
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent response to fluorine-atom reaction dynamics in liquid acetonitrile
G. T.
Dunning
a,
D.
Murdock
a,
G. M.
Greetham
b,
I. P.
Clark
b and
A. J.
Orr-Ewing
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: a.orr-ewing@bristol.ac.uk; Tel: +44 (0)1179287672
bCentral Laser Facility, Research Complex at Harwell, Science and Technology Facilities Council, Rutherford Appleton Laboratory, Harwell Oxford, Didcot, Oxfordshire, OX11 0QX, UK
First published on 5th March 2015
Abstract
Time-resolved infra-red (IR) absorption spectroscopy is used to follow the production of HF from the reaction of fluorine atoms in liquid acetonitrile (CH3CN). Photolysis of dissolved XeF2 using ∼50 fs duration, 267 nm laser pulses generates F atoms and XeF on prompt (sub-ps) timescales, as verified by broadband transient electronic absorption spectroscopy. The fundamental vibrational band of HF in solution spans more than 400 cm−1 around the band centre at 3300 cm−1, and analysis of portions of the time-resolved spectra reveals time constants for the rise in HF absorption that become longer to lower wavenumber. The time constants for growth of 40 cm−1 wide portions of the IR spectra centred at 3420, 3320 and 3240 cm−1 are, respectively, 3.04 ± 0.26, 5.48 ± 0.24 and 7.47 ± 0.74 ps (1 SD uncertainties). The shift to lower wavenumber with time that causes these changes to the time constants is attributed to evolution of the micro-solvation environment of HF following the chemical reaction. The initial growth of the high-wavenumber portion of the band may contain a contribution from relaxation of initially vibrationally excited HF, for which a time constant of 2.4 ± 0.2 ps is deduced from IR pump and probe spectroscopy of a dilute HF solution in acetonitrile.
Introduction
Gas-phase reactions of fluorine atoms with molecular hydrogen and with various organic molecules are exothermic by approximately 130–150 kJ mol−1 and favour production of vibrationally excited HF.1–4 These H-atom transfer reactions exhibit a wealth of fascinating dynamics, including tunnelling through low energy barriers associated with the transition state,5,6 Feshbach resonances that enhance reaction cross sections at specific collision energies,7,8 and non-adiabatic transitions between the ground and low lying potential energy surfaces.9–11 Many features of the gas-phase dynamics persist for scattering of F-atoms from liquid hydrocarbon surfaces.12 Our experimental and computational investigations explore how the mechanisms of reactions such as these are modified in solution in organic solvents. We recently reported first studies of the dynamics of F-atom reactions in the liquid phase and showed that the DF from F + CD3CN and F + CD2Cl2 reactions is highly vibrationally excited at its point of formation.13 We observed vibrational cooling of the nascent DF by coupling to the solvent bath and spectroscopic signatures of the restructuring of the solvent environment around the newly formed reaction products. Our simulations illustrated that the first hydrogen bonding interactions of DF with the solvent occur within a few hundred fs, prior to which the DF undergoes hindered rotational diffusion.13,14 DF vibrational cooling is an order of magnitude slower, with a time constant of 3–4 ps in these solvents, and solvent reorganization persists over more than 10 ps.This article reports an extension of our earlier studies to reaction of F atoms in liquid CH3CN. We generate F atoms photolytically from XeF2 dissolved in acetonitrile,15 and observe the build-up of HF reaction products. Time-resolved electronic absorption spectroscopy (TEAS) in the near ultraviolet (UV) region measures the reactive removal of F atoms, while time-resolved vibrational absorption spectroscopy (TVAS) in the infra-red monitors both the growth of HF products and changes to their microscopic solvation environment. We also report a time constant for vibrational relaxation of HF(v = 1) using IR pump-and-probe experiments that examine the coupling of the vibrational motions of HF solute to the solvent bath. The outcomes provide further evidence for solvent restructuring around the products of a chemical reaction on picosecond timescales.
Experimental
TEAS, TVAS and IR pump-and-probe experiments were carried out using the ULTRA laser system, which is part of the Central Laser Facility at the Rutherford Appleton Laboratory. Comprehensive details of the experimental procedures for time-resolved IR and UV/vis spectroscopy have been presented previously,16,17 and only an overview is included here.TEAS spectra were obtained following 267 nm one-photon photolysis of 0.52 M solutions of XeF2 (99.99%, Sigma Aldrich) in dry CH3CN using a white-light continuum probe dispersed onto a 512-pixel array detector. The photolysis pulses were 50 fs in duration and pulse energies below 1 μJ were used for all measurements presented. IR-pump and IR-probe measurements were conducted on dilute solutions of HF in CH3CN prepared by UV irradiation of XeF2–CH3CN solutions. The presence of HF was confirmed, and the shape of the HF fundamental band determined, by steady-state FTIR spectroscopy of the samples. The IR pump pulse was of ∼2 ps duration, with a bandwidth of ∼12 cm−1 and was tuned either to the centre of the HF fundamental band, or 60 cm−1 to the high wavenumber side of the band centre. The IR probe pulse was <100 fs in duration and spanned the 3000–3700 cm−1 spectral region. After transmission through the sample, the probe pulses were dispersed onto a 128-pixel mercury cadmium telluride array detector. TVAS experiments combined the 267 nm photolysis of XeF2 with the broadband IR probing and detection to monitor the HF products of the F + CH3CN reaction.
In all experiments the excitation and probe pulses were linearly polarized at magic angle to one another to eliminate any effects of rotational anisotropy. Spectra were obtained as a difference between absorptions with and without the pump (UV or IR) laser incident on the sample, so that depletion of ground state molecules appears as negative going bleach signals, and positive going signals are signatures of transient intermediates or stable products.
The TEAS and TVAS experiments used 5 ml samples of the XeF2–CH3CN solutions, circulated through a Harrick cell with CaF2 windows by a peristaltic pump. The solutions passed through a 0.2 mm gap between the cell windows, which were sealed to the cell body by Kalrez o-rings. The cell was continuously rastered in a plane perpendicular to the propagation direction of the laser pulses. Extensive averaging of spectra ensured good signal-to-noise levels even for the very weak (<10−4 OD) IR absorptions typical of the TVAS experiments.
All experiments were conducted in CH3CN that had been carefully dried over a molecular sieve prior to use. Glassware and Harrick cell components were stored in a drying oven when not in use. Water contamination of samples was monitored periodically by FTIR spectroscopy, and only the outcomes of those experiments with minimal water contamination are reported here.
Results and discussion
1. F-atom production by XeF2 photolysis
The XeF product of one-photon, 267 nm photolysis of XeF2 forms in its electronic ground state and has a characteristic absorption band that is centred at 345 nm in solution in acetonitrile.18 Observation of this band allows us to follow the growth and decay of the F-atom co-product of XeF2 dissociation. The near-UV absorption band is assigned to the XeF (B–X) transition, which is perturbed by the presence of an F-atom co-product at short times after XeF2 photolysis.15Fig. 1 shows representative TEAS spectra that can be decomposed into the XeF (B–X) spectrum and the perturbed B–X band attributed to XeF confined in close proximity to an F atom by a solvent cage. We refer to this caged XeF and F-atom pair as an FXe⋯F complex, but do not imply any chemical interaction beyond perturbation of the electronic potentials of the XeF by the geminate F atom. Examination of the long-time (>100 ps) spectra, assigned solely to XeF because of reactive loss of F atoms, allows the two contributions to the time-resolved spectra to be distinguished. Fig. 1 also shows the time-dependence of the integrated band intensities of XeF and the FXe⋯F complexes, together with a fit to a kinetic model that incorporates formation of FXe⋯F and removal by reaction or geminate recombination.15 XeF is known to be stable in acetonitrile for many microseconds.18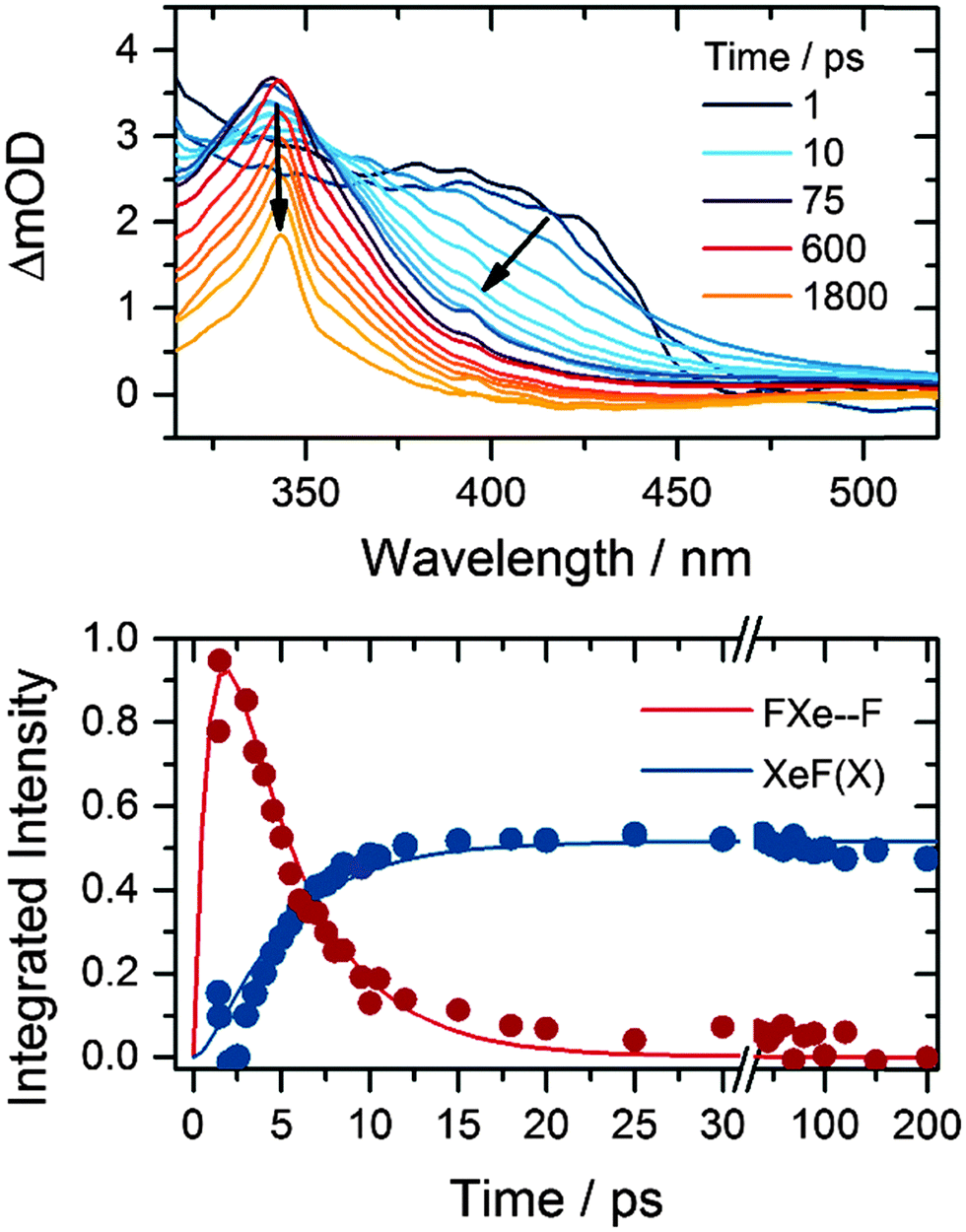 | ||
| Fig. 1 Top: time-resolved electronic absorption spectra of XeF2–CH3CN solutions following 267 nm photoexcitation. The inset key shows the colours corresponding to selected time delays. Bottom: time dependence of the XeF (B–X) band (blue) and the feature to longer wavelength assigned to FXe⋯F complexes (red). Solid lines are fits to a kinetic model previously used to describe the production and loss of FXe⋯F in other solvents.15 | ||
Fits to data of the type shown in Fig. 1 yielded time constants for loss of FXe⋯F complexes (and hence removal of F atoms) and growth of XeF of 3.3 ± 0.6 ps. This value is in keeping with the 4.0 ± 0.2 ps time constant determined for loss of FXe⋯F in CD3CN.13,15 The reciprocal of this time constant is a rate coefficient that accounts for all loss pathways of FXe⋯F complexes including reaction with the solvent and geminate recombination.
2. Vibrational relaxation and spectral diffusion for HF in CH3CN
Our previous investigations of exothermic radical reactions in organic solvents13,17,19–22 suggest that the HF products are likely to be vibrationally excited at the instant of their formation from the F + CH3CN reaction, and that solvent restructuring occurs in the wake of the reaction. We therefore measured the time constants for vibrational relaxation and spectral diffusion for HF in CH3CN using IR pump and probe spectroscopy. The origin of the v = 1 ← v = 0 absorption band in gas-phase HF is located at 3959 cm−1, and at steady state in solution in CH3CN is observed as a broad feature centred at about 3300 cm−1. The large anharmonicity of the HF vibrational mode shifts the gas-phase v = 2 ← v = 1 band 180 cm−1 to lower wavenumber than the v = 1 ← v = 0 band, and a greater anharmonicity and hence larger shift from the fundamental band are expected for hydrogen-bonded HF molecules in solution. Fig. 2 shows examples of time-resolved IR absorption spectra following narrow-bandwidth IR excitation at the centre or to the high wavenumber wing of the fundamental band. Depletion of HF(v = 0) by the IR pump pulse causes the negative change in optical density around 3300 cm−1 at early times, and transfer of population to v = 1 gives rise to a positive transient feature corresponding to the v = 2 ← v = 1 absorption, the high wavenumber wing of which is evident below 3150 cm−1. The considerable breadth of the bands is a consequence of inhomogeneous broadening associated with a range of solvation environments.23 Two time constants are derived from analysis of the recovery of the ground state bleach signal: repopulation of the ground state by vibrational relaxation occurs with τ = 2.4 ± 0.2 ps, whereas regaining the equilibrium band shape takes 2.1 ± 0.3 ps (1 SD uncertainties) following selective excitation of a subset of HF molecules with absorptions in the wing of the broad absorption band. Although these time constants are similar, the former value reflects a re-equilibration of populations of vibrational levels, whereas the latter is controlled by fluctuations in the microsolvation environment of the HF solute.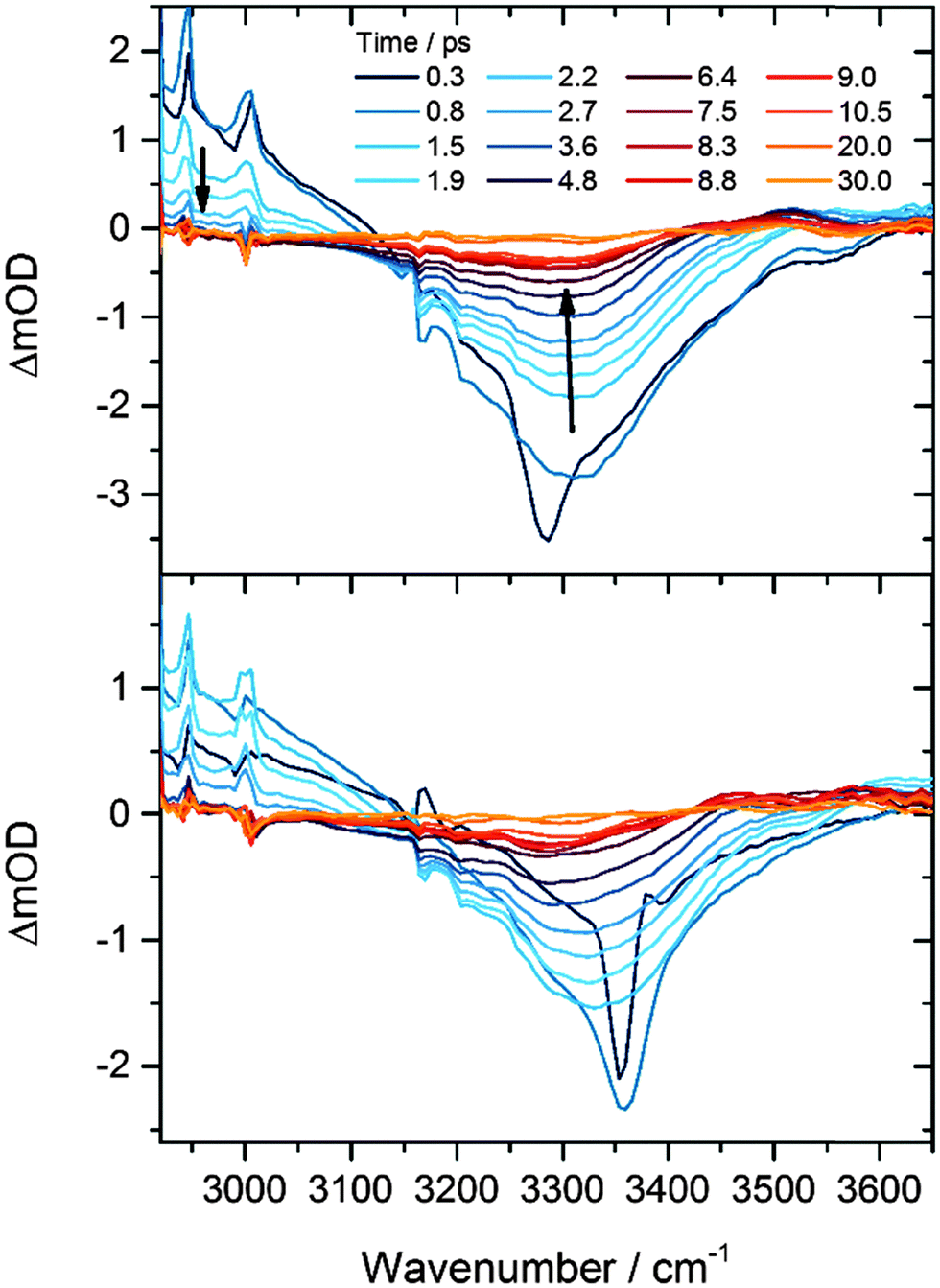 | ||
| Fig. 2 IR pump and IR probe spectra of dilute HF solutions in CH3CN, with the narrow bandwidth pump laser tuned close to the band centre (3300 cm−1, top) or the high-wavenumber wing (3360 cm−1, bottom) of the broad HF fundamental band. The decline in the signal amplitude and a shift of the band centre to lower wavenumber with time result respectively from vibrational relaxation and re-equilibration of the HF solvation environment. | ||
3. HF from bimolecular reaction of F atoms with CH3CN
Fig. 3 presents TVAS spectra obtained in the region of the HF fundamental band from 3075–3550 cm−1 following 267 nm photolysis of XeF2 in CH3CN. The black dashed line is a superimposed steady-state FTIR spectrum of a dilute solution of HF in acetonitrile, which agrees well with previous reports.24,25 The considerable breadth of the fundamental band complicates the analysis of the TVAS spectra for three reasons: (i) the band intensity is spread over all 128 pixels of the detector, so optical density changes measured at any single pixel are low; (ii) the baseline level corresponding to no absorption by HF or the solvent is hard to identify because of the limit of ∼500 cm−1 on the useable bandwidth of the probe IR; and (iii) hot bands associated with vibrationally excited HF molecules will overlap the low wavenumber side of the fundamental band (below 3100 cm−1 from consideration of the spectra in Fig. 2). The spectra have been shifted to a common baseline level using a consistent procedure involving a linear interpolation between the high and low wavenumber sides of the spectrum.26 We recognise that this procedure might skew the spectra, but checks with alternative procedures, such as applying a flat baseline shift to all spectra, show that our chosen method does not significantly alter the outcomes of the kinetic analysis that follows.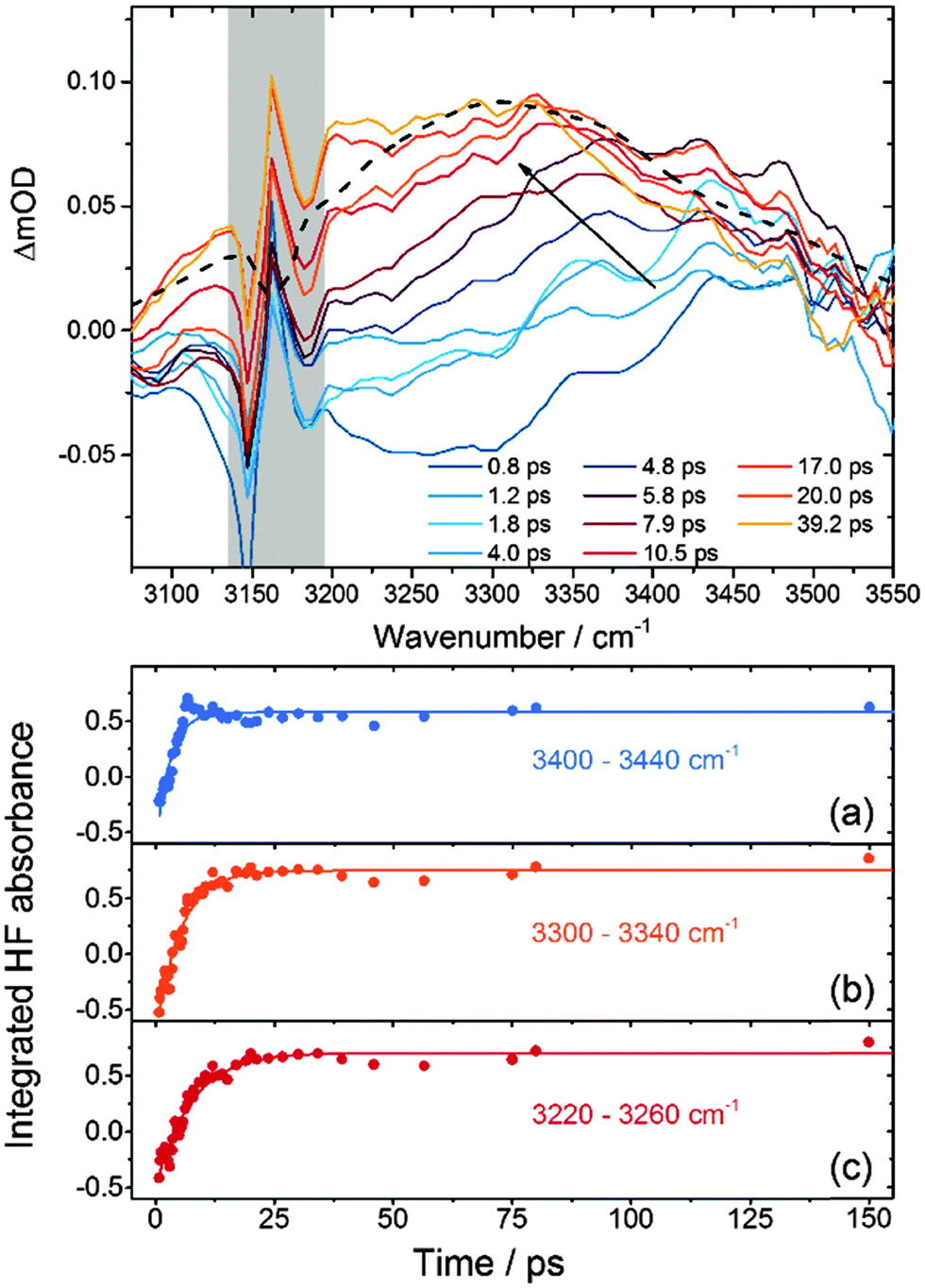 | ||
| Fig. 3 Top: time-resolved IR spectra of HF from the reaction of F atoms with CH3CN. The dashed black line is a steady-state FTIR spectrum of HF in acetonitrile, and the feature masked by the grey shading is a solvent interference. The inset key shows the time delays at which spectra were obtained. Bottom: time-dependence of HF absorbance summed over three wavenumber intervals, (a) 3400–3440 cm−1, (b) 3300–3340 cm−1 and (c) 3220–3260 cm−1. Solid lines are single exponential fits. | ||
The HF IR chromophore is a sensitive reporter of its local environment, and the TVAS spectra in Fig. 3 show initial growth at the high wavenumber side of the HF fundamental band. They then evolve into spectra more closely resembling the steady-state FTIR spectrum on timescales of a few 10 s of ps. Enhancements of the high-wavenumber sides of transient IR spectra have been reported from studies of photo-induced dynamics in hydrogen-bonded solvents such as water and interpreted as solvent heating effects resulting from deposition of excess energy – in our case from a chemical reaction – into the solvent bath.27 However, the band shift we observe is much faster than the reported decay times for these solvent heating effects, and is better interpreted as a solvent relaxation around the reaction products to reach an equilibrated solvent–solute environment.
Our recent study of DF production from F atom reactions in CD3CN revealed two contributions to the evolution of the DF fundamental absorption band beyond the sub-ps formation of hydrogen bonds to the solvent.13,14 The first of these contributions was relaxation of vibrationally hot DF formed in the v = 2 and 1 levels, resulting in a shift of the absorption to higher wavenumber because of the large anharmonicity in the D–F stretch. The second was a slower shift to lower wavenumber that we attributed to the response of the polar solvent in the first few solvation shells around a nascent DF molecule. Similar effects are expected to contribute to the TVAS spectra of HF shown in Fig. 3 but vibrational dynamics are harder to discern because of the greater width of the fundamental band than for DF in solution. We have not succeeded in decomposing the broad and featureless HF bands into contributions from vibrational ground state and vibrationally hot reaction products in the way that was possible for DF, so have applied a more simple method of analysis to extract kinetic information. We integrate the band intensities in three separate regions, 3400–3440 cm−1, 3300–3340 cm−1 and 3220–3260 cm−1 corresponding to the high, central and low wavenumber portions of the spectrum respectively. The low wavenumber portion may contain contributions from the overlapping HF(v = 2 ← v = 1) hot band at early times if reaction produces internally excited HF, and the high wavenumber end preferentially probes HF molecules in weaker hydrogen bonding configurations than those absorbing at the band centre. Fig. 3 shows examples of time-dependent absorbances integrated over these three spectral windows, together with fits to single exponential functions from which time-constants were derived. The dynamics might suggest more complicated fitting functions are appropriate,13 but within the signal-to-noise levels of our data, more complex analysis is not merited.
Separate analysis of 5 independent data sets by this method gives the time constants summarized in Table 1. The specified uncertainties are 1 SD from these 5 measurements. We label these time constants as τ1 (for the 3400–3440 cm−1 range), τ2 (for the 3300–3340 cm−1 range) and τ3 (for the 3220–3260 cm−1 range). An alternative method of analysis in which the broad HF bands were decomposed by fitting to Gaussian functions, each of 70 cm−1 FWHM, centred at 3500, 3420, 3320, and 3240 cm−1 yielded very similar time constants. This decomposition also gave a satisfactory reproduction of the time dependence of the overall HF band shape. The Gaussian function centred at 3500 cm−1 showed an initial rise then decay in intensity, commensurate with a shift of the fundamental band to lower wavenumber.
| Wavenumber range | 3400–3440 cm−1 | 3300–3340 cm−1 | 3220–3260 cm−1 |
| Time constant/ps | τ 1 = 3.04 ± 0.26 | τ 2 = 5.48 ± 0.24 | τ 3 = 7.47 ± 0.74 |
| (τ1 = 3.29 ± 0.17) | (τ2 = 5.24 ± 0.25) | (τ3 = 8.10 ± 0.93) |
A systematic trend is evident from this analysis and can be seen in spectra such as those shown in Fig. 3: the high wavenumber end of the HF fundamental band grows in most quickly, followed by the central region and finally the low wavenumber side. This trend is similar to that we reported for a shift to lower wavenumber of the DF fundamental band with a time constant of 10 ps following F-atom reaction in CD3CN, which we attributed to a reorganization of the solvent environment to accommodate reaction products.13 However, for HF in CH3CN the shift is more rapid: a crude analysis in which a broad single Gaussian function with floated central wavenumber (![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) c) is fitted to the transient HF IR absorption bands, followed by a single-exponential fit to the time-dependence of c, gives a time constant of 5.5 ± 0.8 ps. This value is larger than the spectral diffusion time constant of 2.1 ± 0.3 ps obtained from IR pump and probe experiments described above, indicating a greater perturbation of the solvent environment following the chemical reaction.
c) is fitted to the transient HF IR absorption bands, followed by a single-exponential fit to the time-dependence of c, gives a time constant of 5.5 ± 0.8 ps. This value is larger than the spectral diffusion time constant of 2.1 ± 0.3 ps obtained from IR pump and probe experiments described above, indicating a greater perturbation of the solvent environment following the chemical reaction.
The ∼3 ps time constant for the initial rise in HF absorption agrees well with the 3.3 ps decay of FXe⋯F complexes reported above. However, the reaction might produce vibrationally excited HF (vibrational levels up to v = 3 are energetically accessible), in which case the growth of the HF fundamental band will also be influenced by the time constant for vibrational cooling of HF(v > 0) in solution in CH3CN. The problems outlined earlier for the analysis of the broad HF band prevent us from extracting any definitive evidence for initial formation of vibrationally excited HF and subsequent relaxation, but we see some hints of this behaviour. Our IR pump and probe experiments indicate that any such relaxation should occur with a time constant of 2.4 ± 0.15 ps (for v = 1 → v = 0), which is similar to that observed for the growth of the high-wavenumber side of the fundamental band (with time constant τ1). Other signatures of initial vibrational excitation would include negative-going band intensities in our transient spectra in the case of a vibrational population inversion, and delayed onset to the growth of the fundamental band. Although we see evidence in all TVAS data sets for negative changes in optical density at early times (e.g. for time delays less than 2 ps in Fig. 3), we recognize the aforementioned difficulty in determining the correct baseline for zero absorbance in our spectra and therefore only tentatively propose that the HF is initially formed vibrationally excited. Any delayed onset to growth of the HF fundamental band is hard to resolve within the fluctuations in our integrated spectra over the first few picoseconds of acquisition.
We attribute the observed shift in band intensity to lower wavenumber during the first ∼20 ps from initiation of reaction to secondary changes in the solvation environment of the HF in acetonitrile. The primary solvent effect is the formation of hydrogen bonds from newly formed HF to neighbouring solvent molecules, and evidence from our spectra and from simulations, as well as the known fast solvent response for acetonitrile,28 suggests this process is complete on the sub-ps timescale. The experimental evidence comes from the fact that a non-H-bonded HF molecule will have a vibrational frequency approaching the 3959 cm−1 band origin for isolated HF in the gas phase, whereas all the transient features we observe are in the 3000–3600 cm−1 region. The secondary solvent effect that we observe with 5.5 ps time constant could result from diffusive separation of HF from XeF left behind after initial XeF2 photolysis, or from restructuring of first, second and perhaps further solvation shells about the HF to accommodate this new solute and to dissipate some of the excess energy of reaction. Hynes and coworkers have argued that relaxation of a perturbed distribution of ground state vibrational frequencies in water is associated with making and breaking of hydrogen bonds,23 and we suggest that we may be observing a similar effect here. Alternatively, we may be observing the cooling of initial bending excitation associated with the FH⋯NCCH3 hydrogen bond; recent molecular dynamics simulations of HCl in acetonitrile showed the IR spectrum to shift to higher wavenumber upon bending of the HCl away from collinear to the C3 symmetry axis of CH3CN.29 The significantly shorter time constant than observed for DF in CD3CN in otherwise similar experiments by Dunning et al.13 cannot be accounted for by diffusive separation of XeF and HF or DF; the viscosity of CD3CN differs from that of CH3CN by less than 10% at 298 K,30 and HF and DF diffusion coefficients should differ by 2%. However, exchange of H-bonds will be faster for HF than DF in solution both because of the lower moment of inertia of HF and hence faster (hindered) rotational dynamics, and because tunnelling might contribute to this exchange.
Conclusions
Ultraviolet photolysis of XeF2 in acetonitrile generates fluorine atoms that react with an ∼3 ps time constant to produce HF. We observe the loss of F-atoms through the perturbation they induce in the near-UV XeF absorption spectrum, and formation of HF by the growth of its fundamental vibrational band in the IR. The timescales for the two processes are commensurate. Time-resolved vibrational absorption spectroscopy also reveals a shift of the HF band to lower wavenumber with a 5.5 ps time constant. We attribute this spectral shift to a response by the solvent to the chemical reaction, with structural reorganization to accommodate the newly formed HF.Acknowledgements
The Bristol group thanks the European Research Council (ERC, Advanced Grant 290966 CAPRI) and EPSRC (Doctoral Training Grant studentship for GTD) for financial support. Experimental measurements were conducted at the ULTRA Laser Facility which is supported by the Science and Technology Facilities Council (STFC, Facility Grant ST/501784). We are grateful to J.N. Harvey, D.R. Glowacki and T.J. Preston for valuable discussions.References
- D. M. Neumark, A. M. Wodtke, G. N. Robinson, C. C. Hayden and Y. T. Lee, J. Chem. Phys., 1985, 82, 3045–3066 CrossRef CAS PubMed.
- M. A. Wickramaaratchi, D. W. Setser, H. Hildebrandt, B. Korbitzer and H. Heydtmann, Chem. Phys., 1985, 94, 109–129 CrossRef CAS.
- K. Dehe and H. Heydtmann, Chem. Phys. Lett., 1996, 262, 683–688 CrossRef CAS.
- J. J. Lin, J. G. Zhou, W. C. Shiu and K. P. Liu, Science, 2003, 300, 966–969 CrossRef CAS PubMed.
- D. E. Manolopoulos, K. Stark, H. J. Werner, D. W. Arnold, S. E. Bradforth and D. M. Neumark, Science, 1993, 262, 1852–1855 CAS.
- M. Tizniti, S. D. Le Picard, F. Lique, C. Berteloite, A. Canosa, M. H. Alexander and I. R. Sims, Nat. Chem., 2014, 6, 141–145 CrossRef CAS PubMed.
- W. R. Dong, C. L. Xiao, T. Wang, D. X. Dai, X. M. Yang and D. H. Zhang, Science, 2010, 327, 1501–1502 CrossRef CAS PubMed.
- M. H. Qiu, Z. F. Ren, L. Che, D. X. Dai, S. A. Harich, X. Y. Wang, X. M. Yang, C. X. Xu, D. Q. Xie, M. Gustafsson, R. T. Skodje, Z. G. Sun and D. H. Zhang, Science, 2006, 311, 1440–1443 CrossRef CAS PubMed.
- F. Lique, M. H. Alexander, G. L. Li, H. J. Werner, S. A. Nizkorodov, W. W. Harper and D. J. Nesbitt, J. Chem. Phys., 2008, 128, 084313 CrossRef PubMed.
- S. A. Nizkorodov, W. W. Harper, W. B. Chapman, B. W. Blackmon and D. J. Nesbitt, J. Chem. Phys., 1999, 111, 8404–8416 CrossRef CAS PubMed.
- L. Che, Z. F. Ren, X. G. Wang, W. R. Dong, D. X. Dai, X. Y. Wang, D. H. Zhang, X. M. Yang, L. S. Sheng, G. L. Li, H. J. Werner, F. Lique and M. H. Alexander, Science, 2007, 317, 1061–1064 CrossRef CAS PubMed.
- A. M. Zolot, P. J. Dagdigian and D. J. Nesbitt, J. Chem. Phys., 2008, 129, 194705 CrossRef PubMed.
- G. T. Dunning, D. R. Glowacki, T. J. Preston, S. J. Greaves, G. M. Greetham, I. P. Clark, M. Towrie, J. N. Harvey and A. J. Orr-Ewing, Science, 2015, 347, 530–533 CrossRef CAS PubMed.
- D. R. Glowacki, A. J. Orr-Ewing, J. N. Harvey, 2014, arXiv 1412.4180.
- G. T. Dunning, T. J. Preston, A. J. Orr-Ewing, S. J. Greaves, G. M. Greetham, I. P. Clark and M. Towrie, Phys. Chem. Chem. Phys., 2014, 16, 16095–16102 RSC.
- G. M. Greetham, P. Burgos, Q. Cao, I. P. Clark, P. S. Codd, R. C. Farrow, M. W. George, M. Kogimtzis, P. Matousek, A. W. Parker, M. R. Pollard, D. A. Robinson, Z.-J. Xin and M. Towrie, Appl. Spectrosc., 2010, 64, 1311–1319 CrossRef CAS PubMed.
- S. J. Greaves, R. A. Rose, T. A. A. Oliver, D. R. Glowacki, M. N. R. Ashfold, J. N. Harvey, I. P. Clark, G. M. Greetham, A. W. Parker, M. Towrie and A. J. Orr-Ewing, Science, 2011, 331, 1423–1426 CrossRef CAS PubMed.
- G. Bucher and J. C. Scaiano, J. Am. Chem. Soc., 1994, 116, 10076–10079 CrossRef CAS.
- R. A. Rose, S. J. Greaves, F. Abou-Chahine, D. R. Glowacki, T. A. A. Oliver, M. N. R. Ashfold, I. P. Clark, G. M. Greetham, M. Towrie and A. J. Orr-Ewing, Phys. Chem. Chem. Phys., 2012, 14, 10424–10437 RSC.
- R. A. Rose, S. J. Greaves, T. A. A. Oliver, I. P. Clark, G. M. Greetham, A. W. Parker, M. Towrie and A. J. Orr-Ewing, J. Chem. Phys., 2011, 134, 244503 CrossRef PubMed.
- F. Abou-Chahine, S. J. Greaves, G. T. Dunning, A. J. Orr-Ewing, G. M. Greetham, I. P. Clark and M. Towrie, Chem. Sci., 2013, 4, 226–237 RSC.
- A. J. Orr-Ewing, J. Chem. Phys., 2014, 140, 090901 CrossRef PubMed.
- R. Rey, K. B. Moller and J. T. Hynes, J. Phys. Chem. A, 2002, 106, 11993–11996 CrossRef CAS.
- R. M. Adams and J. J. Katz, J. Mol. Spectrosc., 1957, 1, 306–332 CrossRef CAS.
- N. I. Sushko, V. F. Sukhoverkhov, E. G. Tarakanova and G. V. Yukhnevich, J. Opt. Technol., 2006, 73, 515–518 CrossRef CAS.
- M. P. Grubb, A. J. Orr-Ewing and M. N. R. Ashfold, Rev. Sci. Instrum., 2014, 86, 064104 CrossRef PubMed.
- R. A. Nicodemus, S. A. Corcelli, J. L. Skinner and A. Tokmakoff, J. Phys. Chem. B, 2011, 115, 5604–5616 CrossRef CAS PubMed.
- B. Bagchi and B. Jana, Chem. Soc. Rev., 2010, 39, 1936–1954 RSC.
- N. Bork, V. Loukonen, H. G. Kjaergaard and H. Vehkamaki, Phys. Chem. Chem. Phys., 2014, 16, 24685–24690 RSC.
- P. Sassi, A. Morresi, G. Paliani and R. S. Cataliotti, J. Raman Spectrosc., 1999, 30, 501–506 CrossRef CAS.
| This journal is © the Owner Societies 2015 |