOM (Neaspec GmbH, Germany) in AFM mode for accessing the surface topography of the functionalized substrates and the existence of immobilized Au NPs. Sample areas of 8 μm × 8 μm and 4 μm × 4 μm were scanned with a corresponding pixel number of 400 pixels referring to a resolution of 20 nm (8 μm-scans) and 300 pixels referring to about 13 nm resolution (4 μm-scans). A platinum iridium V coated monolithic silicon AFM tip with a tip radius of curvature with less than 25 nm (NanoWorld AG, Swiss) was used for probing the substrate surface. Image processing (median alignment, pixel column correction) was performed using Gwyddion 2.2.6 (CMI, Czech Republic). The surface topography depicts Au NA and a single NP to be immobilized on the glass slide. The Au NPs showed spherical morphologies and were in the size range of about 40 ± 3 nm.2.4 SR-based FTIR micro-spectroscopy
For FTIR micro-spectroscopy measurements, the SR source (σx = 670 μm, σy = 183 μm, beam current ∼170 mA) was focused through an IR cassegrain objective of 15-fold magnification onto the sample films. MIR-spectra (from 3900 cm−1 to 900 cm−1) were acquired in transflection mode by co-addition of 128 scans at 16 cm−1 resolution. Background scans were collected before each sample measurement from a region free of sample and rationed against the sample spectrum. The use of synchrotron light has many advantages, thanks to its high brilliance, non-invasiveness, coherence and chemical sensitivity compared to conventional IR sources12 and this study illustrates its applicability to the acquisition of large spectral datasets with FPA.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–900 cm−1 spectral range using a MCT (Mercury Cadmium Telluride) detector and an InGaAs photodiode (16 cm−1 spectral resolution, 128 scans). As a reference for immobilized Au NPs, a slide coated with APTMS for each discrete concentration was measured.
000–900 cm−1 spectral range using a MCT (Mercury Cadmium Telluride) detector and an InGaAs photodiode (16 cm−1 spectral resolution, 128 scans). As a reference for immobilized Au NPs, a slide coated with APTMS for each discrete concentration was measured.
Principal component analysis (PCA): the second derivatives of the full dataset of 4400 baseline-corrected absorbance spectra were calculated and consecutively submitted to the Savitzky–Golay algorithm (5 smoothing points) and vector-normalization before PCA using MatlabR2011a (The Mathworks Inc., Natick, USA) and PLS toolbox (Eigenvector Research, Inc., USA). The principle components (PCs) were calculated from PC1 up to PC4 and illustrated as scatter plots; such a high-order approach has been needed for discriminating certain label components.
3. Results and discussion
3.1 Au NA-immobilization on MirrIR low-e-MIR substrates for SEIRA studies
Self-assembled Au NP multilayers have successfully been employed as versatile platforms for molecular sensing approaches.14Fig. 1 displays the spectral features of Au nanoparticulate multilayers that were generated during the drop-coating procedure and analysed in the 22000–900 cm−1 region. Due to aggregation of the Au NPs, a red-shift of the plasmon band is induced, and a broader plasmon band appears, extending from the NIR to the MIR region (compare with Fig. S-1, ESI†). The position of the extended plasmon band shows that pre-resonant excitation by surface plasmons in the MIR region can be exploited for the SEIRA studies. This is in accordance with previous observations,15 where a continuum-like resonance for Au nanoshell array samples was observed in the MIR spectral range.
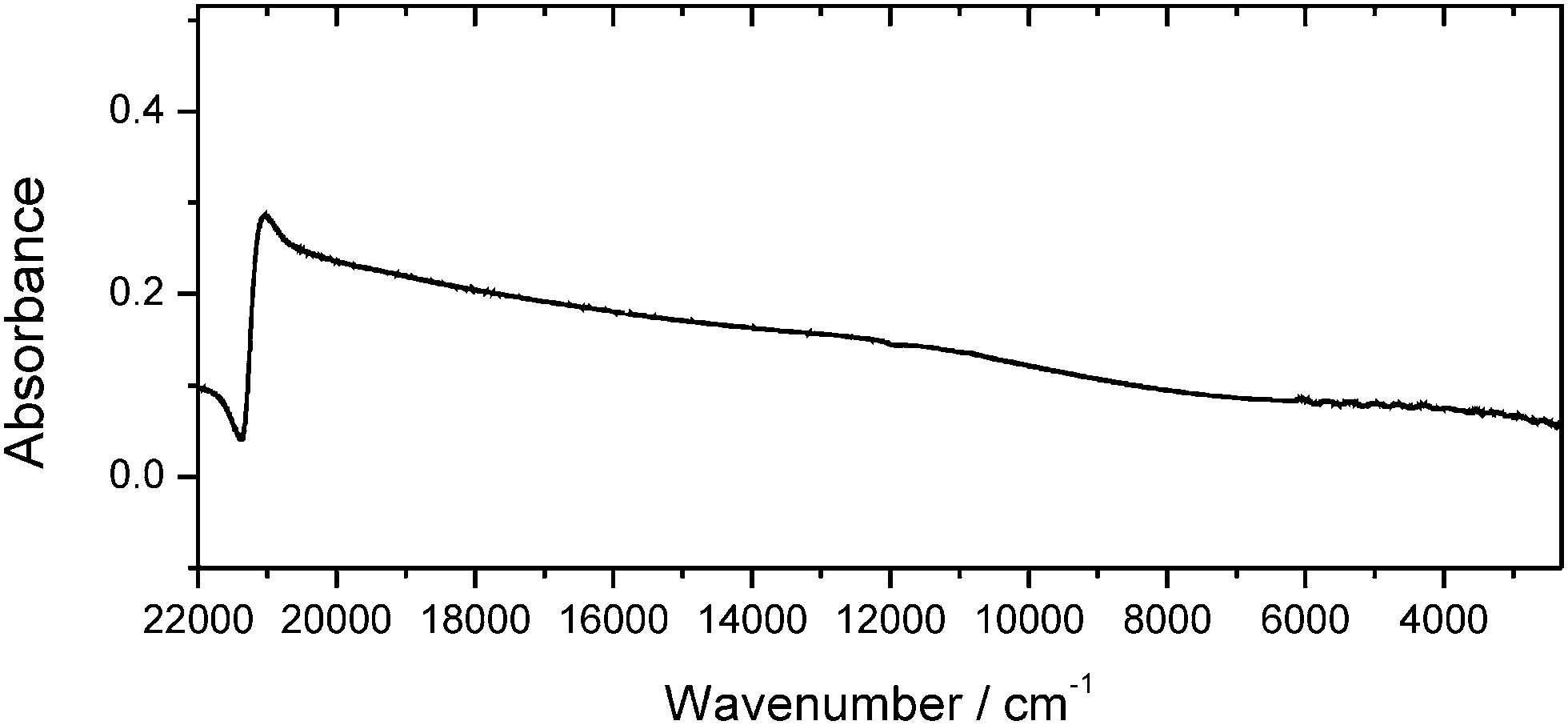 | ||
| Fig. 1 Absorbance spectrum of Au NA showing an extended plasmon band in the mid-infrared region used for the SEIRA experiments. Analysis was performed in the spectral range from 22000 cm−1 to 900 cm−1. | ||
3.2 MIR signatures delivered by fluorophore–antibody NP biolabels
Similar to clinical diagnostics, dried films of serum proteins or antibodies are convenient for FTIR investigations. The study of the secondary structure composition, structural dynamics and/or the study of conformational changes (effects of ligand binding, temperature, and pH) of the antibodies in the liquid phase is not the aim of this work presented here. In the following we discuss the signatures from SEIRA NP-labeled biolabels (such as antibodies and conjugates) that can be used to visualize and ultimately quantify the bound analyte which is not that obvious or detected in the conventional FTIR configuration. Of notice that it is inherent to the SEIRA technique compared to conventional FTIR is the influence of the metal surface that may lead to some spectral shapes, shifts and changes in intensities.16![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and –CC str. vibrations. This can be explained by different binding orientations which are restricted due to the fluorophore–NP incubation procedure during which the chemisorbed FITC molecules tend to orient. In addition, the origin of SEIRA being observed for Au NP-FITC is due to the formation of large colloidal Au NA, presumably induced by the drop-coating process. The NA chemisorbed FITC molecules orient their dipole moments to be in line with the local electric field of the metal NA, resulting in much larger absorption than when they are not in line with the oscillating electric field.22,23 Such larger absorption of FITC molecules can be exploited for building complex conjugates for sensitive multiplexed assay approaches.
N and –CC str. vibrations. This can be explained by different binding orientations which are restricted due to the fluorophore–NP incubation procedure during which the chemisorbed FITC molecules tend to orient. In addition, the origin of SEIRA being observed for Au NP-FITC is due to the formation of large colloidal Au NA, presumably induced by the drop-coating process. The NA chemisorbed FITC molecules orient their dipole moments to be in line with the local electric field of the metal NA, resulting in much larger absorption than when they are not in line with the oscillating electric field.22,23 Such larger absorption of FITC molecules can be exploited for building complex conjugates for sensitive multiplexed assay approaches.
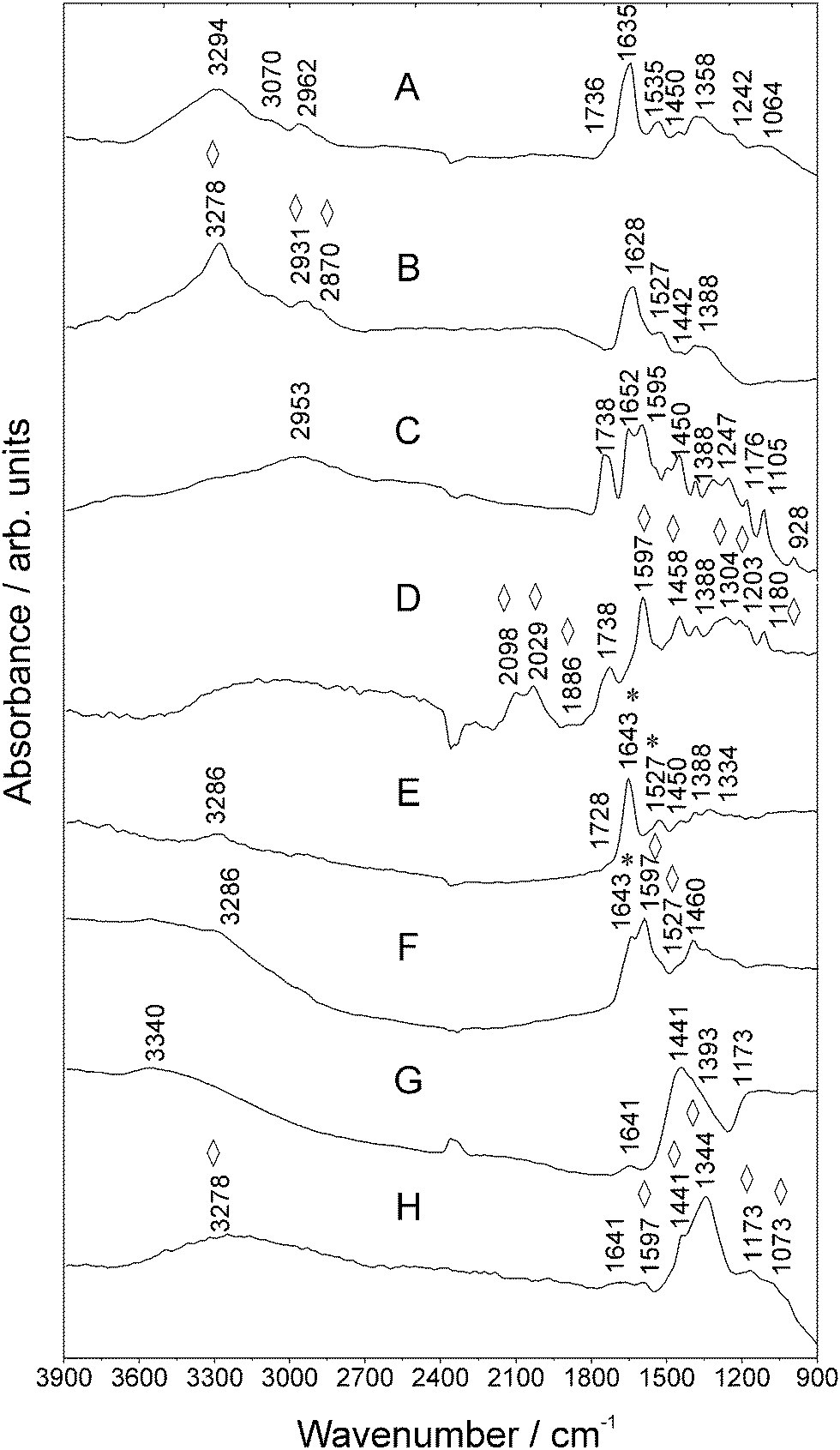 | ||
| Fig. 2 MIR spectra of (a) Goat Anti-Mouse IgG, (b) Au NA incubated with Goat Anti-Mouse IgG, (c) FITC, (d) Au NPs and FITC, (e) Goat Anti-Mouse IgG–FITC, (f) Au NPs and Goat Anti-Mouse IgG–FITC and the antigen (g) Mouse Anti-Goat IgG and (h) Au NPs and Mouse Anti-Goat IgG measured using SR. Vibrations of FITC are marked with asterisks *. Enhanced vibrations are marked with rhombohedra ⋄. For further details, refer to Table S-1 (ESI†). | ||
An enhancement of the modes in the spectral region between 1304 cm−1 and 1180 cm−1, as well as at ca. 1458 cm−1 can be observed as well (compare Fig. 2C and D). The Goat Anti-Mouse IgG–FITC signature in Fig. 2E comprises lines which can be assigned to typical bands of FITC (see asterisks *) and bands between 3720 cm−1 and 1103 cm−1 referring to antibody specific signatures. Fig. 2F shows the SEIRA spectrum of the antibody–FITC conjugate recorded in the presence of Au NA. This signature comprises lines which can be assigned to FITC typical bands (Table S-1, ESI†). For example, in the vicinity of the mode at ca. 1643 cm−1 (str. vibrations of the xanthene skeleton) of the unenhanced antibody–FITC conjugate now a broad shoulder being located at 1597 cm−1 appears in the SEIRA spectrum of the antibody–FITC conjugate. This vibration originates from the aromatic –C–C str. vibrations of the xanthene ring skeleton of FITC molecules which was already observed in the SEIRA spectrum of FITC (compare Fig. 2D). This shows that SEIRA is able to detect with high sensitivity fluorophore molecules in their conjugated states along with their molecular orientation.
Considering the intended use of labeled antibodies in immunoassays, the signatures of the targeting counterpart Mouse Anti-Goat IgG were studied. Fig. 2G illustrates antigen specific lines being typical for antibody protein vibrations. The SEIRA signature of the Mouse IgG antibody is displayed in Fig. 2H that shows enhanced modes in the spectral region between 1800 cm−1 and 900 cm−1. Another possible contribution which may explain the observed spectral differences is due to chemical enhancement. Molecules strongly chemisorbed onto a (noble) metal surface show a larger enhancement than do physisorbed molecules, suggesting a chemical effect between the molecule and the surface due to strong interactions (Au NA–antibody complexation). This mechanism would describe the case that we discuss and correspond to our antibodies that we detect in the presence of Au NA. It is known that the absorption coefficients of chemisorbed molecules are larger than those of condensed overlayers and therefore interactions between adsorbed molecules may also affect the intensity and thus impact and explain the enhancement of some modes. Charge oscillations between molecular orbitals and the metal surface may enlarge the absorption coefficients of adsorbates.2,24
O)H str. vibration.26 The SEIRA spectrum of RITC recorded on Au NA (Fig. 3B) exhibits an enhanced mode at 1589 cm−1. It is remarkable that the relative intensity of the str. mode of the xanthene ring moiety at 1342 cm−1 experiences an enhancement by a factor of five, which proves again the selectivity of revealing one of the most specific molecular groups of the fluorescent dye.
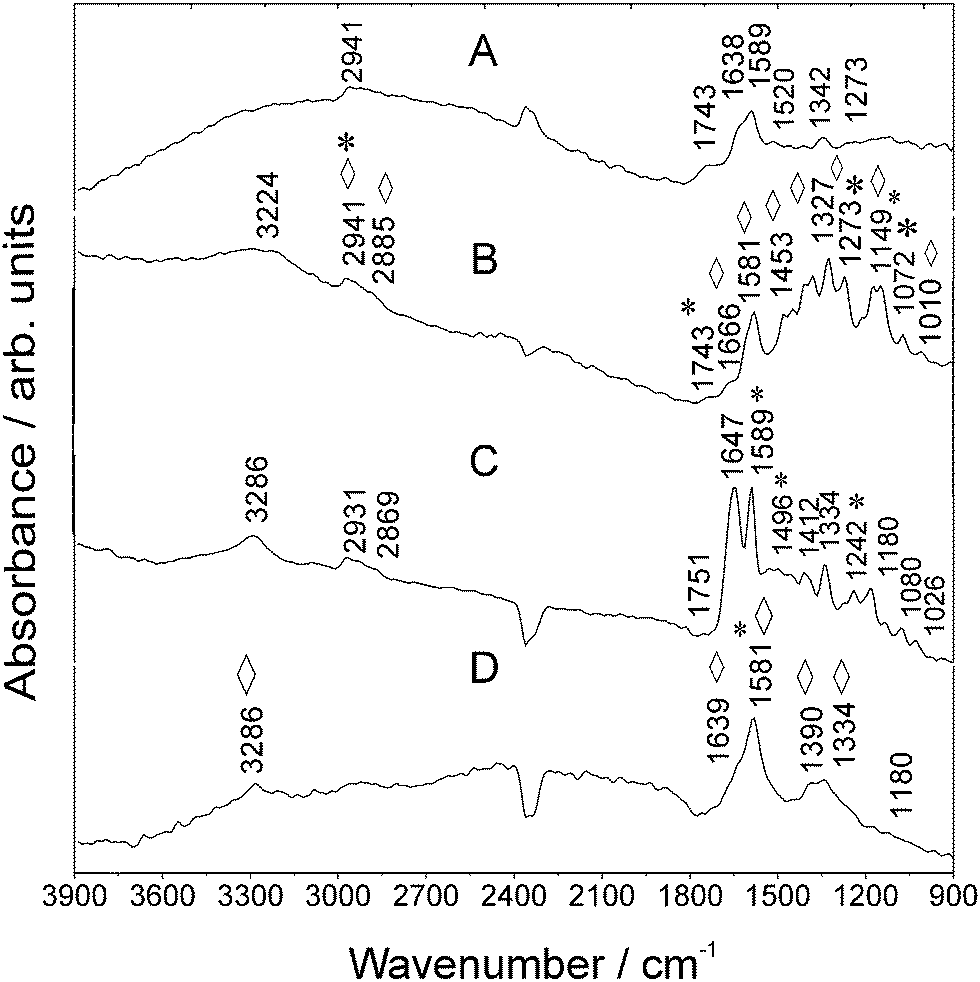 | ||
| Fig. 3 MIR spectra of (a) RITC, (b) Au NPs and RITC, (c) Goat Anti-Mouse IgG–RITC, and (d) Au NPs and Goat Anti-Mouse IgG–RITC measured using SR. Vibrations of RITC are marked with asterisks *. Enhanced vibrations are marked with rhombohedra ⋄. For further details, refer to Table S-2 (ESI†). | ||
In the case of RITC measured on Au NA, further new IR vibrations are observed, for instance, at 1666 cm−1. Further, the mode at 1520 cm−1 disappears which is caused by a change in the molecular orientation. The spectrum of the antibody–RITC conjugate (Fig. 3C) exhibits RITC-typical modes which are also observed in the bare fluorophore spectrum (Fig. 3A). For instance, the mode at 1589 cm−1 originates from aromatic –C–C str. vibrations of RITC molecules.26 In the presence of Au NA, the conjugate signature (Fig. 3B) delivers one broad band at 1589 cm−1 and the mode at 1647 cm−1 is observed as a shoulder being located at ca. 1639 cm−1 (Table S-2, ESI†).
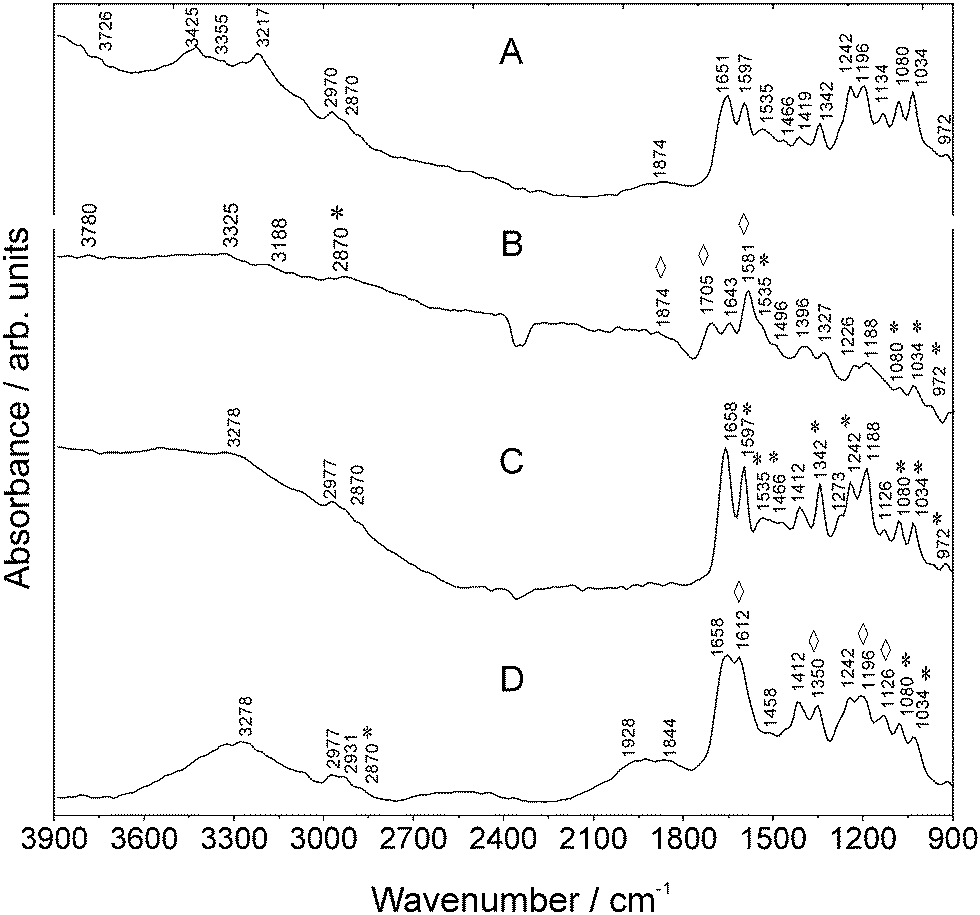 | ||
| Fig. 4 MIR spectra of (a) SULFO, (b) Au NPs and SULFO, (c) Goat Anti-Mouse IgG–SULFO, and (d) Au NPs and Goat Anti-Mouse IgG–SULFO measured using SR. Vibrations of SULFO are marked with asterisks *. Enhanced vibrations are marked with rhombohedra ⋄. For further details, refer to Table S-3 (ESI†). | ||
The univariate study of these different antibody–fluorophore conjugate signatures shows that the electric field enhancing capabilities differ between Au NPs and the type of antibody–fluorophore conjugate which justifies the systematic approach on the spectral properties of such fluorophore–antibody NP complexes. Additionally, this shows that for the bioassay design, SEIRA induced by NPs provides different vibrational information from fluorophore–anibody NP labels that can be used for building up spectral databases.
Summarizing the results, FTIR spectroscopy combined with SEIRA is an important analytical tool for studying the biochemical composition of proteins such as antibodies. Bands of high interest for biological purposes and labeling procedures can be found in the Amide I/II and ester regions. The Amide I region is traditionally exploited for probing the secondary structures of proteins, as an indicator band for monitoring spectral changes due to alterations in the molecular structure, usually due to external perturbations. Our studies show that complex biolabels that are read out by SEIRA deliver distinct spectra that clearly differ from their unenhanced counterparts, the conventional FTIR spectra (please refer to Fig. S-3, and Tables S-1–S-3, ESI†).
3.3 Multivariate discrimination of the signatures of label components
PCA as a powerful chemometric method for finding patterns in complex FTIR datasets enables fast identification and separation of labeled antibodies which are designed for multianalyte detection.30,31Fig. 5 shows score plots (PC1–PC4) that entail spectral differences. All PCA procedures displayed in Fig. 5A–C (and Fig. 6b) were performed by repetitive experiments (data not shown here, 95% reproducibility).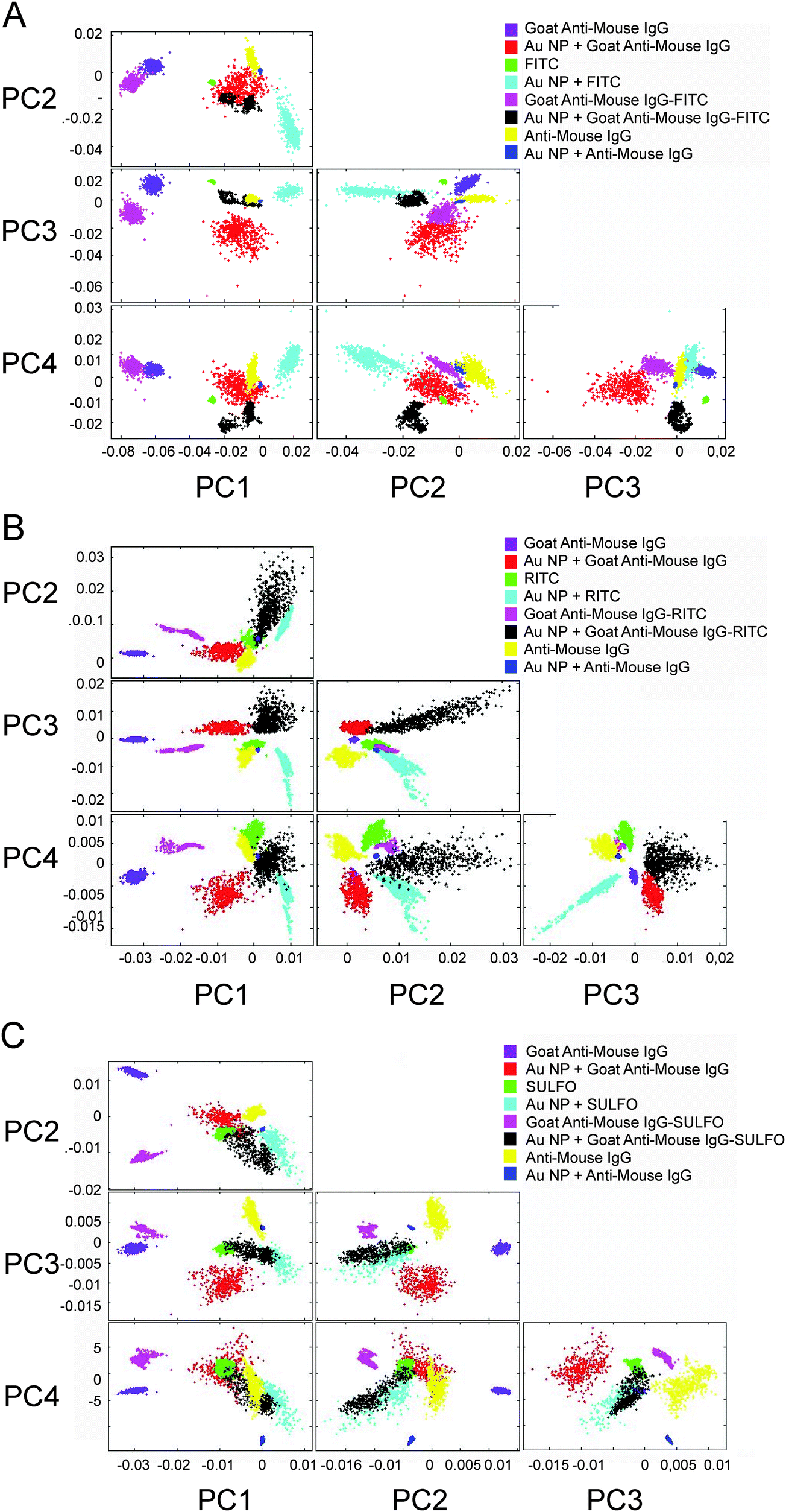 | ||
| Fig. 5 PCA illustrated from PC1 to PC4. PCA was performed with vector-normalized second derivative spectra over the spectral range from 3900 cm−1 to 900 cm−1. PCA clusters are color-coded as follows: Goat Anti-Mouse IgG (violet), Au NPs and Goat Anti-Mouse IgG (red), fluorophore (green), Au NPs and fluorophore (cyan), Goat Anti-Mouse IgG–fluorophore (magenta), Au NP and Goat Anti-Mouse IgG–fluorophore (black), Mouse Anti-Goat IgG and NPs (yellow) and Mouse Anti-Goat IgG with Au NPs (blue). | ||
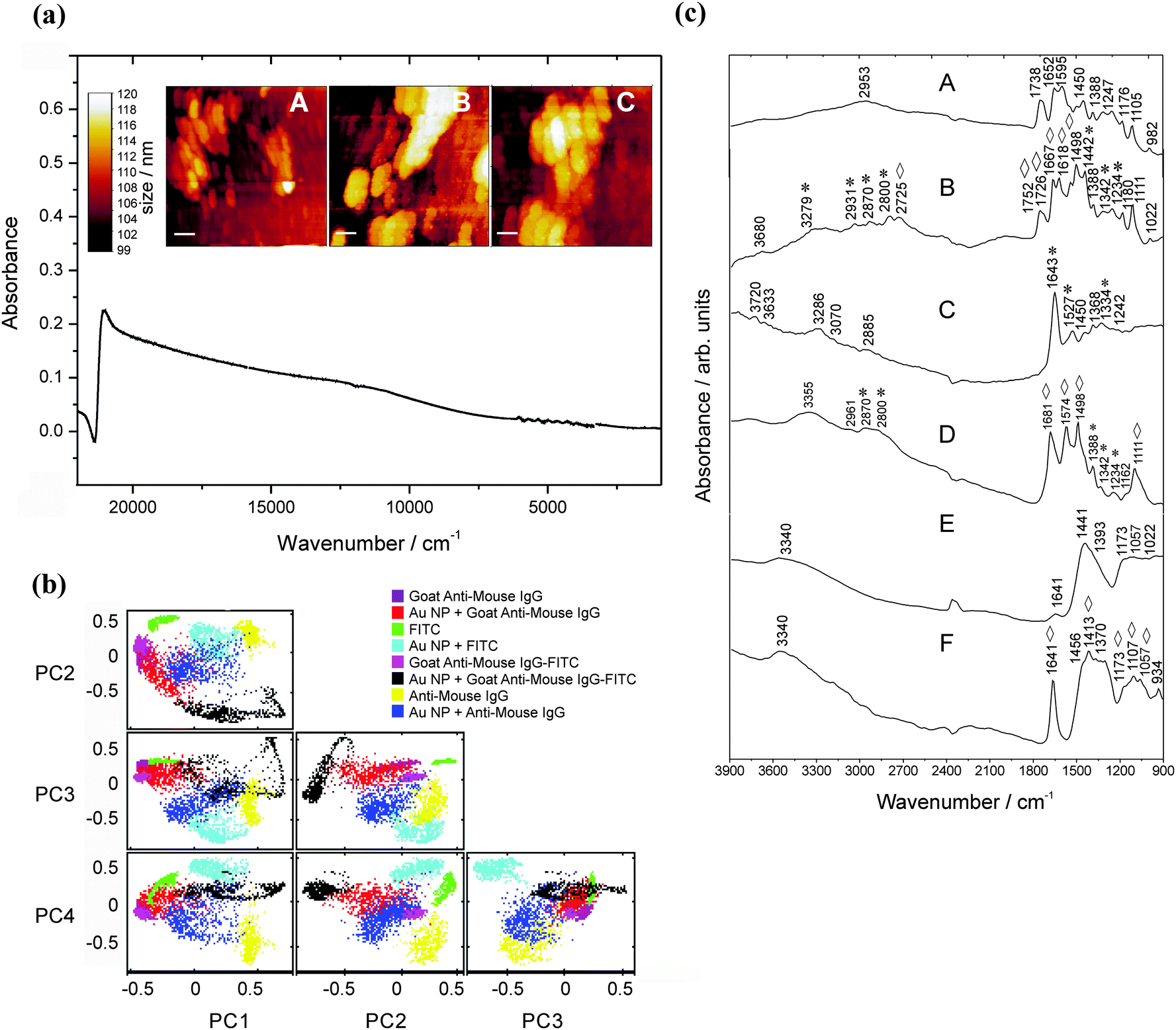 | ||
| Fig. 6 (a) Absorbance spectrum of Au NPs immobilized on an APTMS-functionalized substrate (CAPTMS ∼ 1%, incubation time: 2 h) and the corresponding surface topography (scale bars for (a): 0.1 μm, (b) and (c): 0.05 μm). Au NA are visible as spherical shaped clusters. (b) PCA analysis (3900–900 cm−1). (c) MIR signatures of (a) FITC, (b) FITC on the APTMS-substrate, (c) Goat Anti-Mouse IgG–FITC and (d) Goat Anti-Mouse IgG–FITC on the APTMS-substrate and the antigen (e) Mouse Anti-Goat IgG and (f) Mouse Anti-Goat IgG on the NP-modified APTMS substrate measured using SR. Vibrations of FITC are marked with asterisks *. Enhanced vibrations are marked with rhombohedra ⋄. | ||
The PCA displays datasets of the Goat Anti-Mouse IgG that are well separated from the antigen data in PC1 against PC2. The results of PCA confirm that on average 85% of the variance of the spectral datasets, i.e. the discrimination capability up to the third order, could be explained for the fluorophores. Additionally, we decided to put on display all components up to the fourth order to illustrate the options for discriminating different label and/or label components. The scores (PC1 against PC2) of Goat Anti-Mouse IgG are positive and lie in the second quadrant of the variance-weighted room. In PC1 against PC3, the Goat Anti-Mouse IgG antibody data are well separated from the unenhanced spectra. The spectral differences observed in the SEIRA spectrum of FITC (see Fig. 2D) are also reflected by PC1 against PC2. Fig. 5A illustrates that in PC1 against PC2 FITC is separated from the FITC-labeled antibody. With respect to the design of multiplexed immunoassay formats, this shows that the enhancement capabilities of different Au NA labels provide MIR signatures with different spectral patterns. PCA for studying the multiplexing capability of the other fluorophore conjugates containing RITC molecules was also performed successfully. Similar to the PC scores illustrated in Fig. 5A, a discrimination of the RITC MIR signatures measured on Au NA in PC1 against PC2 was feasible. A variance of about 80% is captured by the components PC1–PC3. The Goat Anti-Mouse IgG–RITC conjugate cluster has quite similar distance towards its unlabeled counterpart as the FITC–antibody conjugate, which is reflected by the good separation in PC1 against PC2.
Compared to all other fluorophore–antibody conjugates discussed so far, the SEIRA signature of the FITC-labeled Goat Anti-Mouse IgG is most similar to the unlabeled antibody sample; for SULFO the distance in PC1 against PC2 is the highest which shows the increase of discrimination capability of the conjugated fluorophores.
The multivariate analysis of spectral features is an important scope for the design and development of bioassay platforms, as functionalization with biolabel components may require several preparation steps before the establishment of a functionally completed assay setup.
3.4 MIR signatures of the antibody–FITC conjugate measured on the Au NP modified low-e-substrate
Fig. 6a shows the extinction spectrum and surface topography of Au NPs being immobilized on an APTMS-functionalized substrate (1% APTMS, time of incubation: 24 h, see Fig. 6a, A–C). A second extended plasmon band is observed in the MIR region and proves the existence of Au NA32 as for the unmodified glass substrate. This is important in the case of the enhancement capabilities of immobilized NPs as large NA reaching microscale size dimensions of bulk materials might drastically lose their enhancement properties.The PCA results confirm that on average 85% of the variance of the spectral datasets up to the third order could be explained. PCA of the Mouse Anti-Goat IgG signatures shows a better separation from the enhanced ones (PC1 against PC2 and PC3 in Fig. 6b). The MIR signatures are illustrated in Fig. 6c. Compared to the FITC signature, spectral differences in the enhanced counterpart can be observed, i.e., the band at 1738 cm−1 (compare with Fig. 3) is split into lines at 1726 cm−1 and 1752 cm−1. The dependence on the substrate configuration and modification is very striking when comparing the antibody–FITC conjugate being adsorbed onto the Au NA on the APTMS-modified slide – the signature exhibits FITC typical bands along with antibody modes (Fig. 6c, C). Compared to the conjugate signature in Fig. 2 where a mode at ca. 1643 cm−1 can be observed, now a new band at 1681 cm−1 (–CO str.) appears after APTMS modification. This shows that the SEIRA signature of FITC and its antibody conjugate differs from the signatures measured with the APMS-modified substrate and the assay configuration itself. It should be noted that the vibrational modes in the 1600 cm−1 to 1300 cm−1 range can be assigned to both aminopropyl and free amino groups as well. However, no peak at 1491 cm−1 associated with NH3+ in the SiO−–H–NH3+ group of APTMS residues33 is noticeable which shows that this substrate enables the overlap-free detection of amino-group modes delivered by the targeting units themselves.
A tremendous impact of this substrate with respect to its enhancement capability is shown for the antigen Mouse Anti-Goat (Fig. 6c, F) in the region from 1460 cm−1 to 1300 cm−1. In particular, the mode at 1641 cm−1 is extremely intensified by a factor of about 8. This is in accordance to the calculated dimension of the average enhancement factor for the MIR spectral region as described in ref. 34.
4. Conclusions
We demonstrate the efficiency of the SEIRA technique to probe the molecular characteristics of different antibody–fluorophore NP conjugates considered in this study. Our measurements show that the spectral signatures of the native antibody Goat Anti-Mouse IgG and the conjugated fluorophores are preserved in the proximity of Au NPs but enhanced by the latter. This implies that the elaboration of specific assay platform spectral databases can be envisaged with SEIRA. Coupled with PCA analysis, this method allows clear discrimination of the functionalized systems, which ascertain their potential for multiplexed bioassay applications and biosensor development of high efficiency with customized responses. Our work highlights the prospective use of NP-assays for biodiagnostics35,36 and as catalysts for selectively enhancing bio-physiological responses of cells or bacteria. Consequently, SEIRA applications37 can be expected to show even higher performance characteristics than the conventional/established methods such as SERS.38As such, a logical extension of our work is to confront the potentialities of SEIRA to discriminate biolabels' spectral features in a real biological environment, which is a subject of another communication.
Altogether, this paper evidences the availability of a novel means in the complex bio-diagnostic micro-assay toolbox to identify the suitability of biolabel systems along with their discrimination potential and their multiplexing capability. Enhancing functionality performances is the key point to obtain higher and targeted screening performance, the ultimate goal of complex bio-diagnostic assay platforms.39
Acknowledgements
We thank Ralf Bienert (BAM Federal Institute for Materials Research and Testing) for use of the DLS apparatus. The machine-physicists team of the Helmholtz-Zentrum Berlin (HZB) is gratefully acknowledged for tuning the MLS according to our experimental needs. We thank Arne Hoehl of the MLS team of the PTB for helpful discussions and use of the beamline instrumentation. Sincere thanks are given to Sören Selve (ZELMI, TU Berlin) for the preparation of the TEM images.References
- G. C. Schatz, THEOCHEM, 2001, 573, 73–80 CrossRef CAS.
- M. Osawa and K.-i. Ataka, Surf. Sci., 1992, 262, L118–L122 CrossRef CAS.
- S. Lal, S. Link and N. J. Halas, Nat. Photonics, 2007, 1, 641–648 CrossRef CAS.
- M. Osawa and M. Ikeda, J. Phys. Chem., 1991, 95, 9914–9919 CrossRef CAS.
- A. Hartstein, J. R. Kirtley and J. C. Tsang, Phys. Rev. Lett., 1980, 45, 201–204 CrossRef CAS.
- H. D. Wanzenböck, B. Mizaikoff, N. Weissenbacher and R. Kellner, Fresenius' J. Anal. Chem., 1998, 362, 15–20 CrossRef.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- C. D. Geddes, H. Cao, I. Gryczynski, Z. Gryczynski, J. Fang and J. R. Lakowicz, J. Phys. Chem. A, 2003, 107, 3443–3449 CrossRef CAS.
- G. T. Hermanson, in Bioconjugate Techniques, ed. A. Press, Elsevier Inc., London, 2008, vol. 2, pp. 396–430 Search PubMed.
- H. Rinderknecht, Nature, 1962, 193, 167–168 CrossRef CAS.
- A. Gottwald, R. Klein, R. Müller, M. Richter, R. Thornagel and G. Ulm, Metrologia, 2012, 49, S146–S151 CrossRef.
- P. Dumas, G. D. Sockalingum and J. Sulé-Suso, Trends Biotechnol., 2007, 25, 40–44 CrossRef CAS PubMed.
- M. Bally, M. Halter, J. Voros and H. M. Grandin, Surf. Interface Anal., 2006, 38, 1442–1458 CrossRef CAS.
- F. Toderas, S. Boca, M. Baia, L. Baia, D. Maniu, S. Astilean and S. Simon, J. Optoelectron. Adv. Mater., 2007, 9, 625–628 CAS.
- F. Le, D. W. Brandl, Y. A. Urzhumov, H. Wang, J. Kundu, N. J. Halas, J. Aizpurua and P. Nordlander, ACS Nano, 2008, 2, 707–718 CrossRef CAS PubMed.
- G. Dovbeshko, O. Fesenko and A. Nazarova, J. Phys. Stud., 2006, 10, 127–134 CAS.
- F. S. Parker and D. M. Kirschenbaum, Nature, 1960, 187, 386–388 CrossRef CAS.
- A. W. Herlinger and T. Veach Long, J. Am. Chem. Soc., 1970, 92, 6481–6486 CrossRef CAS.
- A. A. Kamnev, L. A. Dykman, P. A. Tarantilis and M. G. Polissiou, Biosci. Rep., 2002, 22, 541–547 CrossRef CAS.
- F. Tian, R. Middaugh, T. Offerdahl, E. Munson, S. Sane and H. Rytting, Int. J. Pharm., 2007, 335, 20–31 CrossRef CAS PubMed.
- L. L. Wang, A. Roitberg, C. Meuse and A. K. Gaigalas, Spectrochim. Acta, Part A, 2001, 57, 1781–1791 CrossRef CAS.
- D. Ross and R. Aroca, J. Chem. Phys., 2002, 117, 8095–8103 CrossRef CAS PubMed.
- M. Osawa, K. Ataka, K. Yoshii and Y. Nishikawa, Appl. Spectrosc., 1993, 47, 1497–1502 CrossRef CAS.
- M. Osawa and S. Kawata, Surface-Enhanced Infrared Absorption, Springer, Berlin/Heidelberg, 2001, vol. 81, pp. 163–187 Search PubMed.
- P. Hildebrandt and M. Stockburger, J. Phys. Chem., 1984, 88, 5935–5944 CrossRef CAS.
- M. Majoube and M. Henry, Spectrochim. Acta, Part A, 1991, 47, 1459–1466 CrossRef.
- Y.-F. Fang, Y.-P. Huang, D.-F. Liu, Y. Huang, W. Guo and D. Johnson, J. Environ. Sci., 2007, 19, 97–102 CrossRef CAS.
- R. Markuszewski and H. Diehl, Talanta, 1980, 27, 937–946 CrossRef CAS.
- T. Ohishi, J. Non-Cryst. Solids, 2003, 332, 80–86 CrossRef CAS PubMed.
- R. G. Brereton, Chemometrics: data analysis for the laboratory and chemical plant, John Wiley & Sons Ltd, Chichester, UK, 2006, pp. 183–269 Search PubMed.
- C. A. Sellick, R. Hansen, R. M. Jarvis, A. R. Maqsood, G. M. Stephens, A. J. Dickson and R. Goodacre, Biotechnol. Bioeng., 2010, 106, 432–442 CAS.
- A. M. Schwartzberg, C. D. Grant, A. Wolcott, C. E. Talley, T. R. Huser, R. Bogomolni and J. Z. Zhang, J. Phys. Chem. B, 2004, 108, 19191–19197 CrossRef CAS.
- Y. Zhang, N. Kohler and M. Zhang, Biomaterials, 2002, 23, 1553–1561 CrossRef CAS.
- R. Aroca, Surface-enhanced Vibrational Spectroscopy, John Wiley and Sons Ltd, West Sussex, 2007, p. 198 Search PubMed.
- Y. Ding, X. Chu, X. Hong, P. Zou and Y. Liu, Appl. Phys. Lett., 2012, 100, 013701 CrossRef PubMed.
- A. Quershi, Y. Gurbuza, W. P. Kang and J. L. Davidson, Biosens. Bioelectron., 2009, 25, 877–882 CrossRef CAS PubMed.
- R. Adato and H. Altug, Nat. Commun., 2013, 4, 2154 Search PubMed.
- M. Kahraman, A. I. Zamaleeva, R. F. Fakhrullin and M. Culha, Anal. Bioanal. Chem., 1995, 395, 2559–2567 CrossRef PubMed.
- P. Qiu, E. F. Simonds, S. C. Bendall, K. D. Gibbs Jr, R. V. Bruggner, M. D. Linderman, K. Sachs, G. P. Nolan and S. Plevritis, Nat. Biotechnol., 2011, 29, 886–891 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp05944a |
| This journal is © the Owner Societies 2015 |