Formation of 2- and 1-methyl-1,4-dihydronaphthalene isomers via the crossed beam reactions of phenyl radicals (C6H5) with isoprene (CH2C(CH3)CHCH2) and 1,3-pentadiene (CH2CHCHCHCH3)†
Received
11th October 2014
, Accepted 7th November 2014
First published on 7th November 2014
Abstract
Crossed molecular beam reactions were exploited to elucidate the chemical dynamics of the reactions of phenyl radicals with isoprene and with 1,3-pentadiene at a collision energy of 55 ± 4 kJ mol−1. Both reactions were found to proceed via indirect scattering dynamics and involve the formation of a van-der-Waals complex in the entrance channel. The latter isomerized via the addition of the phenyl radical to the terminal C1/C4 carbon atoms through submerged barriers forming resonantly stabilized free radicals C11H13, which then underwent cis–trans isomerization followed by ring closure. The resulting bicyclic intermediates fragmented via unimolecular decomposition though the atomic hydrogen loss via tight exit transition states located 30 kJ mol−1 above the separated reactants in overall exoergic reactions forming 2- and 1-methyl-1,4-dihydronaphthalene isomers. The hydrogen atoms are emitted almost perpendicularly to the plane of the decomposing complex and almost parallel to the total angular momentum vector (‘sideways scattering’) which is in strong analogy to the phenyl–1,3-butadiene system studied earlier. RRKM calculations confirm that 2- and 1-methyl-1,4-dihydronaphthalene are the dominating reaction products formed at levels of 97% and 80% in the reactions of the phenyl radical with isoprene and 1,3-pentadiene, respectively. This barrier-less formation of methyl-substituted, hydrogenated PAH molecules further supports our understanding of the formation of aromatic molecules in extreme environments holding temperatures as low as 10 K.
1. Introduction
During the past decades, the formation mechanisms of polycyclic aromatic hydrocarbons (PAHs) – organic molecules containing fused benzene rings – have received particular attention due to their toxicity1 and carcinogenic,2–4 mutagenic,4,5 as well as teratogenic properties.4 In the late 1970s, the United States Environmental Protection Agency (USEPA) was engaged in classifying and prioritizing chemicals according to their toxicity, and 16 of them have been identified as PAHs so far.6 On Earth, PAHs are mainly produced by an incomplete combustion of organic materials such as coal, biomass, and fossil fuel or naturally through the biosynthesis of plants, volcanic eruptions, and wood fires.7 PAHs heavier than 500–1000 amu are also regarded as key precursors to soot formation,8,9 with the soot production worldwide estimated be to about 107 tons per year.10 Respiratory particulate matter (PM) is of particular importance since these atmospheric pollutants pose great risk to the human health and can be inhaled by breathing.11–13 Further, PAHs are considered as air and marine pollutants as well,14–16 and can further contribute to global warming.17 Therefore, anthropogenic PAH and soot emission sources need to be controlled by the society; this requires an understanding of the fundamental mechanisms of how PAHs are formed under combustion-like conditions. Besides terrestrial sources, PAHs are also regarded as prominent organic molecules that are supposed to exist in the interstellar medium (ISM).18 Here, PAHs and their (partially) hydrogenated and alkyl-substituted counterparts are thought to account for the ‘unidentified’ infrared (UIR) bands and the diffuse interstellar absorption bands (DIBs).19 PAHs such as naphthalene, anthracene, and phenanthrene as detected in the Murchison meteorite point to an interstellar origin,20 possibly from circumstellar and/or interstellar sources,21 thus providing an elaborate record on the chemical evolution of the universe.
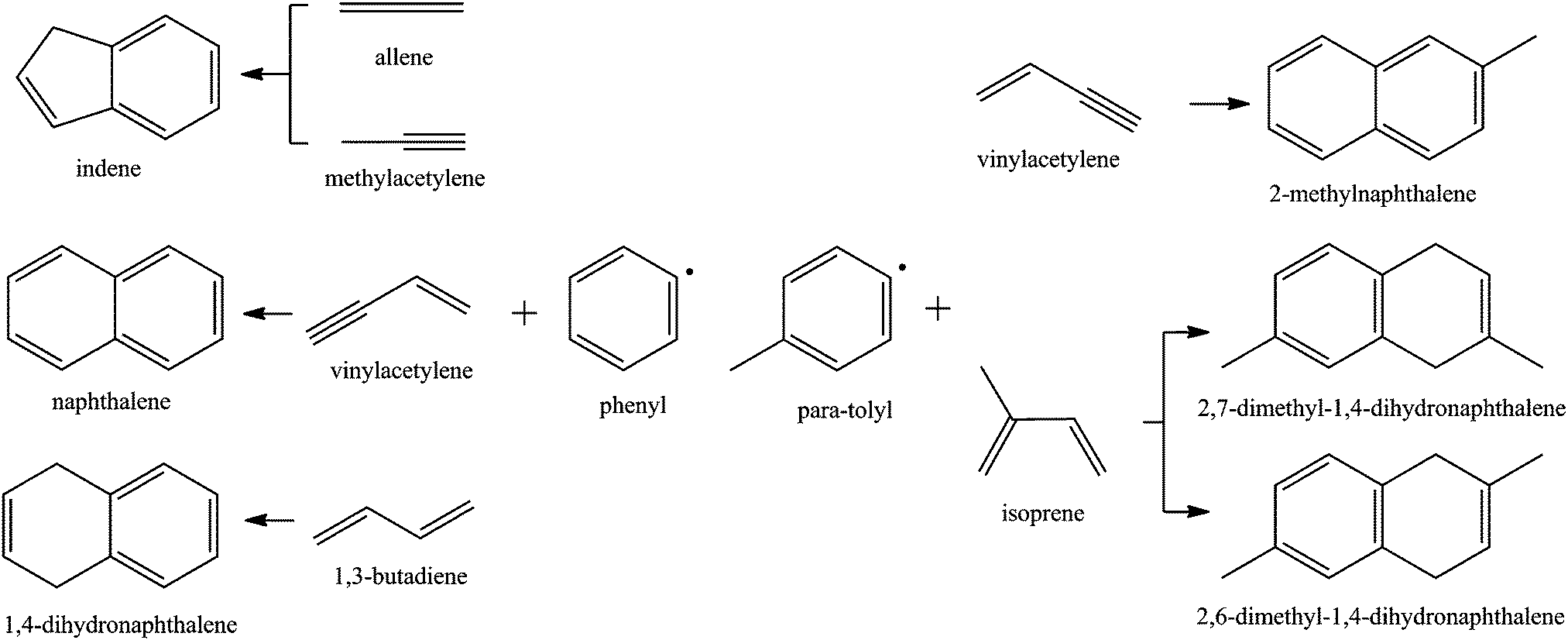
It is well known that small resonantly stabilized free radicals (RSFRs)22–28 as well as small aromatics like benzene (C6H6) and phenyl radicals (C6H5)8,9 play important roles in further PAH growth pathways. The simplest PAHs – indene and naphthalene – can be formed via reactions of single-ring aromatic hydrocarbons such as benzene (C6H6) and the phenyl radical (C6H5) with unsaturated hydrocarbons involving the hydrogen abstraction–acetylene addition (HACA) mechanism,9,29,30 the self-reaction of cyclopentadiene (cyclopentadienyl),31–33 and/or the phenyl (benzene) addition–cyclization (PAC) with unsaturated hydrocarbons such as alkynes,34 olefins,35–37 and aromatic molecules.37–40 In a pyrolysis reactor, Britt et al. found that the pyrolysis of terpenes ((C5H8)n) in a high temperature (600–800 °C) environment resulted in the production of benzene (C6H6), toluene (C7H8), styrene (C8H8), indene (C9H8), naphthalene (C10H8), 2-methylnaphthalene (C11H10), and 1-methylnaphthalene (C11H10). Considering that isoprene (C5H8) presents a fundamental building block of terpenes, the isoprene molecule, which can be considered as a methyl-substituted 1,3-butadiene molecule, can be actively involved in the formation of PAHs.41 Exploiting the crossed molecular beams approach, our group has systematically investigated the formation of PAHs via bimolecular reactions involving phenyl-type radicals with unsaturated hydrocarbons allene (C3H4),42 methylacetylene (C3H4),42 vinylacetylene (C4H4),43 and 1,3-butadiene (C4H6)44 at collision energies up to about 50 kJ mol−1 under single collision conditions leading to indene (C9H8), naphthalene (C10H8), and 1,4-dihydronaphthalene (C10H10) (Fig. 1). Likewise, para-tolyl radicals (C6H4CH3) reacting with vinylacetylene and isoprene (C10H8) barrier-lessly lead to the formation of 2-methylnaphthalene (C11H10)45 and dimethyldihydronaphthalenes46 at a single collision event (Fig. 1). Consequently, bimolecular reactions of phenyl-type radicals (phenyl, para-tolyl) with C4 and C5 hydrocarbons such as vinylacetylene and (methyl-substituted) 1,3-butadiene were found to form polycyclic aromatic hydrocarbons (PAHs) with naphthalene and 1,4-dihydronaphthalene cores in exoergic and entrance barrier-less reactions under single collision conditions. The reaction mechanism involves the initial formation of a van-der-Waals complex and addition of the phenyl-type radical to the C1 position of a vinyl-type group through a submerged barrier. Our investigations indicate that in the hydrocarbon reactant, the vinyl-type group must be in conjugation to a –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH or –HC
CH or –HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 group to form a resonantly stabilized free radical (RSFR) intermediate, which eventually isomerizes to a cyclic intermediate followed by hydrogen loss and aromatization (PAH formation).
CH2 group to form a resonantly stabilized free radical (RSFR) intermediate, which eventually isomerizes to a cyclic intermediate followed by hydrogen loss and aromatization (PAH formation).
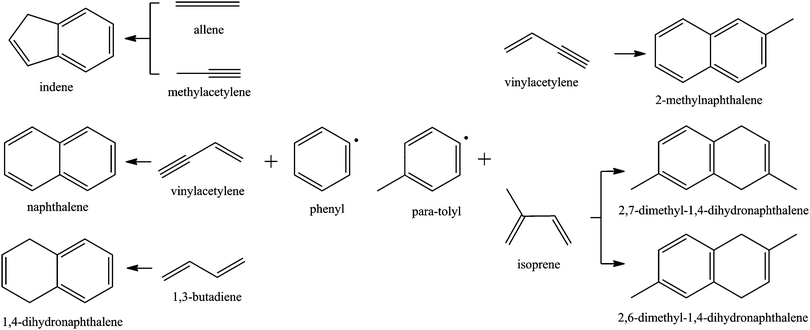 |
| | Fig. 1 Crossed beam reactions of phenyl-type (phenyl, para-tolyl) radicals with unsaturated C3 (allene, methylacetylene), C4 (vinylacetylene, 1,3-butadiene) and C5 (isoprene) hydrocarbons leading to the formation of (methyl-substituted) indene and naphthalene-like structures. | |
Having established that the reaction of phenyl radicals with 1,3-butadiene leads to the formation of 1,4-dihydronaphthalene isomers (Fig. 1), we are probing now the outcome of the bimolecular reactions of the phenyl radical with isoprene (2-methyl-1,3-butadiene) and 1,3-pentadiene (1-methyl-1,3-butadiene) to elucidate if these bimolecular reactions can synthesize methyl-substituted 1,4-dihydronaphthalene isomers, in which the methyl group is bound to 1,4-dihydrogenated ring thus enabling us to control the outcome of a bimolecular reaction leading to distinct PAH isomers by replacing hydrogen atoms selectively by methyl groups.
2. Experimental
The reactions of the phenyl radical (C6H5) with isoprene (CH2C(CH3)CHCH2) and 1,3-pentadiene (CH2CHCHCHCH3) were conducted exploiting a crossed molecular beams machine under single collision conditions.47–50 Briefly, a pulsed supersonic beam of phenyl radicals seeded in helium (99.9999%; Gaspro) at fractions of about 1% was prepared by photodissociation of chlorobenzene (C6H5Cl; 99.9%; Sigma-Aldrich) in the primary source chamber. This gas mixture was formed by passing 1.8 atm helium gas through chlorobenzene stored in a stainless steel bubbler at 293 K. The gas mixture was then released by a Proch–Trickl pulsed valve operated at 120 Hz and −400 V and photodissociated by 193 nm light at 18 ± 2 mJ emitted from an Excimer laser operating at 60 Hz. A four-slot chopper wheel located after the skimmer selected a part of the phenyl beam at a well-defined peak velocity (vp) and speed ratio S (Table 1). This section of the radical beam was perpendicularly intersected in the interaction region of the scattering chamber by a pulsed molecular beam of the hydrocarbon reactant. These were isoprene (CH2CHCCH3CH2; 99%; TCI America) and 1,3-pentadiene (CH2CHCHCHCH3; 97%; TCI America) released at backing pressures of 450 Torr and 420 Torr, respectively, by a pulsed valve in the secondary source chamber at a repetition rate of 120 Hz and pulse duration of 80 μs. In our experiment, the chopper wheel generated the time zero trigger pulse; the primary and the secondary pulsed valve timings were triggered 1894 μs and 1859 μs after the time zero, while the excimer laser was fired 162 μs later than the primary pulsed valve.
Table 1 Primary and secondary beam peak velocities (vp), speed ratios (S), collision energies (Ec) and center-of-mass angles (ΘCM) for the reactions of phenyl with isoprene and 1,3-pentadiene
| Beam |
v
p (ms−1) |
S
|
E
c (kJ mol−1) |
Θ
CM
|
| Phenyl (C6H5) |
1585 ± 18 |
9.3 ± 0.8 |
|
|
| Isoprene (C5H8) |
721 ± 20 |
8.5 ± 0.6 |
56 ± 3 |
21.9 ± 0.5 |
|
|
| Phenyl (C6H5) |
1583 ± 18 |
9.0 ± 0.9 |
|
|
| 1,3-Pentadiene (C5H8) |
711 ± 20 |
8.5 ± 0.7 |
54 ± 3 |
21.6 ± 0.5 |
The reactively scattered products were monitored using a triply differentially pumped quadrupole mass spectrometric detector in the time-of-flight (TOF) mode after electron-impact ionization of the neutral species with an electron energy of 80 eV.51–53 Time-of-flight spectra were recorded over the full angular range of the reaction in the plane defined by the primary and the secondary reactant beams. The TOF spectra were then integrated and normalized to obtain the product angular distribution in the laboratory frame (LAB). To extract information on the reaction dynamics, the experimental data are transformed into the center-of-mass frame utilizing a forward-convolution routine. This method initially assumes an angular flux distribution, T(θ), and the translational energy flux distribution, P(ET) in the center-of-mass system (CM). Laboratory TOF spectra and the laboratory angular distributions (LAB) are subsequently calculated from the T(θ) and P(ET) functions and compared to the experimental data, the functions are iteratively adjusted until the best fit between the two is achieved. In addition, we obtained the product flux contour map I(θ,u) = P(u) × T(θ), which plot represents the differential cross section and generates a vivid ‘image’ of the studied reaction.44 Here, I(θ,u) represents the flux of the reactive scattering products, θ stands for the scattering angle with respect to the CM angle, and u stands for the product velocity in the CM frame.
3. Theory
The geometries and harmonic frequencies of all reaction intermediates, transition states and products on the C11H13 potential energy surfaces (PESs) involved in the reactions of phenyl with isoprene and cis-/trans-1,3-pentadiene were computed at the B3LYP level within the hybrid density functional theory with the 6-311G** basis set.54,55 The final single-point energies were refined using the B3LYP optimized structures at the G3(MP2,CC)//B3LYP/6-311G** theoretical level,56 which is expected to generate relative energies of various species within the accuracy of ±10 kJ mol−1. The GAUSSIAN 0957 and MOLPRO 201058 program packages were employed for all density functional and ab initio calculations. Further, the Rice–Ramsperger–Kassel–Marcus (RRKM) calculations were performed to compute the rate constants k(E) (Table S1, ESI†), considering the internal energy E as a sum of the collision energy and the energy of the chemical activation. The product statistical yields were computed by solving first-order kinetic equations for unimolecular reactions.59,60
4. Experimental results
In the bimolecular reactions of the phenyl radical (C6H5; 77 amu) with isoprene (C5H8; 68 amu) and with 1,3-pentadiene (C5H8; 68 amu), signal was observed at the mass-to-charge ratios (m/z) of 145 (13C10H12+/C11H13+), 144 (C11H12+), 143(C11H11+), 130 (C10H10+) and 129 (C10H9+). After scaling, the time-of-flight (TOF) spectra at m/z = 145, 143, 130 and 129 were superimposable to those taken at m/z = 144. As a matter of fact, signal at m/z = 145 was found to originate from the naturally occurring 13C-substituted C11H12 with the latter formed via the phenyl versus atomic hydrogen exchange pathway. Since the TOF data in the range of 143 to 129 are superimposable to signal taken at m/z = 144, we can conclude that signal at the lower mass-to-charge ratios originates from dissociative electron impact ionization of the parent molecules in the ionizer, and we proceed to collect angular resolved TOF spectra at m/z = 144 (Fig. 2). Further, the molecular hydrogen loss channel is closed and only the phenyl versus atomic hydrogen exchange channels leading to C11H12 isomer(s) are open. The corresponding laboratory angular distributions shown in Fig. 3 were scaled by the primary beam intensities and averaged over the number of the scans. Both distributions peak close to the center-of-mass angle of 21.9° and 21.6° and extend by about 20° within the scattering plane defined by the primary and secondary beams. The peaking of the laboratory angular distributions in vicinity to the corresponding center-of-mass angles and the nearly forward–backward symmetric profile propose indirect scattering dynamics via the formation of C11H13 collision complexes.
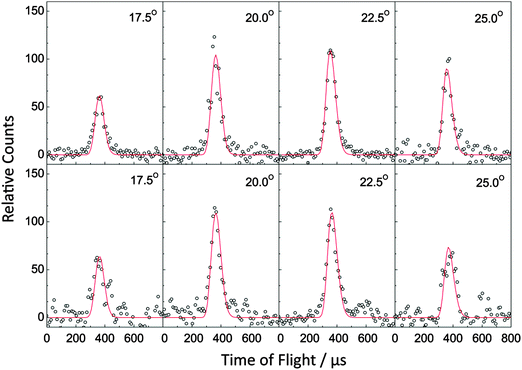 |
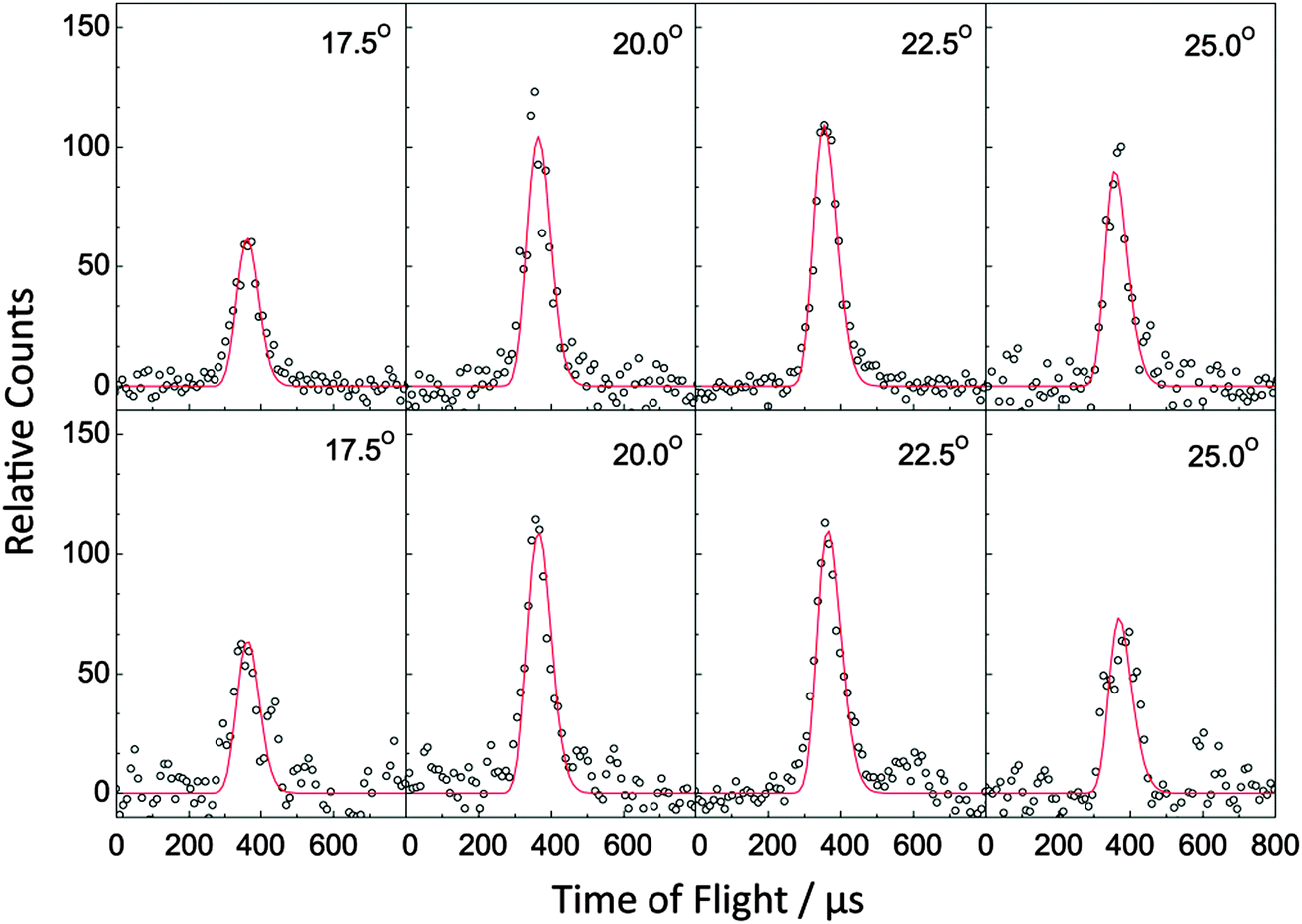
| | Fig. 2 Selected time-of-flight (TOF) spectra recorded at a mass-to-charge ratio (m/z) of 144 (C11H12+) for the reactions of phenyl radicals with isoprene (top) and with 1,3-pentadiene (bottom). The circles present the data points, while the solid lines represent the fits obtained from the forward-convolution routine. | |
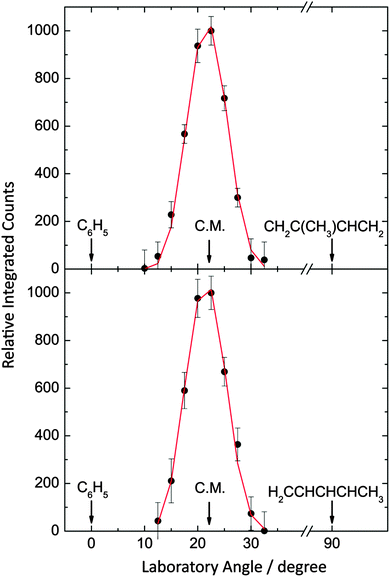 |
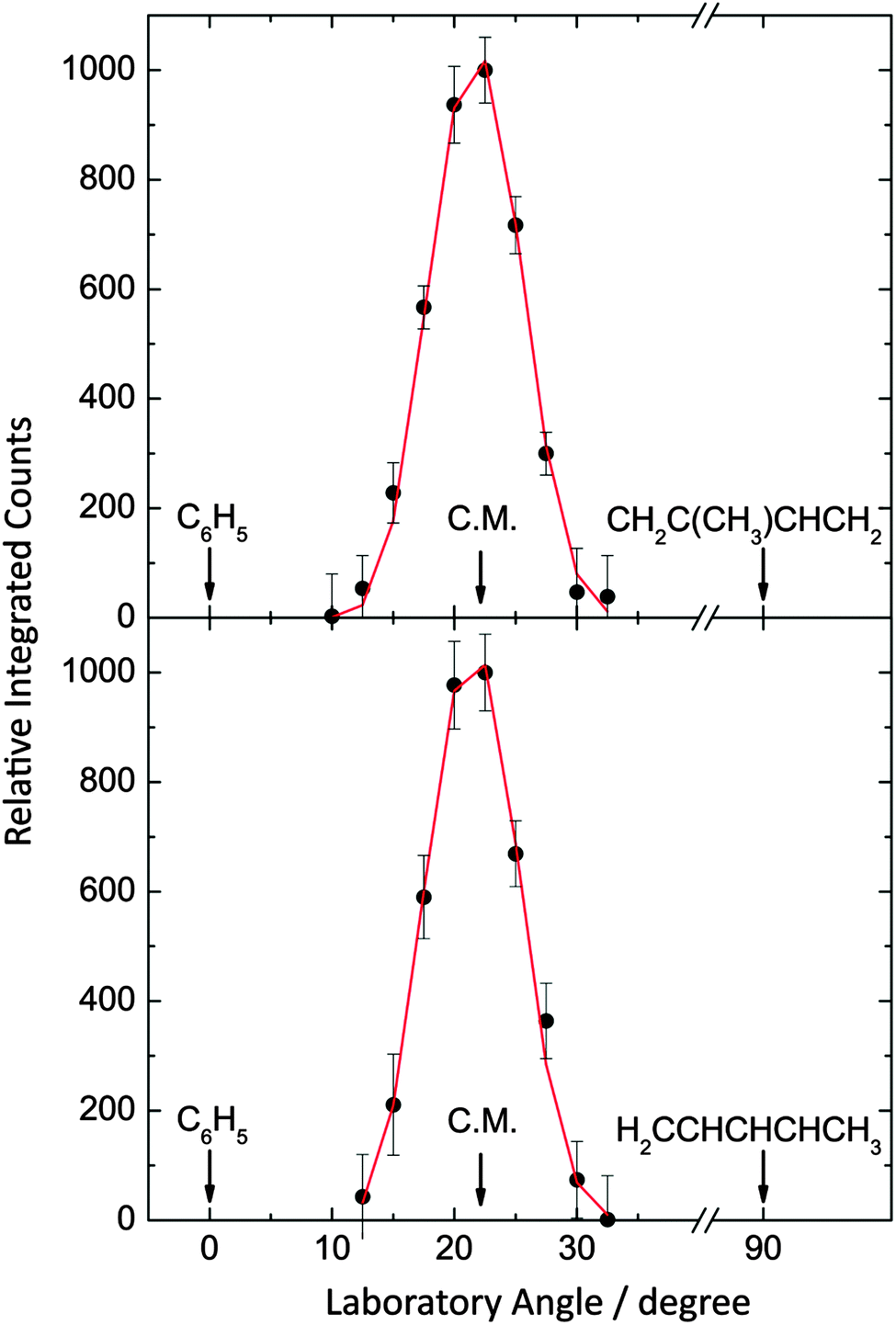
| | Fig. 3 Laboratory angular distributions at a mass-to-charge ratio (m/z) of 144 (C11H12+) for the reactions of phenyl radicals with isoprene (top) and with 1,3-pentadiene (bottom). The circles present the data points, while the solid lines represent the fits obtained from the forward-convolution routine. | |
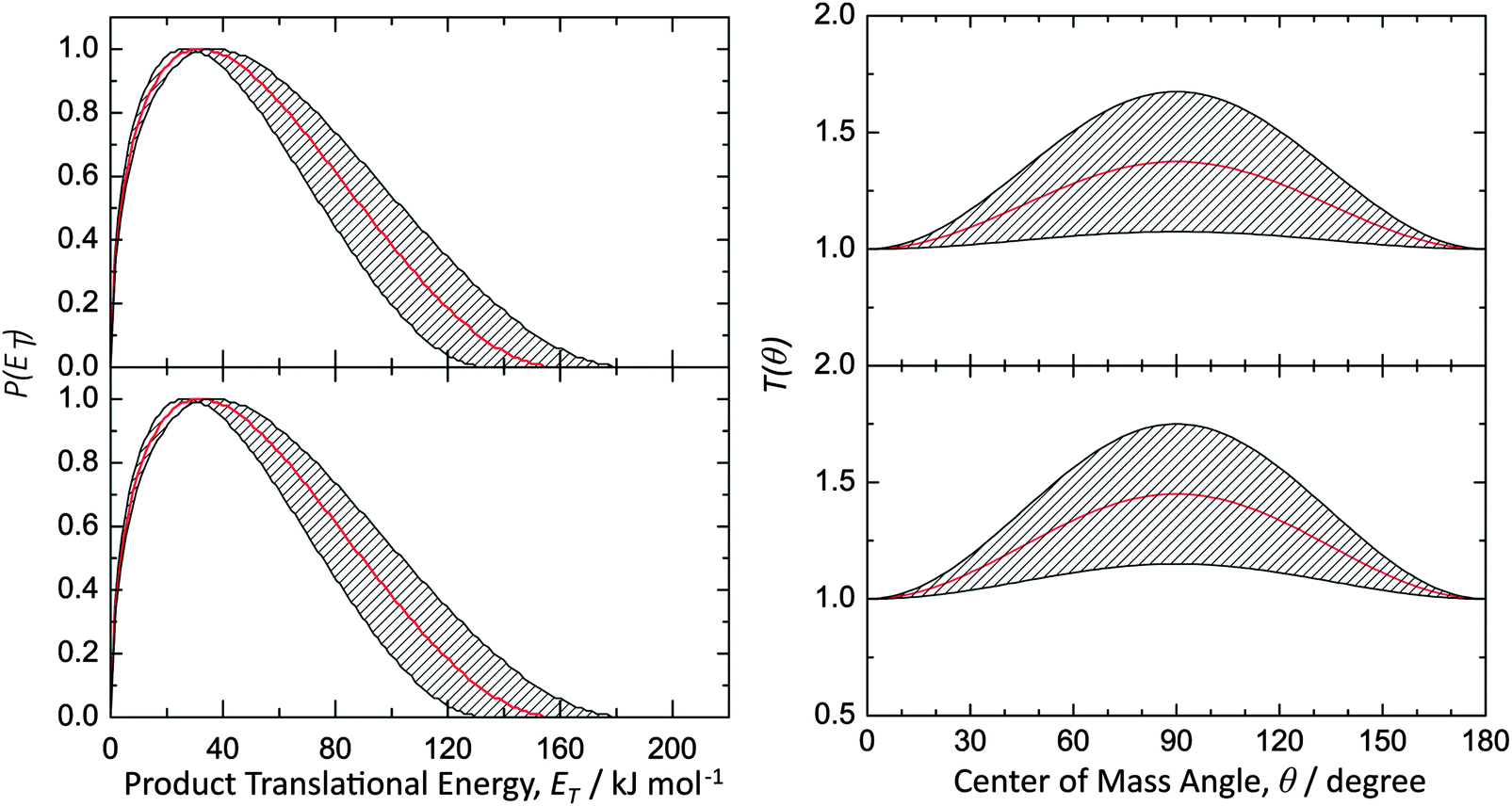
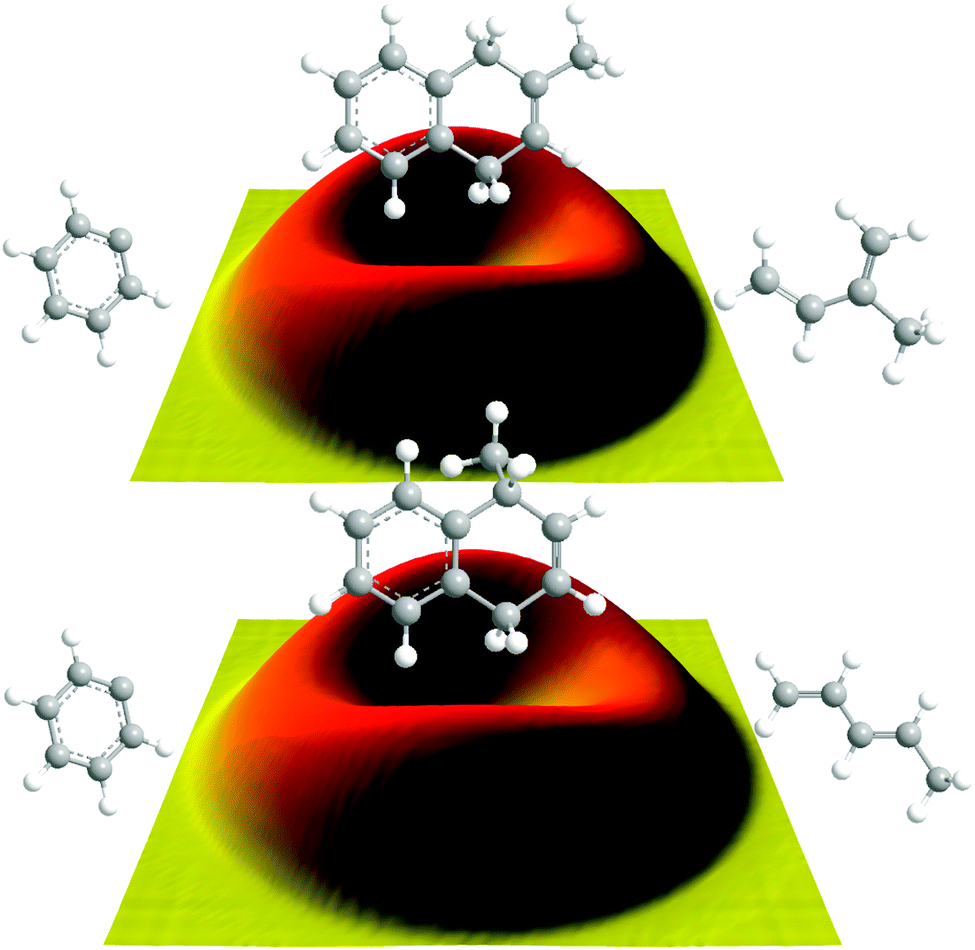
Data at m/z = 144 (C11H12+) could be fit with a single channel accounting for reactive scattering signal at 144 amu (C11H12) plus 1 amu (H) via reaction of the phenyl radical (C6H5; 77 amu) with isoprene (C5H8; 68 amu) and with 1,3-pentadiene (C5H8; 68 amu), respectively. As a matter of fact, the resulting center-of-mass angular and translational energy distribution are very similar (Fig. 4). In detail, the center-of-mass translational energy distributions, P(ET)s, depict maximum translational energy releases of 155 ± 24 kJ mol−1. A subtraction of the collision energy of 55 ± 4 kJ mol−1 from the maximum translational energy release, yields a reaction energy of 100 ± 28 kJ mol−1 for those products formed without internal excitation. Further, the P(ET)s peak distinctively away from zero translational energies at 25 to 40 kJ mol−1; this finding suggests the existence of an exit barrier of this order of magnitude and a tight exit transition state to product formation.61 Here, large exit barriers are often associated with repulsive carbon–hydrogen bond ruptures involving a significant electron rearrangement from the decomposing intermediate to the final products. Considering the concept of microscopic reversibility, in the reversed reaction of a hydrogen atom addition to a closed shell hydrocarbon, we would expect an entrance energy barrier.61 Finally, the average fraction of the available energy channeling into the translational degrees of freedom of the products is derived to be about 54 ± 8%. Note that both center-of-mass angular distributions, T(θ)s, depict intensity over the full angular range indicating an indirect complex formation reaction mechanism forming a bound C11H13 intermediate(s).61 Best fits are achieved with distributions showing distribution maxima at around 90°. These forward–backward symmetric distributions indicate that the life time(s) of the decomposing complex(es) is (are) longer than their rotational period(s). Further, the pronounced maximum around 90° suggest that a hydrogen emission takes place almost parallel to the total angular momentum vector and nearly perpendicularly to the rotational plane of the decomposing complex(es) (so called ‘sideway scattering’),44 which can also be reflected in the flux contour maps (Fig. 5).
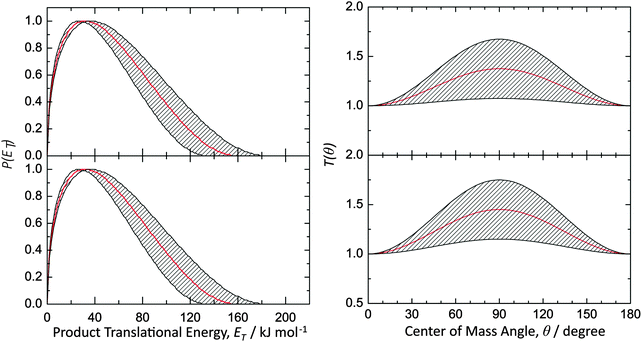 |
| | Fig. 4 Center-of-mass translational energy distributions P(ET)s (left) and angular distribution T(θ)s (right) for the reactions of the phenyl radical with isoprene (top) and with 1,3-pentadiene (bottom) forming C11H12 product isomer(s) via atomic hydrogen emission. The hatched areas show the experimental error limits. | |
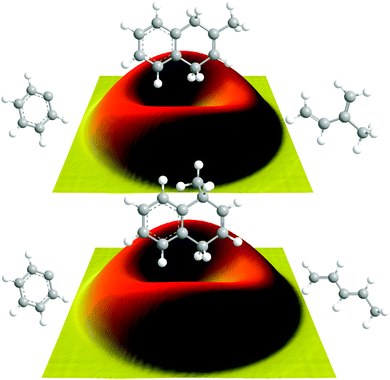 |
| | Fig. 5 Flux contour maps for the reaction of phenyl radicals with isoprene (top) and with 1,3-pentadiene (bottom), respectively. | |
5. Theoretical results
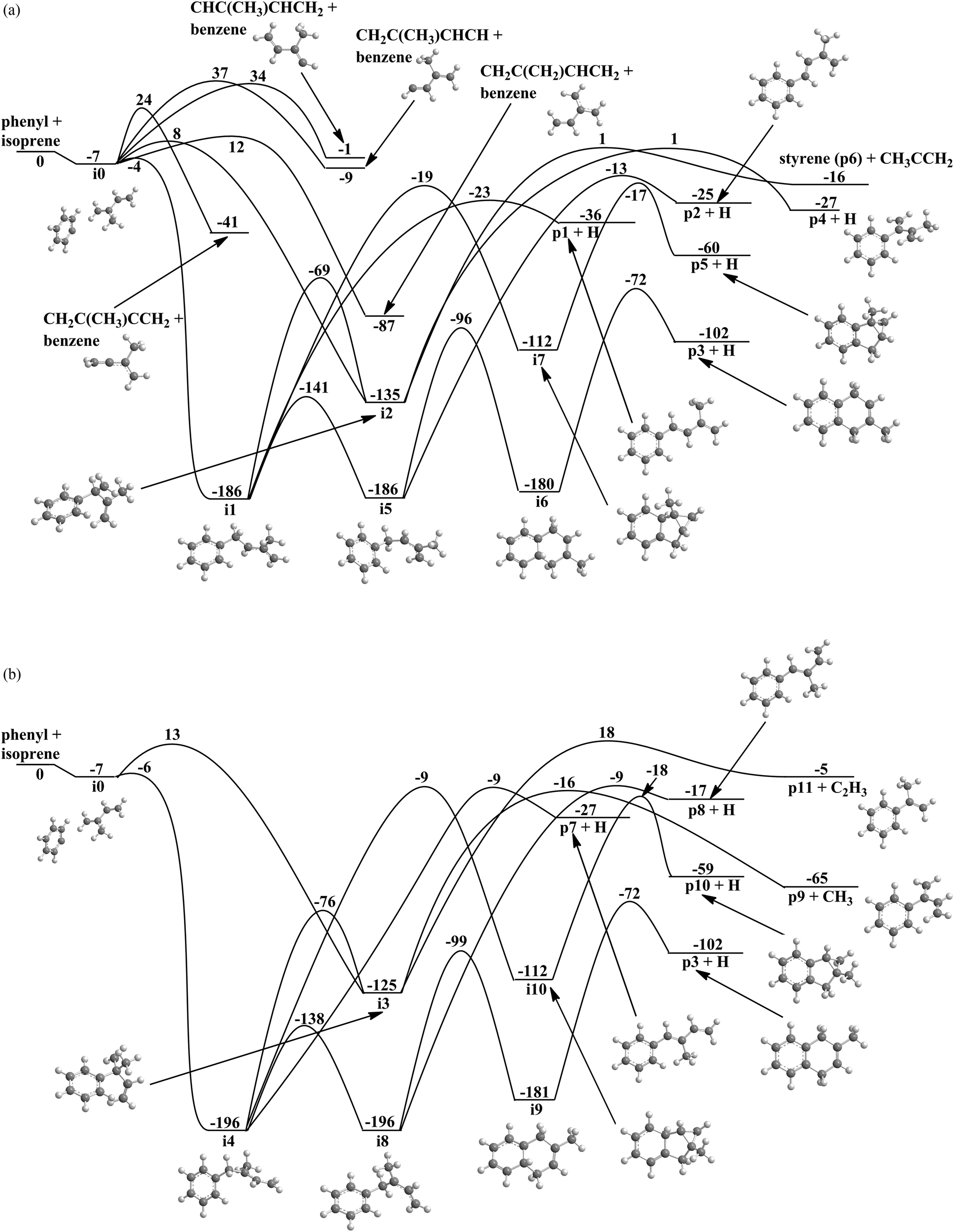
Considering the reaction of the phenyl radical with isoprene (Fig. 6a and b), the ab initio calculations identified 10 intermediates and 15 possible products. The reaction is initiated by the barrier-less formation of a van-der-Waals complex (i0), which is stabilized by 7 kJ mol−1 with respect to the reactants. From here, phenyl can either add to the π electron density or abstract a hydrogen atom from the isoprene molecule. Here, the chemically non-equivalency of the hydrogen atoms translate to four feasible abstraction channels from the C1, C3, C4, as well as CH3 group in overall exoergic reactions (−1 to −87 kJ mol−1) by passing transition states located 12 to 37 kJ mol−1 above the separated reactants. Since the isoprene molecule has four non-equivalent sp2 hybridized carbon atoms at the 1,3-butadiene moiety, four entrance channels to addition exist leading to four distinct intermediates (i1–i4). Note that although all addition pathways have entrance barriers ranging between 1 and 20 kJ mol−1, the additions to the terminal C1 and C4 carbon atoms of the 1,3-butadiene moiety leading to resonantly stabilized free radical intermediates i1 and i4 are de facto barrier-less since these barriers to addition are actually lower than the energy of the separated reactants (submerged barrier). The formation of intermediates i2 and i3 on the other hand exhibit entrance barriers of 8 and 13 kJ mol−1 above the separated reactants. What is the fate if the initial collision complexes? Intermediate i1 can follow a cis–trans isomerization to i5via a low barrier of 45 kJ mol−1, isomerizes to i2, rearranges to an exotic tricyclic intermediate i7via a substantial barrier of 173 kJ mol−1, or decomposes to p1 through an atomic hydrogen emission via a rather loose transition state located only 13 kJ mol−1 above the separated products. Considering the barrier heights, the isomerization to i5 is expected to be favorable. This intermediate either undergoes ring closure to i6 or decomposes via atomic hydrogen emission from the C1 carbon atom of the isoprene molecule to p2. On the other hand, intermediate i6 fragments via hydrogen atom emission to form p3 (2-methyl-1,4-dihydronaphthalene) via an exit barrier of 30 kJ mol−1. What are the fates of i2 and i7? Intermediate i2 either decomposes to p6 (styrene) plus CH3CCH2 radical or p4 plus a hydrogen atom via exit barriers of 17 kJ mol−1 and 28 kJ mol−1, respectively. Intermediate i7 – although unlikely to be formed due to the significant barrier connecting i1 and i7, could fragment to the tricyclic product p5 plus an atomic hydrogen via an exit barrier of 43 kJ mol−1. We are focusing now on the fate of the collision complexes i3 and i4. Intermediate i4 has four possible reaction paths: isomerization to i3,cis–trans isomerization to i8, isomerization to the tricyclic intermediate i10, or decomposition to p7via atomic hydrogen emission with an exit barrier of 18 kJ mol−1. Note that intermediate i3 can lose a methyl group to form p9 or emits a vinyl group yielding p11via an exit barrier of 23 kJ mol−1. Intermediate i8 either isomerizes to the bicyclic intermediate i9, which eventually decomposes via an atomic hydrogen loss from the neighboring carbon atom with an overall exoergicity of 102 kJ mol−1 by overcoming an exit barrier of 30 kJ mol−1; alternatively, i4 decomposes to p8 plus an atomic hydrogen via an exit barrier of 11 kJ mol−1. Note that the tricyclic intermediate i10 can emit a hydrogen atom from the C4 carbon atom of the isoprene moiety and forms the tricyclic product p10via an exit barrier of 41 kJ mol−1.
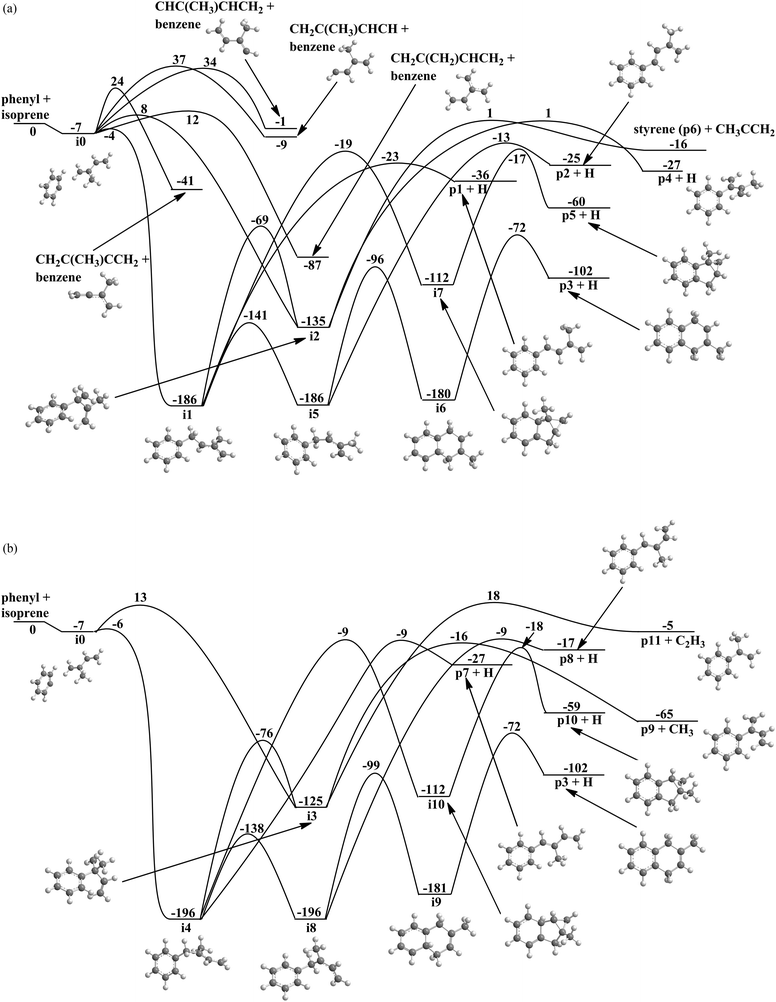 |
| | Fig. 6 (a) Potential energy surface for the reaction of the phenyl radical with isoprene via addition to the C1 and C2 carbon atoms. All energies are given in kJ mol−1. (b) Potential energy surface for the reaction of the phenyl radical with isoprene via addition to the C3 and C4 carbon atoms. All energies are given in kJ mol−1. | |
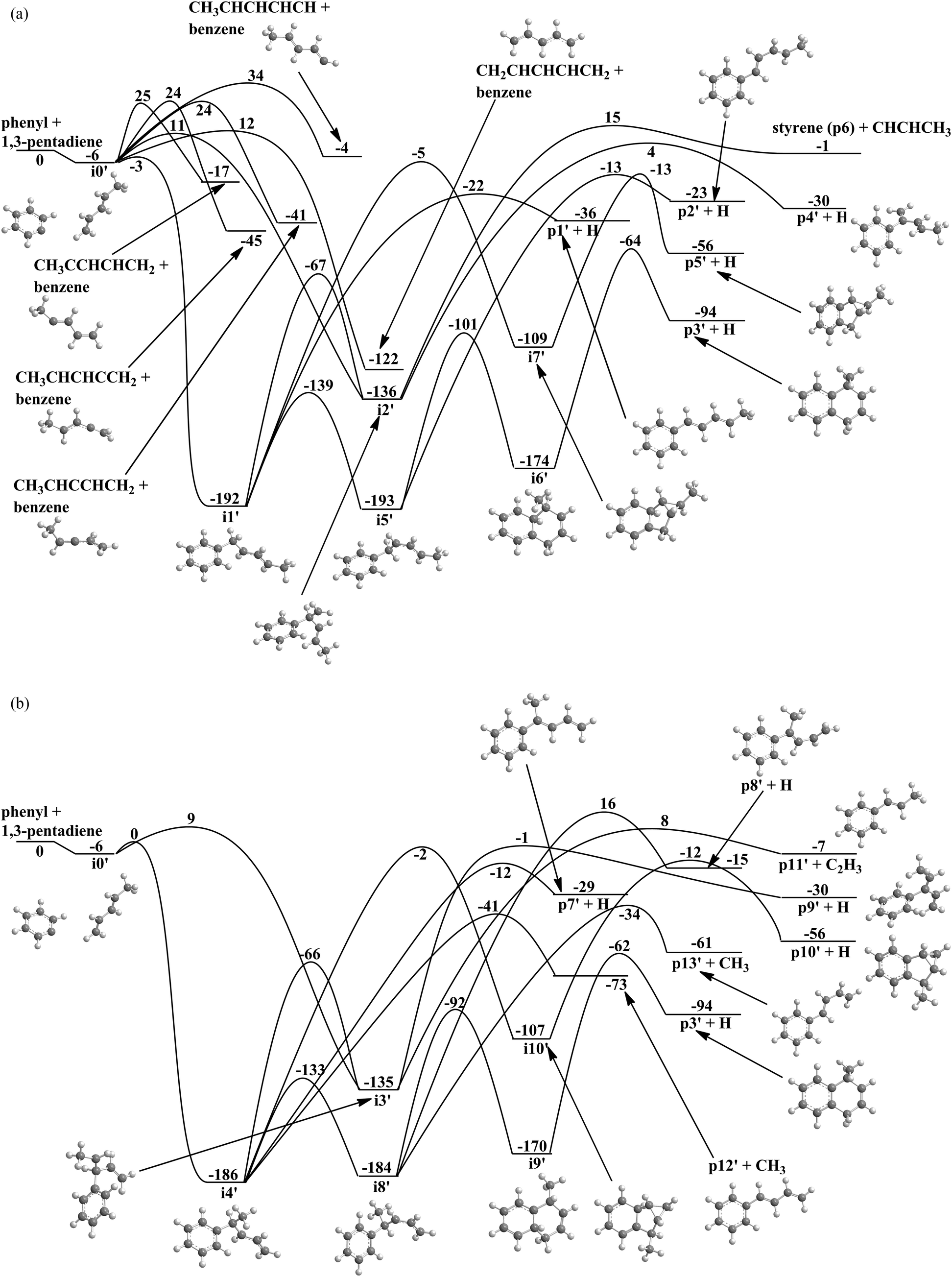
Having unraveled the reaction of phenyl with isoprene, we are shifting now to the related system of phenyl with 1,3-pentadiene. As an isomer of isoprene, 1,3-pentadiene also holds a pair of conjugated carbon–carbon double bonds, while methyl group substitution occurs at the C4 carbon atom of 1,3-butadiene to cis-/trans-1,3-pentadiene rather than at C3 carbon atom for isoprene. Therefore, we project that the PES for the reaction of phenyl with cis-/trans-1,3-pentadiene (Fig. 7a and b) should hold strong similarities compared to the PES of the phenyl–isoprene system (Fig. 6a and b). Here, the ab initio calculations propose 10 intermediates and 18 possible reaction products. Similar to the reaction of phenyl with isoprene, the initial long range interaction between the phenyl radical and 1,3-pentadiene results in the barrier-less formation of a van der Waals complex i0′, which resides 6 kJ mol−1 below the energy of the separated reactants. From here, the phenyl radical can abstract a hydrogen atom by overcoming barriers between 12 and 34 kJ mol−1. The reaction products formed are benzene plus CH3CHCHCHCH, CH3CCHCHCH2, CH3CHCCHCH2, CH3CCHCHCH2, and CH2CHCHCHCH2. Alternatively, the van-der-Waals complex can isomerize via addition of the phenyl radical with its radical center to the C1 to C4 carbon atoms of the 1,3-butadiene moiety leading to the initial collision complexes i1′ to i4′. Similarly to the phenyl–isoprene system, the formation of i1′ and i4′ are de facto barrier-less and proceed via submerged barriers, while i2′ and i3′ are formed through entrance barriers of 11 kJ mol−1 and 9 kJ mol−1, respectively. Thereafter, i1′ can decompose to p1′ plus hydrogen via an exit barrier of 14 kJ mol−1 or isomerize to i2′, i5′ or i7′via barriers of 125 kJ mol−1, 53 kJ mol−1 and 187 kJ mol−1 respectively. Intermediate i2′ will then decompose to either styrene (p6′) plus CHCHCH3 or forms p4′ plus hydrogen. The tricyclic intermediate i7′ can eject an atomic hydrogen from the C2 carbon atom of the phenyl moiety resulting in the formation of the tricyclic product p5′. Intermediate i5′ may isomerize to a bicyclic intermediate i6′ or decomposes to p2′via an exit barrier of 10 kJ mol−1. After that, i6′ emits a hydrogen atom from the C2 carbon atom of the phenyl moiety to generate p3′ (1-methyl-1,4-dihydronaphthalene) via an exit barrier of 30 kJ mol−1 and an overall exoergicity of 94 kJ mol−1. Considering i3′, the latter can decompose to p9′ plus an atomic hydrogen or to p11′ plus a vinyl group via exit barriers of 29 kJ mol−1 and 15 kJ mol−1, respectively. Intermediate i4′ can either decompose to p7′ plus hydrogen or p12′ plus a methyl group via exit barriers of 41 kJ mol−1 and 32 kJ mol−1, or isomerizes to the i3′, i8′, and i10′via barriers of 130 kJ mol−1, 53 kJ mol−1 and 184 kJ mol−1 respectively. The tricyclic intermediate i10′ might form the triyclic product p10′ plus hydrogen by overcoming an exit barrier of 44 kJ mol−1. Intermediate i8′ will either decompose to p8′ plus hydrogen, p13′ plus a methyl group, or undergoes ring-closure to i9′, which eventually decomposes to p3′ plus hydrogen via an exit barrier of 32 kJ mol−1.
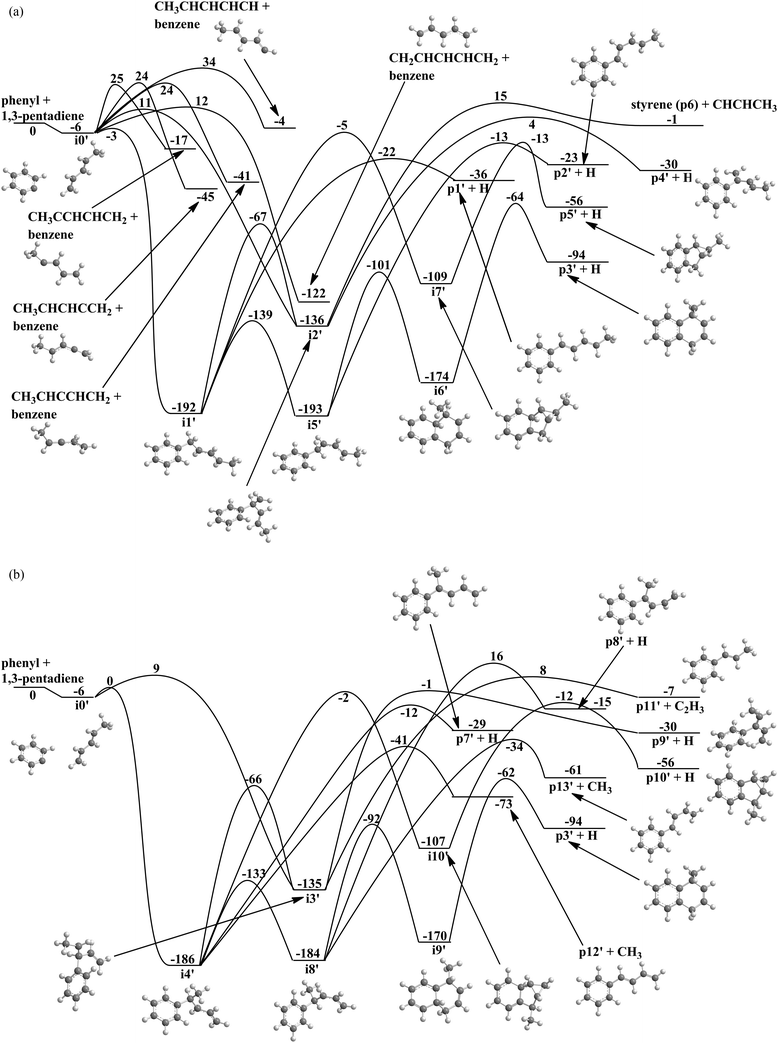 |
| | Fig. 7 (a) Potential energy surface for the reaction of the phenyl radical with 1,3-pentadiene via addition to the C1 and C2 carbon atoms. All energies are given in kJ mol−1. (b) Potential energy surface for the reaction of the phenyl radical with 1,3-pentadiene via addition to the C3 and C4 carbon atoms. All energies are given in kJ mol−1. | |
6. Discussion
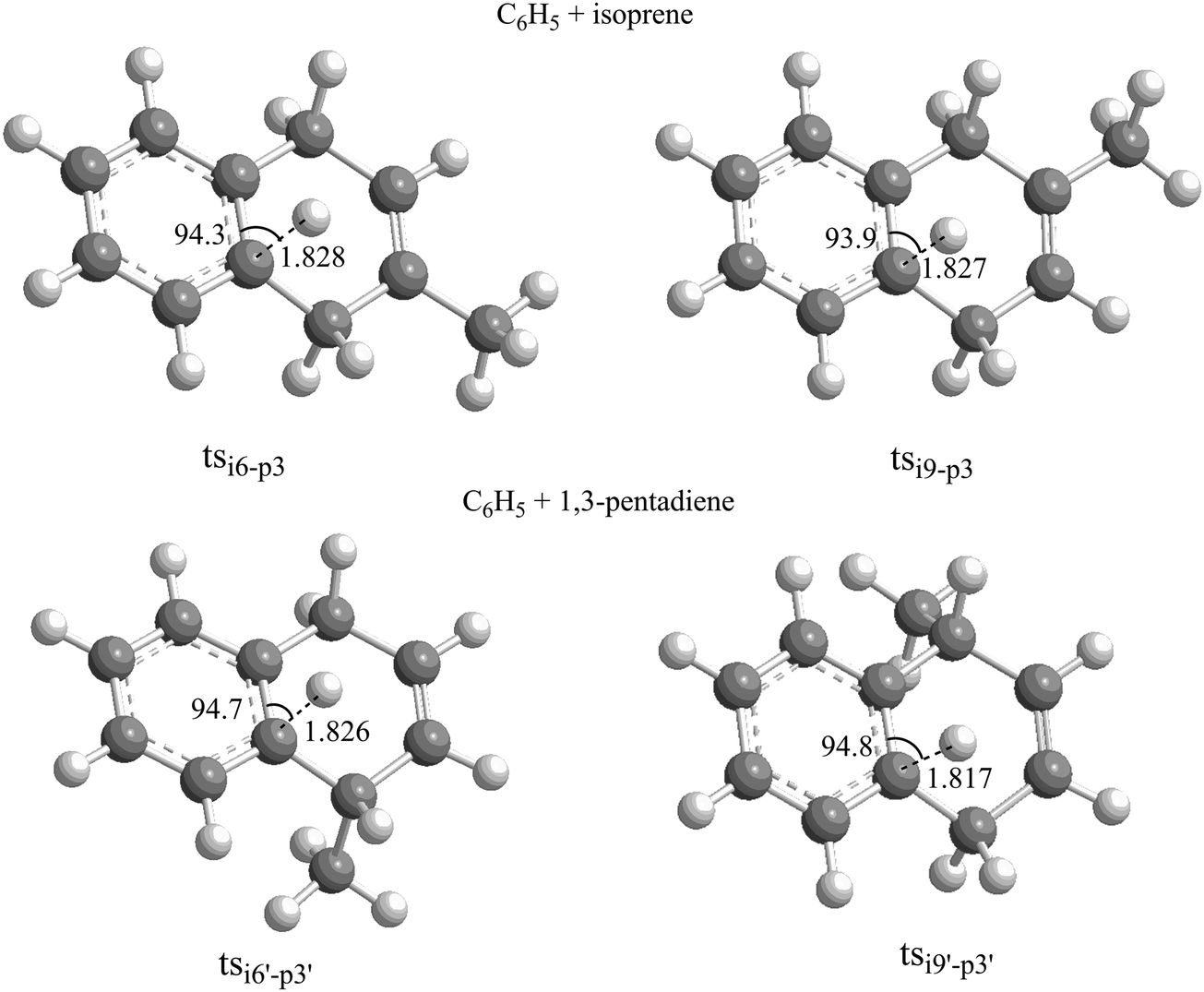
We are now combining our experimental and computational results in an attempt to untangle the underlying reaction dynamics. A comparison of the experimentally derived reaction energies for both systems of 100 ± 28 kJ mol−1 with the computed data (Fig. 6 and 7) suggests at least the formation of p3 (phenyl–isoprene) and p3′ (phenyl–1,3-butadiene) in reactions exoergic by 94 ± 5 kJ mol−1 and 102 ± 5 kJ mol−1, respectively. How are these products formed? For both the phenyl–isoprene and phenyl–1,3-pentadiene systems, the reactions follow indirect scattering dynamics via the involvement of C11H13 complexes. Upon formation of the van-der-Waals complexes i0 and i0′, these complexes isomerized via de facto barrier-less addition of the phenyl radical with its radical center to the C1 and C4 carbon atoms forming resonantly stabilized free radicals i1/i1′ and i4/i4′, respectively; all barriers to addition are below the energy of the separated reactants. The collision complexes undergo cis–trans isomerization (i5/i5′ and i8/i8′) with the latter isomerizing via ring closure yielding i6/i6′ and i9/i9′. These intermediates emit then a hydrogen atom from the ortho-position of the phenyl moiety through tight exit barriers located 30 kJ mol−1 above the separated reactants. Note that the computed barrier heights agrees very well with the estimated ones based on the off-zero peaking of the center-of-mass translational energy distributions of 25 to 40 kJ mol−1. Further, it is important to highlight that the electronic structure calculations predict angles of the hydrogen emission from 93.9° to 94.8° (Fig. 8), which correlates nicely with the experimentally found ‘sideways scattering’. In other words, in the reversed reaction, the hydrogen atom adds perpendicularly to the molecular plane of the 2- and 1-methyl-1,4-dihydronaphthalene molecule. It is important to recall that at our collision energy, the phenyl radical can also add to the C2 and C3 carbon atoms of isoprene and 1,3-pentadiene by passing the barriers to addition. The resulting complexes i2/i2′ and i3/i3′ can isomerize to i1/i1′ and i4/i4′, respectively. These conclusions also gain full support from our RRKM calculations depicting that 2- and 1-methyl-1,4-dihydronaphthalene are the dominating reaction products formed at levels of 97% and 80% in the reactions of the phenyl radical with isoprene and 1,3-pentadiene, respectively (Table 2).
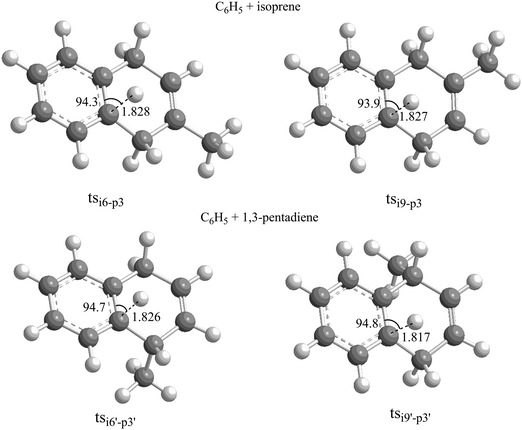 |
| | Fig. 8 Geometries of the decomposing complexes tsi6–p3, tsi9–p3, tsi6′–p3′ and tsi9′–p3′ leading to methyl-substituted 1,4-dihydronaphthalene isomers via the reactions of phenyl radicals with isoprene and 1,3-pentadiene. | |
Table 2 Calculated product branching ratios (%) in the reaction of phenyl radical (C6H5) with C5H8 isoprene (a) and 1,3-pentadiene (b)
| Products |
Collision energy/kJ mol−1 |
| 0 |
10 |
20 |
30 |
40 |
50 |
55 |
60 |
| (a) |
|
p1
|
0.00 |
0.00 |
0.01 |
0.02 |
0.05 |
0.10 |
0.13 |
0.17 |
|
p2
|
0.00 |
0.00 |
0.00 |
0.00 |
0.01 |
0.02 |
0.03 |
0.04 |
|
p3
|
99.70 |
99.68 |
99.42 |
98.98 |
98.40 |
97.69 |
97.29 |
96.86 |
|
p4
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
p5
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
p6
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
p7
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.01 |
0.01 |
|
p8
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.01 |
0.02 |
0.02 |
|
p9
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.01 |
|
p10
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
p11
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
| CH2C(CH3)CCH2 |
0.01 |
0.01 |
0.01 |
0.02 |
0.04 |
0.06 |
0.08 |
0.10 |
| CH2CHC(CH3)CH |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.01 |
0.01 |
| CH2C(CH3)CHCH |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
| CH2C(CH2)CHCH2 |
0.29 |
0.31 |
0.56 |
0.97 |
1.50 |
2.11 |
2.44 |
2.79 |
|
|
| (b) |
|
p1′
|
0.05 |
0.20 |
0.55 |
1.22 |
2.31 |
3.97 |
4.92 |
6.06 |
|
p2′
|
0.00 |
0.01 |
0.04 |
0.12 |
0.28 |
0.58 |
0.79 |
1.04 |
|
p3′
|
98.54 |
96.66 |
93.88 |
90.49 |
86.63 |
82.12 |
79.74 |
77.12 |
|
p4′
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
p5′
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
p6′
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
p7′
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
p8′
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
p9′
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
p10′
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
p11′
|
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
|
p12′
|
0.49 |
2.31 |
3.64 |
4.66 |
5.36 |
5.88 |
6.06 |
6.22 |
|
p13′
|
0.06 |
0.35 |
0.67 |
0.99 |
1.31 |
1.60 |
1.73 |
1.86 |
| CH3CHCHCCH2 |
0.08 |
0.03 |
0.03 |
0.04 |
0.07 |
0.11 |
0.14 |
0.16 |
| CH3CHCCHCH2 |
0.13 |
0.04 |
0.05 |
0.07 |
0.11 |
0.16 |
0.20 |
0.24 |
| CH3CCHCHCH2 |
0.15 |
0.05 |
0.06 |
0.08 |
0.13 |
0.20 |
0.25 |
0.30 |
| CH3CHCHCHCH |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
0.01 |
0.01 |
0.01 |
| CH2CHCHCHCH2 |
0.50 |
0.36 |
1.09 |
2.33 |
3.80 |
5.36 |
6.16 |
6.96 |
It is also appealing to compare the present results with those of the phenyl–1,3-butadiene system studied earlier in our group.44 Here, the approach of the phenyl radical to 1,3-butadiene toward the terminal C1 atom is attractive until a van-der-Waals complex is formed, which is stabilized by 25 kJ mol−1 with respect to the separated reactants. As the carbon–carbon distance continues to decrease, the system proceeds via a submerged barrier located 3 kJ mol−1 above the complex, but 22 kJ mol−1 below the reactants. Hence, the overall reaction from phenyl plus 1,3-butadiene to form a resonantly stabilized free radical (RSFR) intermediate C6H5H2CCHCHCH2 is de facto barrier-less. This intermediate undergoes a cis–trans isomerization followed by ring closure to a bicyclic intermediate, which eventually ejects a hydrogen atom to form the 1,4-dihydronaphthalene product via a tight exit transition state located 31 kJ mol−1 above the separated products. Also, the phenyl radical can attack through a barrier of 12 kJ mol−1 at the C2/C3 carbon atom of 1,3-butadiene; this intermediate can isomerize via phenyl group shift to C6H5H2CCHCHCH2. It is important to note that in this system, the ‘sideways scattering’ as verified by a pronounced maximum of the center-of-mass angular distribution at 90° indicates geometrical constraints upon decomposition of the bicyclic intermediate, i.e. an emission of the hydrogen atom perpendicularly to the rotation plane of the fragmenting intermediate. Finally, the product branching ratios computed at collision energies lower than 55 kJ mol−1 reveal that 1,4-dihydronaphthalene clearly dominates the product distribution giving up to about 90% of the total yield.
7. Conclusion
We investigated the crossed molecular reactions on the phenyl radical with isoprene and with 1,3-pentadiene at a collision energy of 55 ± 4 kJ mol−1. Both reactions are dictated by the indirect scattering dynamics and involve the formation of a van-der-Waals complex in the entrance channel. The latter was found to isomerize via addition of the phenyl radical to the terminal C1/C4 carbon atoms through submerged barriers forming resonantly stabilized free radicals, which then undergo cis–trans isomerization followed by ring closure. These bicyclic intermediates undergo unimolecular decomposition via an atomic hydrogen loss through tight exit transition states in overall exoergic reactions forming 2- and 1-methyl-1,4-dihydronaphthalene isomers. It is important to note that the hydrogen atoms are emitted almost perpendicularly to the plane of the decomposing complex and almost parallel to the total angular momentum vector (‘sideways scattering’) which is in strong analogy to the phenyl–1,3-butadiene system studied earlier. Electronic structures and RRKM calculations confirm that 2- and 1-methyl-1,4-dihydronaphthalene are the dominating reaction products formed at levels of 97% and 80% in the reactions of the phenyl radical with isoprene and 1,3-pentadiene, respectively. The barrier-less formation of the methyl-substituted, hydrogenated PAH molecules further supports our understanding of the formation of aromatic molecules in extreme environments holding temperatures as low as 10 K.
Acknowledgements
We acknowledge the support from the US Department of Energy, Basic Energy Sciences, via the grants DE-FG02-03ER15411 (Hawaii) and DE-FG02-04ER15570 (FIU). A. M. M. would like to acknowledge the Instructional & Research Computing Center (IRCC, web: http://ircc.fiu.edu) at Florida International University for providing HPC computing resources that have contributed to the research results reported within this paper.
References
- I. C. Nisbet and P. K. LaGoy, Regul. Toxicol. Pharmacol., 1992, 16, 290–300 CrossRef CAS PubMed.
- P. P. Fu, F. A. Beland and S. K. Yang, Carcinogenesis, 1980, 1, 725–727 CrossRef CAS PubMed.
- W. F. Busby, E. K. Stevens, E. R. Kellenbach, J. Cornelisse and J. Lugtenburg, Carcinogenesis, 1988, 9, 741–746 CrossRef CAS PubMed.
- K. H. Kim, S. A. Jahan, E. Kabir and R. J. C. Brown, Environ. Int., 2013, 60, 71–80 CrossRef CAS PubMed.
- J. L. Durant, W. F. Busby, A. L. Lafleur, B. W. Penman and C. L. Crespi, Mutat. Res., Genet. Toxicol., 1996, 371, 123–157 CrossRef CAS.
- L. Keith and W. Telliard, Environ. Sci. Technol., 1979, 13, 416–423 CrossRef.
- S. Baek, R. Field, M. Goldstone, P. Kirk, J. Lester and R. Perry, Water, Air, Soil Pollut., 1991, 60, 279–300 CrossRef CAS.
- H. Richter and J. Howard, Prog. Energy Combust. Sci., 2000, 26, 565–608 CrossRef CAS.
- M. Frenklach, Phys. Chem. Chem. Phys., 2002, 4, 2028–2037 RSC.
- Z. Mansurov, Combust., Explos. Shock Waves (Engl. Transl.), 2005, 41, 727–744 CrossRef.
- M. Zheng, L. G. Salmon, J. J. Schauer, L. Zeng, C. Kiang, Y. Zhang and G. R. Cass, Atmos. Environ., 2005, 39, 3967–3976 CrossRef CAS.
- H. Guo, S. Lee, K. Ho, X. Wang and S. Zou, Atmos. Environ., 2003, 37, 5307–5317 CrossRef CAS.
- M. del Rosario Sienra, N. G. Rosazza and M. Préndez, Atmos. Res., 2005, 75, 267–281 CrossRef.
- K. J. Hylland, J. Toxicol. Environ. Health, Part A, 2006, 69, 109–123 CrossRef CAS PubMed.
- B. J. Finlayson-Pitts and J. N. Pitts Jr., Science, 1997, 276, 1045–1051 CrossRef CAS PubMed.
- J. H. Seinfeld and J. F. Pankow, Annu. Rev. Phys. Chem., 2003, 54, 121–140 CrossRef CAS PubMed.
- R. J. Andres, T. A. Boden, F. M. Breon, P. Ciais, S. Davis, D. Erickson, J. S. Gregg, A. Jacobson, G. Marland and J. Miller,
et al.
, Biogeosciences, 2012, 9, 1845–1871 CrossRef CAS.
- A. G. Tielens, Annu. Rev. Astron. Astrophys., 2008, 46, 289–337 CrossRef CAS.
- F. Salama, Proc. Int. Astron. Union, 2008, 4, 357–366 CrossRef.
- P. Schmitt-Kopplin, Z. Gabelica, R. D. Gougeon, A. Fekete, B. Kanawati, M. Harir, I. Gebefuegi, G. Eckel and N. Hertkorn, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2763–2768 CrossRef CAS PubMed.
- D. Beintema, M. Van den Ancker, F. Molster, L. Waters, A. Tielens, C. Waelkens, T. De Jong, T. de Graauw, K. Justtanont and I. Yamamura, Astron. Astrophys., 1996, 315, L369–L372 CAS.
- R. I. Kaiser, D. Stranges, H. M. Bevsek, Y. T. Lee and A. G. Suits, J. Chem. Phys., 1997, 106, 4945–4953 CrossRef CAS.
- F. Stahl, P. v. R. Schleyer, H. F. Schaefer III and R. I. Kaiser, Planet. Space Sci., 2002, 50, 685–692 CrossRef CAS.
- R. I. Kaiser, N. Balucani, D. O. Charkin and A. M. Mebel, Chem. Phys. Lett., 2003, 382, 112–119 CrossRef CAS.
- N. Balucani, A. M. Mebel, Y. T. Lee and R. I. Kaiser, J. Phys. Chem. A, 2001, 105, 9813–9818 CrossRef CAS.
- R. I. Kaiser, T. N. Le, T. L. Nguyen, A. M. Mebel, N. Balucani, Y. T. Lee, F. Stahl, P. v. R. Schleyer and H. F. Schaefer III, Faraday Discuss., 2002, 119, 51–66 RSC.
- R. I. Kaiser, I. Hahndorf, L. C. L. Huang, Y. T. Lee, H. F. Bettinger, P. v. R. Schleyer, H. F. Schaefer and P. R. Schreiner, J. Chem. Phys., 1999, 110, 6091–6094 CrossRef CAS.
- R. Kaiser, C. Chiong, O. Asvany, Y. Lee, F. Stahl, P. v. R. Schleyer and H. Schaefer III, J. Chem. Phys., 2001, 114, 3488–3496 CrossRef CAS.
- H. Wang and M. Frenklach, J. Phys. Chem., 1994, 98, 11465–11489 CrossRef CAS.
- V. V. Kislov, N. I. Islamova, A. M. Kolker, S. H. Lin and A. M. Mebel, J. Chem. Theory Comput., 2005, 1, 908–924 CrossRef CAS.
- D. Wang, A. Violi, D. H. Kim and J. A. Mullholland, J. Phys. Chem. A, 2006, 110, 4719–4725 CrossRef CAS PubMed.
- V. Kislov and A. Mebel, J. Phys. Chem. A, 2008, 112, 700–716 CrossRef CAS PubMed.
- N. Marinov, W. Pitz, C. Westbrook, M. Castaldi and S. Senkan, Combust. Sci. Technol., 1996, 116, 211–287 CrossRef.
- I. V. Tokmakov, J. Park and M. C. Lin, ChemPhysChem, 2005, 6, 2075–2085 CrossRef CAS PubMed.
- T. Yu and M. C. Lin, Combust. Flame, 1995, 100, 169–176 CrossRef CAS.
- J. Park, G. J. Nam, I. V. Tokmakov and M. C. Lin, J. Phys. Chem. A, 2006, 110, 8729–8735 CrossRef CAS PubMed.
- S. Fascella, C. Cavallotti, R. Rota and S. Carra, J. Phys. Chem. A, 2004, 108, 3829–3843 CrossRef CAS.
- J. Park, S. Burova, A. S. Rodgers and M. C. Lin, J. Phys. Chem. A, 1999, 103, 9036–9041 CrossRef CAS.
- M. Shukla, A. Susa, A. Miyoshi and M. Koshi, J. Phys. Chem. A, 2008, 112, 2362–2369 CrossRef PubMed.
- P. Lindstedt, L. Maurice and M. Meyer, Faraday Discuss., 2001, 119, 409–432 RSC.
- P. F. Britt, A. Buchanan and C. V. Owens Jr, Prepr. Pap. - Am. Chem. Soc., Div. Fuel Chem., 2004, 49, 868 CAS.
- D. S. N. Parker, F. T. Zhang, R. I. Kaiser, V. V. Kislov and A. M. Mebel, Chem. – Asian J., 2011, 6, 3035–3047 CrossRef CAS PubMed.
- D. S. N. Parker, F. T. Zhang, Y. S. Kim, R. I. Kaiser, A. Landera, V. V. Kislov, A. M. Mebel and A. G. G. M. Tielens, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 53–58 CrossRef CAS PubMed.
- R. I. Kaiser, D. S. N. Parker, F. Zhang, A. Landera, V. V. Kislov and A. M. Mebel, J. Phys. Chem. A, 2012, 116, 4248–4258 CrossRef CAS PubMed.
- D. S. Parker, B. B. Dangi, R. I. Kaiser, A. Jamal, M. N. Ryazantsev, K. Morokuma, A. Korte and W. Sander, J. Phys. Chem. A, 2014, 118, 2709–2718 CrossRef CAS PubMed.
- B. B. Dangi, T. Yang, R. I. Kaiser and A. M. Mebel, Phys. Chem. Chem. Phys., 2014, 16, 16805–16814 RSC.
- Y. Guo, X. B. Gu, E. Kawamura and R. I. Kaiser, Rev. Sci. Instrum., 2006, 77, 034701 CrossRef.
- X. B. Gu, Y. Guo, F. T. Zhang, A. M. Mebel and R. I. Kaiser, Faraday Discuss., 2006, 133, 245–275 RSC.
- R. I. Kaiser, P. Maksyutenko, C. Ennis, F. T. Zhang, X. B. Gu, S. P. Krishtal, A. M. Mebel, O. Kostko and M. Ahmed, Faraday Discuss., 2010, 147, 429–478 RSC.
- F. T. Zhang, S. Kim and R. I. Kaiser, Phys. Chem. Chem. Phys., 2009, 11, 4707–4714 RSC.
-
J. D. Bittner, PhD thesis, Massachusetts Institute of Technology, Cambridge, MA 02139, USA, 1981.
-
P. S. Weiss, PhD thesis, University of California at Berkeley, Berkeley, California 94720, USA, 1986.
- R. I. Kaiser, T. N. Le, T. L. Nguyen, A. M. Mebel, N. Balucani, Y. T. Lee, F. Stahl, P. V. Schleyer and H. F. Schaefer, Faraday Discuss., 2001, 119, 51–66 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- L. A. Curtiss, K. Raghavachari, P. C. Redfern, A. G. Baboul and J. A. Pople, Chem. Phys. Lett., 1999, 314, 101–107 CrossRef CAS.
-
M. Frisch, G. Trucks, H. B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian, Inc., Wallingford, CT, 2009.
-
H. Werner, P. Knowles, R. Lindh, F. Manby, M. Schütz, P. Celani and T. Korona, Version 2010.1, a package of ab initio programs, 2010 Search PubMed.
- V. V. Kislov, T. L. Nguyen, A. M. Mebel, S. H. Lin and S. C. Smith, J. Chem. Phys., 2004, 120, 7008–7017 CrossRef CAS PubMed.
- V. V. Kislov and A. M. Mebel, J. Phys. Chem. A, 2010, 114, 7682–7692 CrossRef CAS PubMed.
-
R. D. Levine, Molecular Reaction Dynamics, Cambridge University Press, Cambridge, UK, 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A table giving RRKM calculated energy-dependent rate constants (s−1) for unimolecular reaction steps in the reaction systems of phenyl with isoprene and 1,3-pentadiene at different collision energies. See DOI: 10.1039/c4cp04612a |
|
| This journal is © the Owner Societies 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.