
Chemical imaging of single catalyst particles with scanning μ-XANES-CT and μ-XRF-CT†
S. W. T.
Price
*a,
K.
Ignatyev
a,
K.
Geraki
a,
M.
Basham
a,
J.
Filik
a,
N. T.
Vo
a,
P. T.
Witte
b,
A. M.
Beale
*cd and
J. F. W.
Mosselmans
a
aScience Division, Diamond Light Source, Harwell Science and Innovation Campus, Didcot, Oxon, OX11 0DE, UK. E-mail: stephen.price@diamond.ac.uk
bCatalysis Research GCC/PB, BASF Nederland B.V. Strijkviertel 67, 3454 ZG, De Meern, The Netherlands
cUK Catalysis Hub, Research Complex at Harwell, Harwell Science and Innovation Campus, Harwell, Didcot, Oxon, OX11 0FA, UK
dUniversity College London, Department of Chemistry, 21 Gordon Street, London, WC1H 0AJ, UK. E-mail: andrew.beale@ucl.ac.uk
First published on 5th November 2014
Abstract
The physicochemical state of a catalyst is a key factor in determining both activity and selectivity; however these materials are often not structurally or compositionally homogeneous. Here we report on the 3-dimensional imaging of an industrial catalyst, Mo-promoted colloidal Pt supported on carbon. The distribution of both the active Pt species and Mo promoter have been mapped over a single particle of catalyst using microfocus X-ray fluorescence computed tomography. X-ray absorption near edge spectroscopy (XANES) and extended X-ray absorption fine structure revealed a mixed local coordination environment, including the presence of both metallic Pt clusters and Pt chloride species, but also no direct interaction between the catalyst and Mo promoter. We also report on the benefits of scanning μ-XANES computed tomography for chemical imaging, allowing for 2- and 3-dimensional mapping of the local electronic and geometric environment, in this instance for both the Pt catalyst and Mo promoter throughout the catalyst particle.
Introduction
Hydrogenation of nitrobenzene and functionalised nitrobenzene compounds to form aniline and haloanilines is a key industrial process. The products are important intermediate compounds used in the production of pesticides, pigments, pharmaceuticals and other speciality chemicals.1–3 Typical catalysts have included nickel borides4 and precious metal powders supported on oxides or carbon.3,5 Modern day nanocatalysts are prepared in such a way as to yield nanoparticles with a narrow particle size distribution, and the utilisation of colloids has proven particularly beneficial in controlling this.6–12 One such example is the use of hexadecyl(2-hydroxyethyl)dimethyl ammonium dihydrogen-phosphate (HHDMA) as a water soluble stabiliser/reductant for such purposes, and includes the synthesis of both monometallic d8 and doped d8 metal nanoparticles, leading to the development of colloidal Pt/C promoted with Mo (as ammonium heptamolybdate tetrahydrate). This catalyst (cPt + Mo/C, NanoSelect™) shows remarkable performance for the selective hydrogenation of (non)functionalised nitroarenes.10,11 Whilst information on the colloid size is available, little is known about the 3-dimensional distribution of Pt and Mo, the extent to which they alloy and whether this is a likely cause of the impressive catalytic performance, something that is true for heterogeneous catalysis in general. This type of information can be obtained by performing “chemical imaging” experiments and could lead to better understanding of relevant catalytic structure–function relationships. Given the low loadings and small particle sizes, most common laboratory-based techniques are not capable of providing this information on the catalyst ex situ, or under operating conditions.The highly tuneable, bright X-ray beams provided by synchrotrons are ideal for measuring samples with the low metal concentrations, typical of industrial catalysts, and are extensively used for studying catalytic systems.13–19 However, recent developments in synchrotron source brightness and detector technology have enabled the development of ‘chemical imaging’ in which a 2-dimensional or 3-dimensional image of the sample is recorded and where each pixel in the image contains either a full spectrum/pattern allowing for the extraction of more detailed information on the sample by post hoc analysis. Examples include the application of diffraction-based X-ray imaging methods such as pencil-beam and microfocus X-ray diffraction computed tomography (μ-XRD-CT) which have led to greater appreciation of the importance of spatial distribution on crystalline phase formation and how this can impact upon the final properties of a material.20–25 Alternatively spectroscopic techniques such as X-ray fluorescence (XRF) or X-ray absorption near edge spectroscopy (XANES) provide information on the elemental distribution and the oxidation state of each active element present in the catalyst, as well as any molecular (e.g. heavy metal) poisons.19,21 With careful experimental design these data can be collected under reaction conditions to provide the structure function relationships.22
Recently, μ-XANES-CT collected in full field (FF) mode have been reviewed, mapping the 3-dimensional variation of chemical environment.13 Collection in FF mode offers excellent spatial resolution in the 10’s nm domain for transmission measurements and use of a CCD allows for fast data collection. One complication of FF measurements is that the focal length of the zone plates that focus the image changes with energy. The zone plate can be moved to compensate for this however this results in a change in the optical focus of the image, and so the position of the CCD must also change with energy.
An alternative to collecting in FF mode is to use a scanning microfocus X-ray beam.26 When collecting μ-XANES-CT in scanning mode the resolution is determined by the focal size of the beam, typically 2 to 100 microns depending on the beamline.14 In scanning mode, data is usually collected with a fluorescence detector perpendicular to the X-ray beam. The beam is translated across the sample and the sample rotated by a suitable angle step; this translation and rotation process is repeated until enough projections (for a reliable reconstruction the rotation angle should cover 180° and the number of rotation steps should equal the number of translational steps) are collected to enable a reconstruction of the data, visualised as a slice through the sample. Each slice is collected at a different incident X-ray wavelength, unlike in XRF-CT or XRD-CT.
Reconstructed spectra from a μ-XANES-CT experiment can be compared with those of known compounds or fitted to a combination of them (linear combination fitting – LCF) to determine the oxidation state(s) and ligands present in the sample and the local coordination geometry. Current hard-X-ray beamlines with FF transmission XANES-CT capabilities offer an energy range of ∼5–14 keV.13 The benefit of using a scanning X-ray microscopy beamline is the larger energy range available, in the case of I18 it is 2.05–20.5 keV.
In this study we report XANES, EXAFS and XRF characterisation of a commercial hydrogenation catalyst. The superior spatial resolution and detection limits available at a synchrotron reveal a more detailed representation of the catalyst and promoter distribution on the support, the local coordination environment of the catalyst and the degree of physicochemical interaction between the catalyst and promoter. We also report on the benefit of scanning μ-XANES-CT, allowing 2- and 3-dimensional imaging of the oxidation state and ligands of the catalyst on through the support.
Experimental
Sample preparation
The cPt + Mo/C catalyst (0.8 wt% Pt, 0.3 wt% Mo) was supplied by BASF and used as received. The catalyst preparation procedure has been detailed previously.10,11 Briefly, 30% HHDMA solution (84 g, 10 equivalents) was added to Pt (as H2PtCl6, 1.2 g) in H2O (2.4 L) at 95 °C and pH 5.0 to form the colloidal solution. This was added to an activated carbon slurry (120 g) followed by addition of the Mo promoter (as (NH4)6Mo7O24·4H2O) followed by filtering and washing of the catalyst. Pellets were prepared by mixing the catalyst with boron nitride for bulk XAS measurements. (NH4)6Mo7O24·4H2O (≥99.0% Sigma Aldrich) was mixed as received with boron nitride and pressed into a pellet for use as a XAS reference sample. A single particle of the supported catalyst was mounted on a MiTeGen micromount with 50 μm Kapton grid using epoxy resin. The particle was aligned over the centre of rotation using a manual Huber 1001 goniometer head. The particle chosen was slightly larger than the average particle size, however the dimensions (ca. 30 × 45 × 80 μm) were suitable with respect to the focussed X-ray beam size (2 × 2 μm) to map the particle with a sufficient number of pixels for analysis in the time available. A second smaller particle (ca. 12 × 15 × 20 μm) was mapped with μ-XRF-CT only to observe the elemental distribution.X-ray absorption spectroscopy (XAS) and data analysis
XAS data on the cPt + Mo/C pellet, Mo and Pt reference foils, and Mo reference samples were recorded at the Pt L3 absorption edge (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 564 eV) and the Mo K absorption edge (20000 eV) at beamlines B18 and I18 of Diamond Light Source. Beamline I18 was operating with a fixed exit Si(111) double crystal monochromator, focussed to a 2 μm × 2 μm spot on the sample using Kirkpatrick–Baez focussing mirrors. Beamline B18 was operating in QuXAFS mode with a fixed exit Si(111) double crystal monochromator. The Mo reference sample was collected on I18 in transmission mode using an ionisation chamber, all other XAS measurements were collected on B18 in fluorescence mode using a 9-element Ge solid-state detector. Energy calibration of the monochromator on each beamline was performed using Pt and Mo foils measured in transmission mode.
564 eV) and the Mo K absorption edge (20000 eV) at beamlines B18 and I18 of Diamond Light Source. Beamline I18 was operating with a fixed exit Si(111) double crystal monochromator, focussed to a 2 μm × 2 μm spot on the sample using Kirkpatrick–Baez focussing mirrors. Beamline B18 was operating in QuXAFS mode with a fixed exit Si(111) double crystal monochromator. The Mo reference sample was collected on I18 in transmission mode using an ionisation chamber, all other XAS measurements were collected on B18 in fluorescence mode using a 9-element Ge solid-state detector. Energy calibration of the monochromator on each beamline was performed using Pt and Mo foils measured in transmission mode.
The XAS data were processed (calibration, normalisation etc.) and analysed using the programs Athena and Artemis27 which implement the FEFF6 and IFEFFIT codes.28,29 The AUTOBK method30 was used to isolate the k-space EXAFS data from the raw data and a theoretical EXAFS signal was constructed using FEFF6. The influence of the fitted variables varies with the k-weighting used. The fit was co-refined with multiple k weighting over the entire k range making better use of all available data during the evaluation of χ2. Data from the metal foils were fit to determine the amplitude reduction factor (S02) for both Pt and Mo. These were found to be 0.85 and 0.83 respectively, and all coordination numbers reported have been corrected accordingly. LCF of the XANES was carried out using Athena, between −30 and +50 eV over the Pt L3 absorption edge and using the following reference standard spectra: Pt foil, 10 wt% Pt/C (nanoparticles), 10 wt% PtCl2/Al2O3, 10 wt% K2PtCl4/Al2O3, 10 wt% K2PtCl6/Al2O3, 10 wt% PtO2/Al2O3 and 1 wt% Pt/Al2O3 (nanoparticles).
X-ray fluorescence computed tomography (XRF-CT), X-ray absorption near edge spectroscopy computed tomography (XANES-CT) and data analysis
XRF maps were collected using an incident X-ray energy of 20300 eV with a spot size of 2 μm × 2 μm. The sample was rastered across the beam, with the XRF spectrum collected every 1 μm with a collection time of 0.3 s per pixel. A total of 41 translations and 71 rotations were used for the larger particle. The smaller particle required 30 translations and 71 rotations. The data was recorded using a 9-element Ge solid-state detector and XSPRESS-2 electronics.31
A script was written in Python 2.7 to extract the regions of interest (ROI) from the total fluorescence spectrum (9250 to 9600 eV for Pt, and 17300 to 17700 eV for Mo), sum the total counts for within each ROI and generate TIFF images of the elemental distribution. The centre of mass of each TIFF was then aligned with the centre of the image, to compensate for any misalignment of the centre of the particle above the centre of rotation whilst mounting the sample in the X-ray beam. The stack of TIFFs were loaded into ImageJ32 and the TomoJ plugin33 used to reconstruct the data in 3-dimensions. From this, orthogonal projections allowed for the 3-dimensional distribution of each element to be visualised.
To collect the XANES-CT data, the X-ray beam energy was set to 100 eV below the absorption edge of interest and the sample was rastered across it with a time per pixel of 0.3 s. Once the imaging of the row was completed the sample was then rotated by 5° and the imaging repeated. The process was repeated 37 times until particle had been rotated by 180° forming a sinogram with sufficient rotations to enable an accurate reconstruction. Four further sinograms were collected at 20 eV intervals approaching the absorption edge, at 1 eV intervals over the edge for 50 eV, at 2 eV intervals for a further 30 eV and finally at 5 eV intervals with the last sinogram collected at 130 eV past the absorption edge. A total of 80 sinograms were collected for both the Pt L3 and Mo K absorption edges.
The data was processed initially in a similar manner to before: the fluorescence signal in the relevant energy window was summed to give the total fluorescent intensity at each pixel of the sinogram. In the raw data, the edges of each row in the sinograms (Fig. 1a) do not align neatly; this is due to hysteresis in the position of the mechanical stage (<1 μm) moving back and forth between each rotation. To correct for this, the centre of mass of (the fluorescence signal for) each row of the sinogram was determined and a sine function fitted through these points (ESI,† Fig. S1).
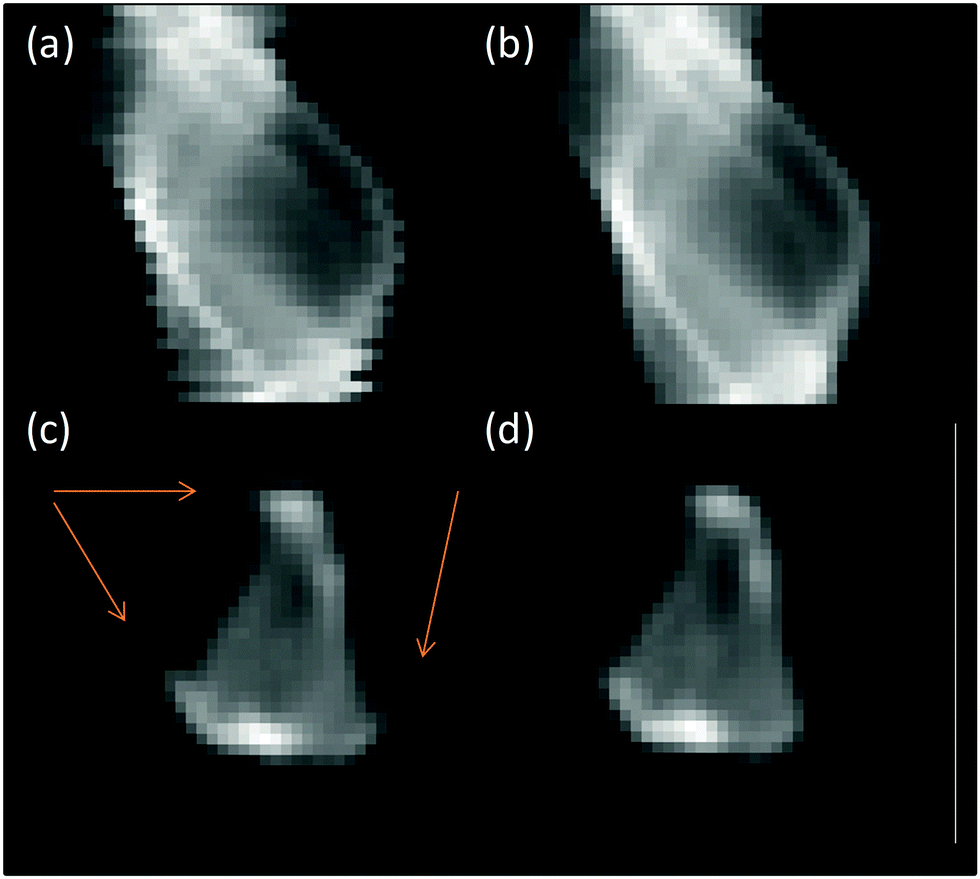 | ||
| Fig. 1 Processing stages of XANES tomography sinograms (a) raw sinogram data, (b) aligned and centred sinogram, (c) reconstruction of raw sinogram with artefacts highlighted and (d) final reconstruction. Sinograms shown correspond to slice at 11564 eV from μ-XANES-CT (cf.Fig. 7). | ||
For each row in the sinogram, the residual between the centre of mass and fitted function was used to align the rows (Fig. 1b). The artefacts observed in Fig. 1c are a result of the sinogram not being aligned with the centre of the image before the back-projection. This was corrected by shifting the centre of rotation of the sinogram to the centre of the image. The sinogram is then back-projected using an inverse Radon transform with the simultaneous algebraic reconstruction technique (SART) to produce a real-space image of the Pt fluorescence at the chosen slice through the sample (Fig. 1c and d). The process is repeated for the remaining sinograms over the energy range collected and the final images stacked in energy saved as a single HDF5 file.34 The data processing and alignment code were written in Python 2.7. The reconstructed fluorescence maps that form each energy step of the XANES tomogram have a spatial resolution of 2 μm – the same as the X-ray beam spot size, and at any given pixel location, a XANES spectrum can be plotted through (the energy dimension of) the data stack. To analyse this further, the data stack was loaded into MANTIS35,36 for cluster analysis; generating a map of where clusters of a particular component (e.g. spectral intensity or shape) are present on the map of the sample, and giving the average spectrum associated with each cluster. The computed XANES spectra were processed (energy calibration, normalisation, etc.) in the same manner as the standard XANES spectra, detailed in the previous section.
Results and discussion
XAS
The Mo XANES features (Fig. 2) of the catalyst pellet are slightly less intense just after the edge jump (at 20024 and 20045 eV) when compared with the ammonium heptamolybdate reference compound, and may be a result of the different energy resolutions of the two beamlines used to record the spectra. The pre-edge peak in the Mo XANES is a result of the 1s to 4d transition, and the main absorption edge due to the 1s to 5d transition. No Pt neighbours could be fitted surrounding the Mo (the reverse is true from fitting the Pt edge EXAFS) indicating that the Mo complex does not directly interact with the colloidal Pt particles on the support. The EXAFS data of the catalyst pellet recorded at the Mo K edge was fitted to a model of distorted octahedrally coordinated MoO3 (Fig. 3 and Table 1), consistent with the heptamolybdate structure.37
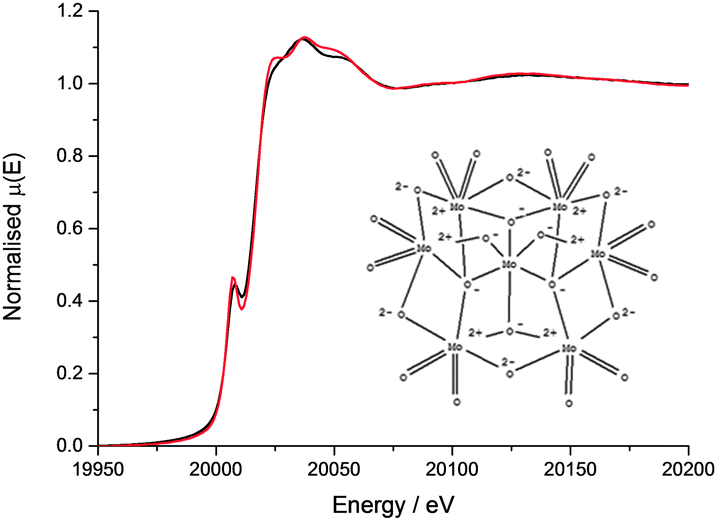 | ||
| Fig. 2 Mo K edge XAS comparison of ammonium heptamolybdate reference sample (black) with cPt + Mo/C pellet (red). | ||
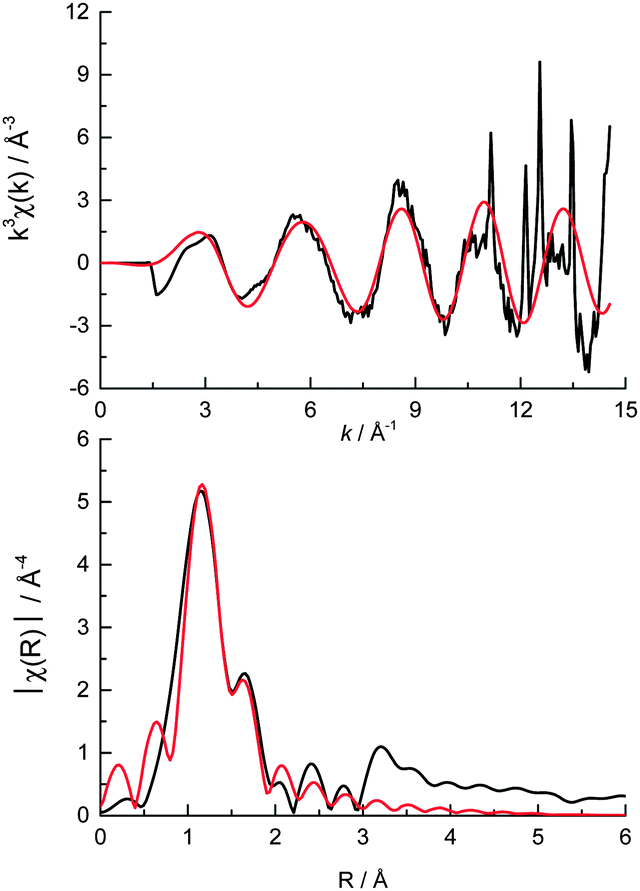 | ||
| Fig. 3 (top) k3 weighted experimental data (black) and fit (red) and (bottom) the corresponding k3 weighted Fourier transform of cPt + Mo/C at the Mo K edge. | ||
| Shell | N | R/Å | σ 2 × 104/Å2 | ΔE0/eV | R f |
|---|---|---|---|---|---|
| a N is the coordination number, R is the coordination distance, σ2 is the Debye–Waller (disorder) term, ΔE0 is the shift in the Fermi energy, and Rf is a measure of the goodness of fit.50 b The crystal structure for the Mo fitting model contained 5 separate Mo–O bond distances (classed as different if ΔR > 0.03 Å). This was simplified to 3 Mo–O distances to minimise the number of variables in the fit (by classifying different bond lengths if ΔR > 0.05 Å). | |||||
Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ob Ob |
1.9 (0.3) | 1.68 (0.02) | 75 (13) | −7.6 (3.5) | 0.014 |
| Mo–O2−b |
1.9 (0.3) | 1.92 (0.02) | 126 (33) | ||
| Mo–O−b |
1.9 (0.3) | 2.26 (0.02) | 210 (205) | ||
| Pt—Cl | 2.1 (0.3) | 2.30 (0.02) | 40 (14) | 12.2 (2.5) | 0.012 |
| Pt—Pt | 1.5 (0.7) | 2.76 (0.02) | 57 (21) | ||
The Pt L3 absorption edge is a result of the 2p3/2 to 5d5/2 transition. LCF of the Pt L3 edge XANES reveals that the Pt local environment is approximately 44% metallic Pt, 47% Pt chloride and 9% Pt oxide (Fig. 4, Table 2). It is well known that small Pt nanoparticles readily oxidise in air and so it might be assumed that the Pt–O interactions were due to air oxidation of the nanoparticles,38 however recent DFT calculations of similar HHDMA colloidal Pd systems indicate that the three O atoms from the dihydrogen phosphate anion of the HHDMA ligand strongly adsorb to the metal surface within the colloid.39 Similar behaviour is anticipated on Pt surfaces, and therefore the oxide contribution identified from the LCF may in fact be a result of adsorbed dihydrogen phosphate anions within the colloidal shell, rather than passivation.
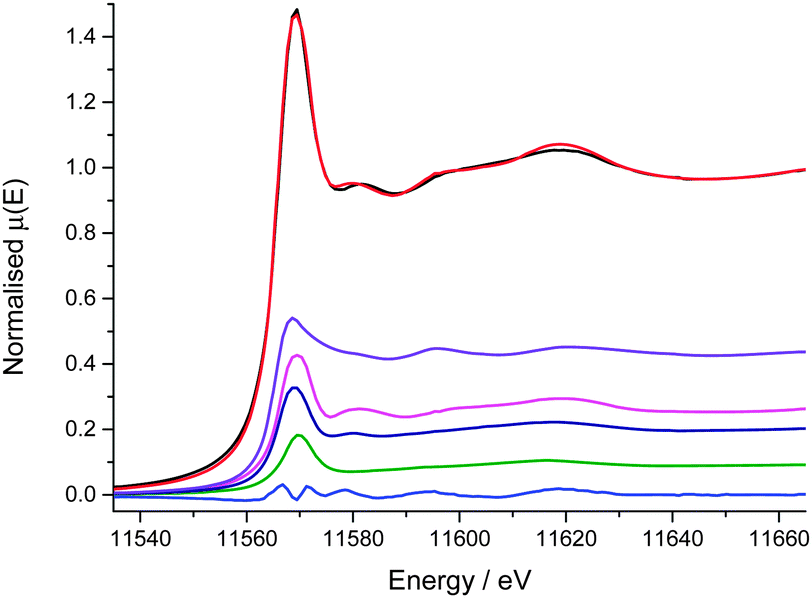 | ||
| Fig. 4 Results of Pt L3 edge XANES LCF (−30 to +50 eV over Pt L3 edge); experimental data (black), fit (red), residual (blue), K2PtCl6 (pink), PtO2 (green), PtCl2 (navy) and Pt/C nanoparticles (purple). | ||
| Component | Weight/% | R f | Reduced χ2 |
|---|---|---|---|
| Pt/C nano | 44 (2) | 0.0007 | 0.0001 |
| PtO2 | 9 (5) | ||
| PtCl2 | 21 (3) | ||
| K2PtCl6 | 26 (3) |
The Pt chloride contribution in the XANES is split between K2PtCl6 (the Pt precursor during synthesis10) and PtCl2. Given that 10 equivalents of HHDMA are added during the reduction process, it seems unlikely that any completely unreacted H2PtCl6 is contained within the colloids. The LCF fit suggests that the Pt has an average oxidation state of +2 in the as received catalyst. Pt chloride compounds have a wide range of reported bond lengths for similar oxidation states.40–44 The EXAFS fitted Pt–Cl bond length of 2.30 Å falls in the middle of this range, broadly consistent with a +4 oxidation state, and given the ratio of first shell coordination numbers of Pt-metal to Pt-chloride (relative to each other and to bulk), this is in agreement with the average +2 oxidation state of the sample, as are the number of Cl neighbours present.
Two equivalents of HHDMA have been reported to be enough to reduce a similar Pd precursor, with 10 equivalents enough to stabilise the colloid, with use of a Pt precursor requiring a higher reduction temperature.11 Given the presence of partially reduced Pt chloride species determined from both XANES and EXAFS measurements, it may be the case that the colloid formation process ensues with sufficient speed to trap partially reduced Pt chloride within the HHDMA shell along with the Pt nanoclusters, as it is unlikely that there was not sufficient HHDMA resent to fully reduce the precursor.
It is possible that the HHDMA stabiliser may have a chloride anion instead of the phosphate anion, something not considered in the DFT calculations. The presence of an oxide contribution in the XANES indicates that if this has occurred, it has not been a complete anion exchange of phosphate for chloride. In this case, the Pt colloid would consist of an inner Pt cluster surrounded by a shell of chloride, coordinated to the Pt surface as well as the HHDMA stabiliser. Both of these potential models are not distinguishable from the XANES and EXAFS data as they result in very similar chloride bond distances and coordination numbers. However, given the large excess of HHDMA used in the synthesis, it is likely that all precursor is reduced, and so the exchange of phosphate for chloride on the HHDMA is likely the cause of the Pt–Cl observed, forming a similar structure as suggested by Bucher et al.45 It should be noted that the presence of this residual chloride does not hamper the performance of the catalyst.
The ratio of the Pt first shell coordination numbers fitted from the Pt L3 edge EXAFS (Fig. 5, Table 1) corroborates the amount of metallic Pt present (ca. 41%) however the number of Pt–Pt scattering contributions is low. A 9% Pt–O contribution in the XANES corresponds to an average of ca. 0.4 O neighbours per Pt atom and as such is too low to be reasonably fitted from the data. The very low Pt–Pt coordination number indicates that metallic Pt is present in very small clusters within the colloids rather than as nanoparticles (where a 1st shell coordination number of 8–9 would be expected for particles of 2.3 ± 0.3 nm diameter as measured by TEM).10,46–49 The EXAFS data after 10 Å−1 is quite noisy (a result of the very low Pt loading and fast scanning mode), resulting in very poor fitting quality beyond the first coordination shells (i.e. Pt–Cl and Pt–Pt).
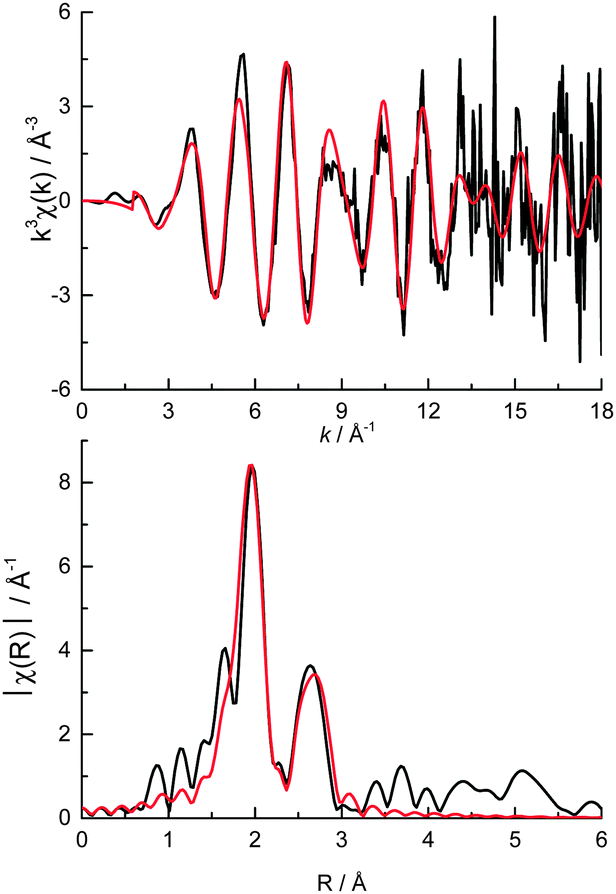 | ||
| Fig. 5 (top) k3 weighted experimental data (black) and fit (red) and (bottom) the corresponding k3 weighted Fourier transform of cPt + Mo/C at the Pt L3 edge. | ||
The lack of any measureable diffraction by powder XRD (ESI,† Fig. S2) reveals that the colloids are not crystalline and also suggests that the TEM may be overestimating the average particle size as (1) EXAFS is a per atom average technique and therefore will be weighted towards smaller average sizes if there is a sizeable amount of atomic Pt present, and (2) similar sized Pt nanoparticles (ca. 2 nm) without a stabilising colloid are known to diffract.48 Based on previous studies relating the average first shell coordination number with approximate nanoparticle size,46–48 an average nanoparticle size of around 1 nm can be inferred (the Pt chloride is 50% under-coordinated and Pt over 85% under-coordinated). If some Pt atoms/clusters were adsorbed on the surface of the carbon outside of a colloid this would skew the average coordination numbers measured by EXAFS, resulting in a lower apparent particle size, and would be missed by both TEM and XRD due to the near-atomic of the species.
Repeated XANES measurements on a single point on the sample revealed a reduction in the rising absorption edge intensity and increase in the post edge features intensity (ESI,† Fig. S3) indicating that the Pt is partially reduced by prolonged exposure to the X-ray beam.51 In general, organic compounds are far more susceptible to beam damage than inorganic compounds. The reduction of the Pt signal therefore is attributed to the partial loss of the HHDMA/Cl colloid, caused by the X-ray beam, whereas the inorganic Mo complex does not appear to be reduced by the X-ray beam. This ‘beam-induced’ reduction process could conceivably occur during TEM measurements (under vacuum) and may have resulted in the comparatively larger particle sizes observed in the microscopy images.10
XRF tomography
Fig. 6 shows horizontal and vertical cross-sections taken from the XRF tomography reconstruction. The Mo is distributed evenly throughout the carbon support whereas the Pt distribution is clearly uneven with a more concentrated shell around the outside of the particle, and several areas on the inside that have very low relative concentrations; the outer 6 μm layer of the particle account for approximately 70% of the deposited Pt. Of particular interest are several bright spots on the outside and a horizontal slice through these was chosen as the ROI for the XANES tomography A very low concentration of Fe (ca. 0.08 wt%) was also present in the XRF tomography, evenly distributed throughout the carbon support, probably as remnants from the C-support production process (ESI,† Fig. S4). | ||
| Fig. 6 Horizontal and vertical cross-sections from XRF tomography showing Pt (red) and Mo (green) distributions throughout the carbon support. | ||
Overlaying the distribution of both elemental maps reveals that the Pt is distributed over a larger area of the support than the Mo, the extra area being the more concentrated Pt shell. Indeed it appears that the outermost 2 μm of the particle is almost exclusively Pt, and that it is within this shell and throughout the support that the Mo is deposited (as heptamolybdate).
Given the preparation procedure of the catalyst whereby the Pt is first added to the carbon slurry,10,11 it appears to be the case that the colloidal Pt first deposits on the surface of the carbon before the remaining Pt colloid diffuses through the pores and deposits within the carbon particle. The bulk of the HHDMA colloid likely prevents the colloid diffusing through the smallest nano-pores in the activated carbon (<2 nm), akin to that proposed for similar colloidal Pd catalysts.12 The Mo promoter subsequently added would find the majority of the surface of the carbon covered with Pt colloid, but being smaller in size (ca. 1 nm) than the Pt colloid may more easily diffuse into the <2 nm pores of the carbon and thus deposit throughout the support. A second smaller particle (ca. 20 × 10 × 10 μm) was also mapped using XRF-CT, reveals a very similar elemental distribution with even Mo throughout the support however the Pt outer layer is less distinct (ca. 1 μm thick, ESI,† Fig. S5).
Previously, focussed ion beam scanning electron microscopy (FIB-SEM) was used to image the colloidal (Pd) catalyst distribution on the support.12 This revealed that the colloids did not penetrate into the pores of the support, and concluded that the egg-shell distribution this resulted in was the cause of the superior selectivity of the catalyst. In the previous study, the carbon support was spheres 50–200 μm diameter and the colloidal particles also larger than the Pt colloids in this study. The improved sensitivity of synchrotron XRF reveals that the smaller Pt colloids do penetrate through the pore system to the inside of the support, albeit in lower amounts than that seen on the surface. The fact that the Pt is in the same mixed metallic/chloride environment as the Pt on the surface of the support (see XANES tomography below) suggests that it is colloidal particles that have penetrated the pores of the support rather than nanoclusters not stabilised by HHDMA. The relative amount of Pt inside the support is low, the rate of diffusion of the nitrobenzene through the pore structure is unknown, and given that the Mo is deposited after the Pt, the Pt inside the support may not be accessible as there may not be a clear path through the pore network (the nano-pore structure of the carbon is below the micron spatial resolution of the beamline).12 Therefore, it cannot be definitively stated if this internal Pt colloid also participates in the catalysis.
The bulk XAS measurements revealed a mixture of Pt chemical environments, and the XRF tomography an uneven distribution of Pt across the support, however neither of these techniques is able to provide information about any possible distribution of these different chemical environments within the particle at the micro-scale.
XANES tomography
The region chosen for the μ-XANES-CT slice corresponds to the horizontal cross-sections displayed in Fig. 6 from the μ-XRF-CT. The region was chosen due to the large variation in intensity in Pt signal and both Pt L3 edge and Mo K edge tomograms were collected at the same horizontal section. Of the 3 major clusters representing the particle, there is no change in the edge position between each cluster indicating that the Pt oxidation state is uniform on the micro-scale across support (discussion of the total number of clusters analysed is included in the ESI†). The major difference between the different clusters is the intensity of the Pt fluorescence signal. The Pt is deposited in greatest amounts at the top and bottom of the particle however the XANES for this cluster (Fig. 7, purple) is far noisier than that for majority of the particle (Fig. 7, red and yellow clusters).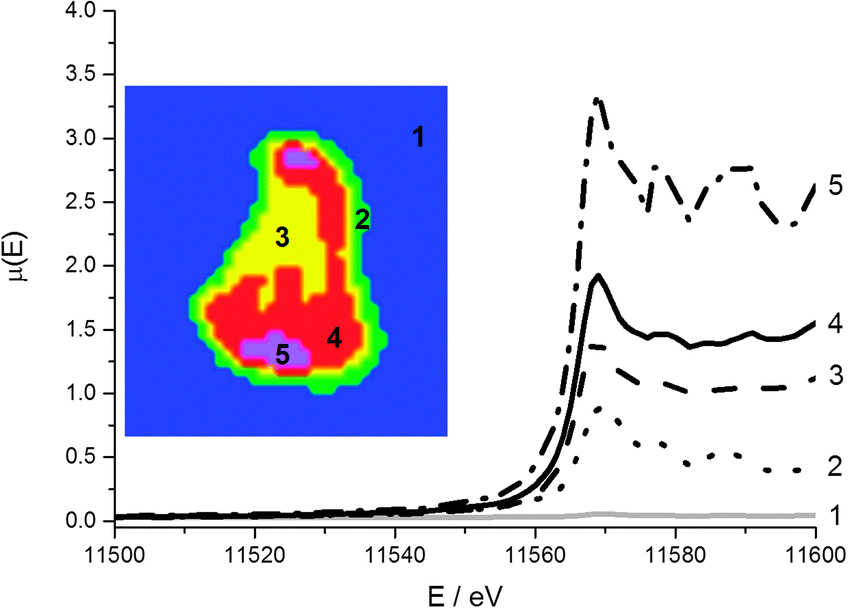 | ||
| Fig. 7 Cluster map of reconstructed Pt XANES tomography data stack (inset) and associated XANES spectra. The clusters and corresponding spectra are matched (1 to 5). | ||
Whilst the dwell time per point is very low per row of the sinogram, once the data is reconstructed to give the 2-dimensional slice through the particle, the signal at any given pixel is the sum of all the projections passing through it and therefore comparable to a XANES spectrum collected by traditional point measurement. The main region of noise in the Pt XANES spectra is the increased amplitude of post edge oscillations beyond 11580 eV, and this is most apparent in the 5th cluster (Fig. 7, purple). These artefacts in the reconstructed spectra, compared with the ex situ pellet spectrum, are attributed to the low concentration of the metals of interest.
The intensity of the rising absorption edge for all 4 clusters in the Pt XANES tomography is reduced compared with bulk spectra; this implies the Pt throughout the particle is in a more reduced state than an “average” particle based on the bulk XAS. Based on spot XANES measurements of the particle, it is probable that this slight reduction in the white line intensity is the result of beam damage of the sample. One advantage of collecting a series of sinograms at single energies rather than collecting a single sinogram of the full XANES spectra at each point is that the beam passes quickly over the sample (0.3 s per pixel vs. ca. 15 min for a XANES spectrum), reducing chances of beam damage by sustained exposure on the same point. Whilst radiation damage appears to be proportional to the accumulated dose,52 there are indications that damage occurs both on very fast and slow timescales,53 and therefore by reducing the exposure time per point, some radiation damage is avoided.54–56 This effective dose thresholding method mitigated the amount of reduction of the Pt relative to collecting a full XANES spectrum on a single point (ESI,† Fig. S3), and would also be relevant when imaging other sensitive samples. Even though the Pt XANES spectra collected in this case are not of sufficient quality for LCF over the whole range due to the large residual from the fit, useful information on the oxidation state distribution is still available. The consistent position and shape of the edge jump in all the major clusters reveals that the metallic Pt and Pt chloride are well mixed at the micron scale. This supports the conclusion that both Pt species are within the HHDMA shell and not separate species on the support. The Mo XANES tomography (Fig. 8) revealed one major cluster in the centre of the particle, with an edge position and pre- and post-edge features consistent with the XANES of ammonium heptamolybdate tetrahydrate (Fig. 2).
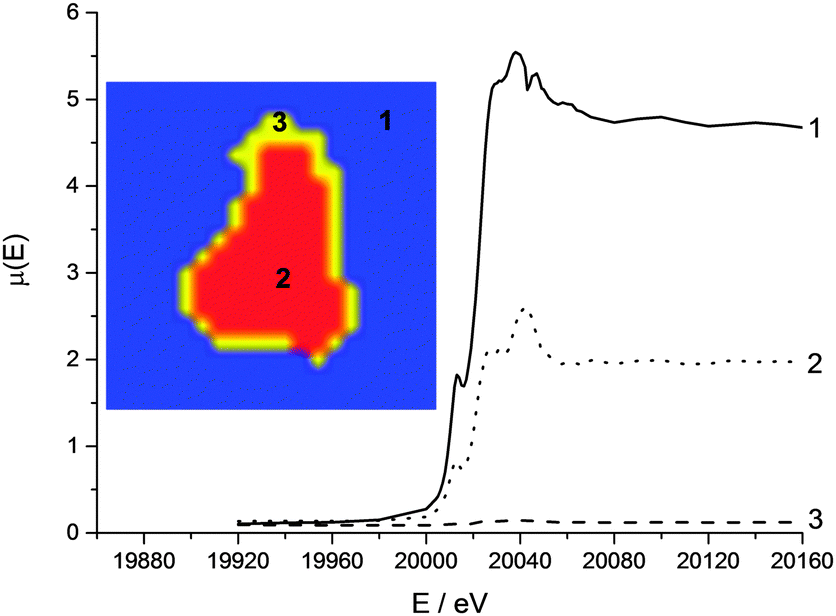 | ||
| Fig. 8 Cluster map of reconstructed Mo XANES tomography data stack (inset) and associated XANES spectra. A single glitch is present at 20050 eV. The clusters and corresponding spectra are matched (1 to 3). | ||
Conclusions
A combination of XAS and XRF revealed more clearly the elemental and chemical distribution of colloidal catalyst particles on a carbon support. Synchrotron XRF-CT has revealed details regarding the metal location and speciation within the support that had not been previously observed. The concentration of Pt on the surface of the support was confirmed, and utilisation of the internal pore structure by a small proportion of Pt colloid as well as the Mo complex was revealed. The use of scanning XANES microtomography, in combination with bulk XAS measurements, has shown that the as received catalyst has a more complex local coordination structure than previously thought with both metallic Pt and Pt chloride present, however the chloride is present as a result of anion exchange with the HHDMA stabiliser and not an incomplete reaction of the precursor. A schematic of the proposed colloid inner structure is illustrated in Fig. 9.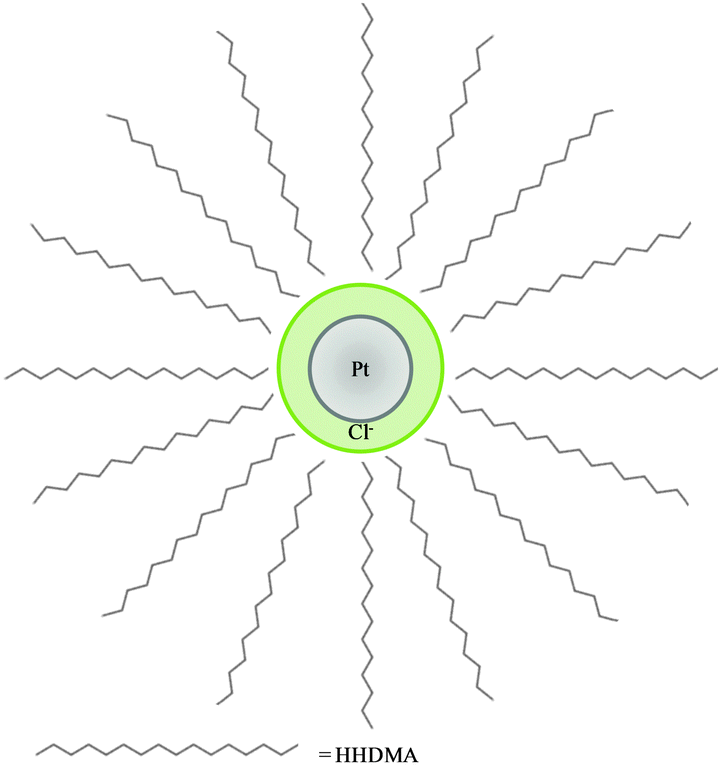 | ||
| Fig. 9 Representation of the proposed Pt colloid structure of Pt nanoparticle (grey) surrounded by HHDMA shell with chloride anions (green). The double layer nature of the HHDMA shell is not shown for simplicity.12 | ||
The lack of bimetallic interactions in the EXAFS in both the Pt and Mo data indicates that there is no direct interaction between the two metals during catalyst preparation, and suggest that a close proximity of the Mo complex is enough for the promotion effects observed.
Collection of the data as sinograms provides a more powerful approach than 2-dimensional XANES mapping as it enables a slice through a sample to be observed rather than collecting a surface map. The technique has an excellent spatial resolution of 2 μm, and complements the current XRF microtomography already available, allowing for chemical environment tomography as well as 3-dimensional elemental distribution. One useful benefit of the XANES tomography processing procedure developed here is that it can take as little as 10 s to process a data stack (of 80 sinograms) running on a standard computer, making on the fly data analysis possible, e.g. looking for variations in pre-edge features or white line intensity in the early maps, whilst the maps at the higher energies are being collected.
A microreactor is currently being developed at the beamline to enable the imaging of the evolution of elemental, chemical and phase distribution of catalysts under operating conditions. That will help to reveal the key structure–function relationships, and in turn facilitate the design and preparation of improved catalysts.
Acknowledgements
We thank Diamond Light Source for providing access to synchrotron beamtime on B18 and I18 (proposal SP4939) that contributed to the results presented here. EPSRC are acknowledged for providing funding for Andrew M. Beale. The authors would like to thank Dr Giannantonio Cibin for collecting additional XAS measurements on beamline B18 and Dr Tim Hyde for supplying platinum XANES reference spectra.Notes and references
- H.-U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner and M. Studer, Adv. Synth. Catal., 2003, 345, 103–151 CrossRef CAS.
- H.-U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210–221 CrossRef CAS.
- K. Möbus, D. Wolf, H. Benischke, U. Dittmeier, K. Simon, U. Packruhn, R. Jantke, S. Weidlich, C. Weber and B. Chen, Top. Catal., 2010, 53, 1126–1131 CrossRef.
- D. J. Collins, A. D. Smith and B. H. Davis, Ind. Eng. Chem. Prod. Res. Dev., 1982, 21, 279–281 CrossRef CAS.
- M. Takasaki, Y. Motoyama, K. Higashi, S.-H. Yoon, I. Mochida and H. Nagashima, Org. Lett., 2008, 10, 1601–1604 CrossRef CAS PubMed.
- H. Bonnemann, W. Brijoux, K. Siepen, J. Hormes, R. Franke, J. Pollmann and J. Rothe, Appl. Organomet. Chem., 1997, 11, 783–796 CrossRef CAS.
- H. Bonnemann, W. Brijoux, A. S. Tilling and K. Siepen, Top. Catal., 1997, 4, 217–227 CrossRef.
- V. I. Pârvulescu, V. Pârvulescu, U. Endruschat, G. Filoti, F. E. Wagner, C. Kübel and R. Richards, Chem. – Eur. J., 2006, 12, 2343–2357 CrossRef PubMed.
- J. Roelofs and P. H. Berben, Chem. Commun., 2004, 970–971 RSC.
- E. Boymans, S. Boland, P. T. Witte, C. Mueller and D. Vogt, ChemCatChem, 2013, 5, 431–434 CrossRef CAS.
- P. T. Witte, P. H. Berben, S. Boland, E. H. Boymans, D. Vogt, J. W. Geus and J. G. Donkervoort, Top. Catal., 2012, 55, 505–511 CrossRef CAS.
- P. T. Witte, S. Boland, F. Kirby, R. van Maanen, B. F. Bleeker, D. A. M. de Winter, J. A. Post, J. W. Geus and P. H. Berben, ChemCatChem, 2013, 5, 582–587 CrossRef CAS.
- J. C. Andrews and B. M. Weckhuysen, ChemPhysChem, 2013, 14, 3655–3666 CrossRef CAS PubMed.
- A. M. Beale, S. D. M. Jacques and B. M. Weckhuysen, Chem. Soc. Rev., 2010, 39, 4656–4672 RSC.
- I. D. Gonzalez-Jimenez, K. Cats, T. Davidian, M. Ruitenbeek, F. Meirer, Y. Liu, J. Nelson, J. C. Andrews, P. Pianetta, F. M. F. deGroot and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2012, 51, 11986–11990 CrossRef CAS PubMed.
- J. D. Grunwaldt, J. B. Wagner and R. E. Dunin-Borkowski, ChemCatChem, 2013, 5, 62–80 CrossRef CAS.
- M. A. Newton and W. van Beek, Chem. Soc. Rev., 2010, 39, 4845–4863 RSC.
- M. G. O’Brien, A. M. Beale, S. D. M. Jacques, M. Di Michiel and B. M. Weckhuysen, Appl. Catal., A, 2011, 391, 468–476 CrossRef PubMed.
- J. Ruiz-Martinez, A. M. Beale, U. Deka, M. G. O'Brien, P. D. Quinn, J. F. W. Mosselmans and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 5983–5987 CrossRef CAS PubMed.
- M. Alvarez-Murga, P. Bleuet, L. Marques, C. Lepoittevin, N. Boudet, G. Gabarino, M. Mezouar and J.-L. Hodeau, J. Appl. Crystallogr., 2011, 44, 163–171 CrossRef CAS.
- F. Basile, P. Benito, S. Bugani, W. De Nolf, G. Fornasari, K. Janssens, L. Morselli, E. Scavetta, D. Tonelli and A. Vaccari, Adv. Funct. Mater., 2010, 20, 4117–4126 CrossRef CAS.
- A. M. Beale, S. D. M. Jacques, E. K. Gibson and M. Di Michiel, Coord. Chem. Rev., 2014, 277, 208–233 CrossRef PubMed.
- P. Bleuet, E. Welcomme, E. Dooryhee, J. Susini, J.-L. Hodeau and P. Walter, Nat. Mater., 2008, 7, 468–472 CrossRef CAS PubMed.
- S. D. M. Jacques, M. Di Michiel, A. M. Beale, T. Sochi, M. G. O'Brien, L. Espinosa-Alonso, B. M. Weckhuysen and P. Barnes, Angew. Chem., Int. Ed., 2011, 50, 10148–10152 CrossRef CAS PubMed.
- M. G. O'Brien, S. D. M. Jacques, M. Di Michiel, P. Barnes, B. M. Weckhuysen and A. M. Beale, Chem. Sci., 2012, 3, 509–523 RSC.
- C. G. Schroer, M. Kuhlmann, T. F. Günzler, B. Lengeler, M. Richwin, B. Griesebock, D. Lützenkirchen-Hecht, R. Frahm, E. Ziegler, A. Mashayekhi, D. R. Haeffner, J.-D. Grunwaldt and A. Baiker, Appl. Phys. Lett., 2003, 82, 3360–3362 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. J. Rehr, R. C. Albers and S. I. Zabinsky, Phys. Rev. Lett., 1992, 69, 3397–3400 CrossRef CAS.
- M. Newville, J. Synchrotron Radiat., 2001, 8, 96–100 CrossRef CAS.
- M. Newville, P. Līviņš, Y. Yacoby, J. J. Rehr and E. A. Stern, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 14126–14131 CrossRef CAS.
- R. Farrow, G. E. Derbyshire, B. R. Dobson, A. J. Dent, D. Bogg, J. Headspith, R. Lawton, M. Martini and K. Buxton, Nucl. Instrum. Methods Phys. Res., Sect. B, 1995, 97, 567–571 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- C. Messaoudil, T. Boudier, C. O. S. Sorzano and S. Marco, BMC Bioinf., 2007, 8, 288–296 CrossRef PubMed.
- Hierarchical Data Format, http://www.hdfgroup.org/HDF5/.
- M. Lerotic, C. Jacobsen, T. Schäfer and S. Vogt, Ultramicroscopy, 2004, 100, 35–57 CrossRef CAS PubMed.
- M. Lerotic, http://https://code.google.com/p/spectromicroscopy/, 2014.
- H. T. Evans, B. M. Gatehouse and P. Leverett, J. Chem. Soc., Dalton Trans., 1975, 505–514 RSC.
- P. Dietrich, T. Wu, A. Sumer, J. Dumesic, J. Jellinek, W. N. Delgass, F. Ribeiro and J. Miller, Top. Catal., 2013, 56, 1814–1828 CrossRef CAS PubMed.
- G. Vilé, N. Almora-Barrios, S. Mitchell, N. López and J. Pérez-Ramírez, Chem. – Eur. J., 2014, 20, 5926–5937 CrossRef PubMed.
- A. L. Ankudinov, J. J. Rehr and S. R. Bare, Chem. Phys. Lett., 2000, 316, 495–500 CrossRef CAS.
- E. Curis, K. Provost, D. Bouvet, I. Nicolis, S. Crauste-Manciet, D. Brossard and S. Benazath, J. Synchrotron Radiat., 2001, 8, 716–718 CrossRef CAS.
- T. I. Hyde, P. W. Ash, D. A. Boyd, G. Randlshofer, K. Rothenbacher and G. Sankar, Platinum Met. Rev., 2011, 55, 233–245 CrossRef CAS.
- R. J. Williams, D. R. Dillin and W. O. Milligan, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 1369–1372 CrossRef CAS.
- A. Jentys, G. L. Haller and J. A. Lercher, J. Phys. Chem., 1993, 97, 484–488 CrossRef CAS.
- S. Bucher, J. Hormes, H. Modrow, R. Brinkmann, N. Waldöfner, H. Bönnemann, L. Beuermann, S. Krischok, W. Maus-Friedrichs and V. Kempter, Surf. Sci., 2002, 497, 321–332 CrossRef CAS.
- R. E. Benfield, J. Chem. Soc., Faraday Trans., 1992, 88, 1107–1110 RSC.
- A. Jentys, Phys. Chem. Chem. Phys., 1999, 1, 4059–4063 RSC.
- S. W. T. Price, N. Zonias, C.-K. Skylaris, T. I. Hyde, B. Ravel and A. E. Russell, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 075439 CrossRef.
- A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2010, 12, 5562–5574 RSC.
- S. D. Kelly, D. Hesterberg and B. Ravel, Methods of Soil Analysis. Part 5. Mineralogical Methods, Soil Science Society of America, Madison, 2008, ch. 14, p. 444 Search PubMed.
- J. G. Mesu, A. M. Beale, F. M. F. de Groot and B. M. Weckhuysen, J. Phys. Chem. B, 2006, 110, 17671–17677 CrossRef CAS PubMed.
- P. Sliz, S. C. Harrison and G. Rosenbaum, Structure, 2003, 11, 13–19 CrossRef CAS.
- M. Warkentin, J. B. Hopkins, R. Badeau, A. M. Mulichak, L. J. Keefe and R. E. Thorne, J. Synchrotron Radiat., 2013, 20, 7–13 CAS.
- K. M. Davis, I. Kosheleva, R. W. Henning, G. T. Seidler and Y. Pushkar, J. Phys. Chem. B, 2013, 117, 9161–9169 CrossRef CAS PubMed.
- E. G. Allan, M. C. Kander, I. Carmichael and E. F. Garman, J. Synchrotron Radiat., 2013, 20, 23–36 CAS.
- J. W. Murray, E. Rudino-Pinera, R. L. Owen, M. Grininger, R. B. G. Ravelli and E. F. Garman, J. Synchrotron Radiat., 2005, 12, 268–275 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp04488f |
| This journal is © the Owner Societies 2015 |