
Interaction of gold nanoparticles mediated by captopril and S-nitrosocaptopril: the effect of manganese ions in mild acid medium
Emilia
Iglesias
*a and
Rafael
Prado-Gotor
b
aDepartamento de Química Física e E. Q. I. Facultade de Ciencias, Universidad de La Coruña, 15071-A Coruña, Spain. E-mail: emilia.iglesias@udc.es
bDepartamento de Química Física, Facultad de Química, Universidad de Sevilla, 41012–Sevilla, Spain
First published on 7th November 2014
Abstract
We report herein results regarding reactivity and assembly of citrate-capped gold nanoparticles (AuNPs) mediated by captopril (cap) and S-nitrosocaptopril (NOcap), two angiotensin converting enzyme inhibitors and antihypertensive agents. The results were compared with that of cysteine (Cys), a thiol-containing amino acid found in plasma. The interparticle interactions were characterized by monitoring the evolution of the surface plasmon resonance band using the spectrophotometric method. The original gold nanoparticles were efficiently modified by small amounts of Mn+2 ions, which are adsorbed onto the surface of 15.4 nm citrate-capped gold nanoparticles, giving rise to manganese–gold nanoparticles (Mn–AuNPs) that, in mild acid medium, have proved to be highly sensitive and a rapid colorimetric detection method for thiols. Depending on the concentration of the Mn+2 ions the aggregation of AuNPs can be rapidly induced. The kinetics of the assembly process has been studied. Good first-order kinetics has been observed, with the exception of captopril-mediated nanoparticle aggregation at low concentration of either cap or acid. The rate of Cys-mediated assembly of gold nanoparticles in aqueous 10 mM acetic acid is more than 20-times faster than pure AuNPs and concentrations of Cys as low as 34 nM can be detected in less than 40 min under conditions of stable Mn–AuNPs. Similar effects were observed with cap or NOcap. The assembly–disassembly reversibility is shown with cap and NOcap and depends highly on pH.
Introduction
Noble metallic nanoparticles, especially gold nanoparticles, have unique properties of enormous interest for biological and chemical sensing applications.1–5 The fact that AuNPs absorb and scatter light at their surface plasmon resonance (SPR) band makes nanometric gold a valuable optical probe for detection methods. The optical properties of AuNPs depend strongly on the surface chemistry and on the interparticle interactions.6 Gold nanoparticles form stable chemical bonds with S- and N-containing groups. Consequently, small organic ligands may be conveniently attached to the surface of AuNPs through their thiol or amine functionalities.Localized surface plasmon resonance peaks of gold nanoparticles are strongly governed by their size, shape and dielectric constant of the surrounding medium.3 The preparation of colloidal spherical AuNPs with diameters of 5–50 nm can be done from the reduction of HAuCl4 with sodium citrate in boiling water, according to the well-known Turkevich method.7,8 The procedure affords well-dispersed colloidal gold aqueous solutions of red colour, whereas surface plasmon coupling results in a colour change from red to blue at nanomolar concentrations.3,9,10 Encapsulation of AuNPs by adsorption or binding organic molecules, such as thiols, on the particle surface and subsequent cross-linking at the binding sites of the encapsulating shells are of crucial importance in determining the colour change, which is the basis of colorimetric methods that use SPR sensors.
Thiols are among the organic molecules most studied in this sense. Homocysteine (Hcys) rapidly adsorbs onto the AuNP surface via –S–Au bond formation by displacing the original citrate molecules that capped the gold surface; this process is followed by NP assembly and the interparticle reactivity is accelerated by the addition of electrolytes, as well as, by lowering the pH.11 Cysteine (Cys) also interacts with gold nanoparticles, but the SPR band evolution shows a much slower reaction rate than Hcys, ∼10 h being necessary to see the maximum change.12 The presence of 1 mM Cu+2 drastically enhances the rate of gold nanoparticle aggregation.13 Glutathione (GSH) is even less reactive than Hcys or Cys. The simultaneous presence of GSH and 40 mM sodium nitrate yields a quasi-stable state of AuNPs whose solution remains red, contrarily to that observed with other thiols, a fact that serves as a basis for colorimetric method of detection for GSH analysis with very low detection limit of 0.5 μM.14 Nevertheless, in organic solvents such as THF, the aggregation of gold organosols can be controlled using GSH, a pH sensitive biomolecule, as a molecular linker of the ‘parent’ gold nanoparticles.15 Penicillamine (PEN) and N-acetylpenicillamine (NAP) modify gold nanoparticles in a few seconds at very low pH (1.9 and 1.7, respectively). From isothermal titration calorimetry measurements the equilibrium constants for thiol binding to the gold surface have been determined to be of the order of 107 M−1, and nearly 40 or 60% of gold surface coverage by PEN or NAP, respectively, was estimated.16
Reeves et al.17 carried out a systematic and detailed study on the ability of small molecules that include thiol-, amine-, or carboxyl-functionalities, or their combinations, to bind and aggregate gold nanoparticles at variable pH. The main goal of this study is to present models for mechanisms of aggregation of colloidal gold solutions, including the possible reduction in the zeta potential that leads to nanoparticle instability and bridging linkers from one nanoparticle to another, or even by forming the so-called surface micelles.18 The interesting study of Reeves shows that the as-synthesized colloidal gold solutions have minimal absorption in the red spectral region despite considerable variation in the pH of the solution ranging from pH 3.1 to 11.2, and after incubation of as long as 24 h, which evidences the non-aggregated nanoparticle solution. The study also verifies that the protonation state of the carboxylic groups, in general, and citrates, in particular, plays a crucial role in driving the aggregation kinetics. Consequently, low (acidic) pH values accelerate the aggregation rate and, on the other hand, the replacement of adsorbed anionic citrates with molecules not containing carboxylate groups or bearing positively charged groups, such as amino acids, favours nanoparticle aggregation.
Our aim in this research work was to find suitable conditions for AuNP aggregation mediated by captopril (cap) and S-nitrosocaptopril (NOcap). Therefore, we carried out a systematic study of the interaction of cap and NOcap with citrate stabilized gold nanoparticles, and the results were compared with the well-studied Cys. Scheme 1 shows the corresponding molecular structures along with that of other thiols already studied and their pKa values.19–21 NOcap is a nitric oxide donor and NO is a biological transmitter that mediates vasodilation and angiogenesis.21,22 By coupling the exciting properties of AuNPs with paramagnetic properties of Mn+2 ions we present new sensors of improved response time in aqueous acetic acid medium. Consequently, the interactions of AuNPs with both acetic acid (AcH) and Mn+2 ions have been analysed. The experimental data verify the models of gold nanoparticle assembly proposed by Reeves. In this sense, the high efficiency of Mn+2 ions in reducing the zeta potential of citrate-capped gold nanoparticles strongly affects their stability even at neutral pH. Moreover, the tuned protonation of carboxyl citrate by the presence of small amounts of AcH accelerates the rate of nanoparticle assembly promoted by bridging linkers, such as Cys, cap or NOcap, from one nanoparticle to another.
 | ||
| Scheme 1 Molecular structure of model organothiols and the pKa values of the ionisable carboxyl-, thiol-, and amino-groups (notice that the ionisable amine group must be –NH3+). | ||
Experimental
Materials
Captopril, cysteine and other chemicals, such as hydrogen tetrachloroaurate trihydrate (HAuCl4·3H2O), sodium citrate, manganese(II) chloride, acetic acid, and sodium nitrite were used as received from Sigma-Aldrich Chemicals.S-Nitrosocaptopril was synthesized by mixing 300 μL of 20 mM NaNO2 with 800 μL of 7.5 mM cap and 1.9 mL of 6.7 mM acetic acid. The rapid NOcap formation is observed by the evolution of the absorption band at 332 nm.21 Solutions of NOcap were used within the next 4 or 5 hours; nevertheless, at low temperature they are stable for several days.
Gold nanoparticles were prepared following the standard procedures.7,8 Briefly, aqueous HAuCl4 was first heated to boiling followed by rapid addition of an excess of aqueous sodium citrate. Characterization of gold nanoparticles capped by citrate shells resulted in [AuNPs] = 2.82 nM of 15.4 nm diameter that show the maximum wavelength absorption at 520 nm. The Mn–AuNPs were prepared by diluting aliquots (30–200 μL) of 4.0 mM Mn+2 (MnCl2) into 1 mL final volume of citrate capped 0.85 nM AuNPs, resulting in final concentrations of Mn+2 varying between 0.12 and 0.8 mM.
Water was firstly deionised and subsequently distilled twice (the first distillation is over potassium permanganate).
Methods
The UV-visible (UV-vis) spectra and kinetics were recorded on a Kontron-Uvikon double beam spectrophotometer fitted with thermostated multicell holders. Spectra were collected over the 200–800 nm range. In a typical experiment, the stock solution of nanoparticles was diluted with distilled water to reach a final colloidal gold concentration of 0.85 nM. Quartz cuvettes of 1 cm path-length and 1.4 mL capacity were used. In studying the kinetics of the evolution of the surface plasmon resonance band of AuNPs, measurements of absorbance (A) versus time (t) were recorded and fitted to the first-order integrated rate equation, A = A∞ − (A0 − A∞)·exp(−k0t), unless otherwise indicated. The non-linear regression analysis of A–t data gives k0, A0, and A∞ as optimizable parameters, with k0 being the pseudo-first order rate constant and A, A0, and A∞ being the absorbance values at time t, zero, and at the end of the reaction respectively. All experiments were conducted at 25 °C.The pH was measured using a Crison 2001 pH-meter equipped with a GK2401B combined glass electrode and calibrated using commercial buffers of pH 4.01, 7.02, and 9.26 (Crison).
Transmission electron microscopy (TEM) was carried out using a JEOL JEM 1010 electron microscope operating at an acceleration voltage of 100 kV and equipped with a Mega View III camera controlled using Analysis software. Samples of TEM analysis were prepared by placing drops of gold colloidal solutions on a carbon-coated copper grid sample holder followed by evaporation in air at room temperature. The reported TEM images are representative of the entire grid sample.
The ζ-potential was determined using a Malver Zetasizer Nano-ZS90. Measurements were recorded at 25 °C as a function of Mn+2 ion concentration. The values of the ζ-potential were automatically calculated from electrophoretic mobility based on the Smoluchowski equation.
Results and discussion
1. Manganese–gold nanoparticles, Mn–AuNPs
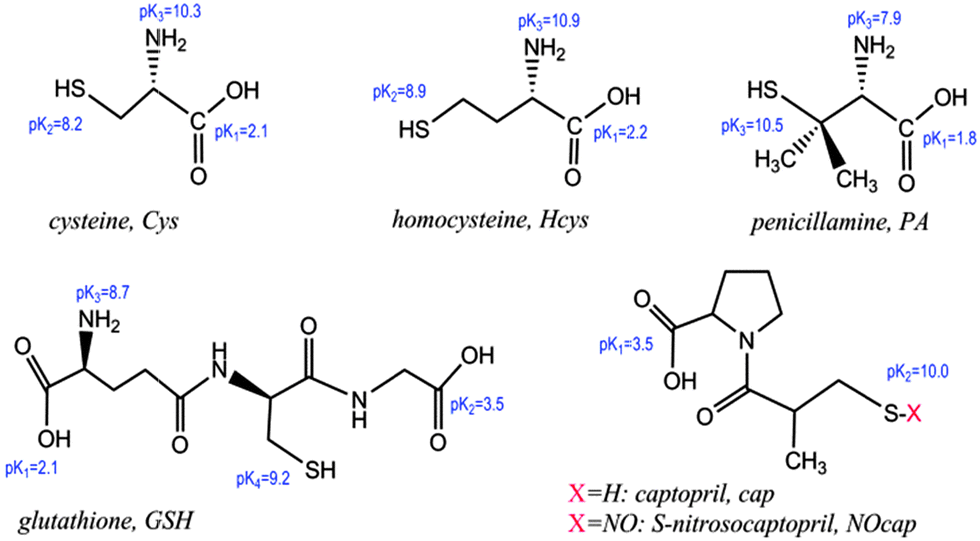
Fig. 1(a and b) show the UV-vis spectra comparing the SPR band evolution for the as-synthesized solutions of gold nanoparticles in 10 mM AcH upon the addition of 0.20 mM Mn+2 ions. Aqueous solution of AuNPs exhibits a characteristic absorption band at 520 nm that corresponds to the plasmon resonance band of well-dispersed spherical AuNPs. In 10 mM acetic acid the spectra remain practically unchanged (spectra recorded during 2 h, Fig. 1(a)), and the corresponding TEM image, shown in Fig. 1(c), does not indicate any aggregation effects.23 After the addition of Mn+2 the intensity of the SPR band goes down a bit while a new plasmon band (600–800 nm) appears, Fig. 1(b).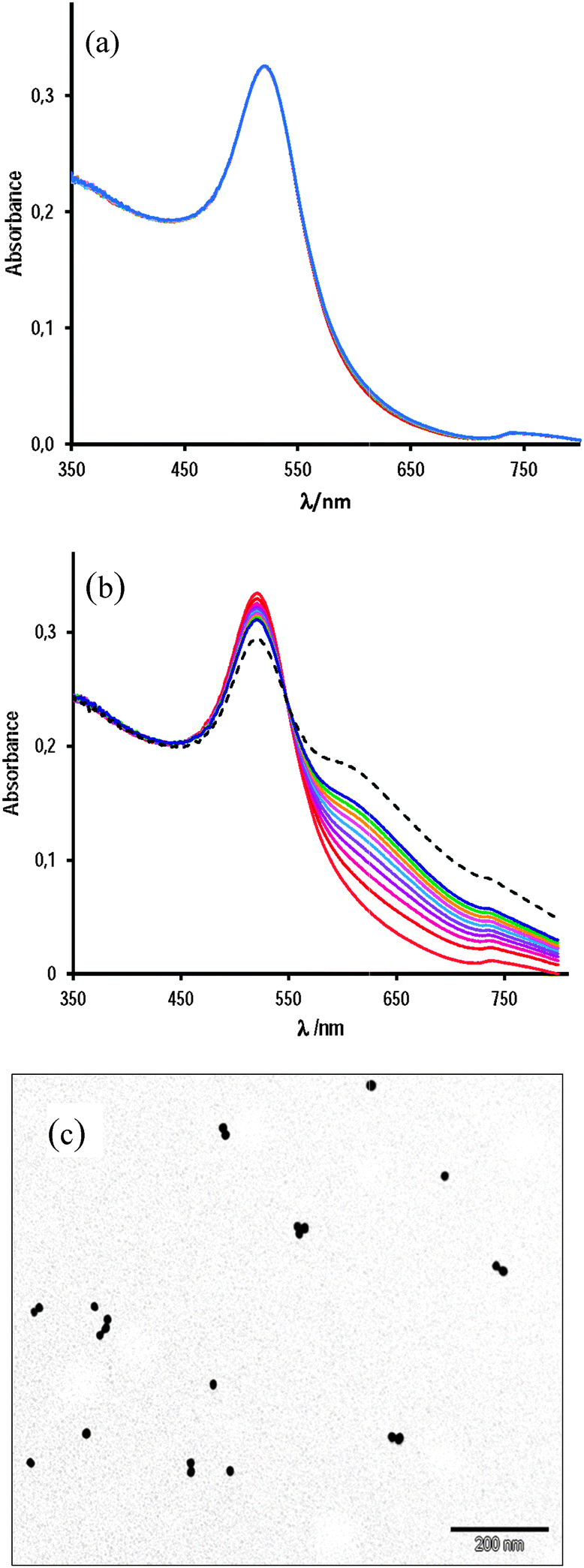 | ||
| Fig. 1 UV-vis spectra of AuNPs (0.85 nM) in the presence of 10 mM AcH at (a) [Mn+2] = 0 and (b) [Mn+2] = 0.20 mM; scans at 6 min interval during 120 min; (---) scan at 120 min; and (c) the TEM image of AuNPs under conditions of (a) after 90 min of mixing the reactants. | ||
In the absence of acetic acid, the spectrum of aqueous AuNPs recorded at 0.20 mM Mn+2 remains unchanged during at least 30 min; by doubling the Mn+2 concentration, a very slow increase of the absorbance in the 600–800 nm range is clearly observed, whereas at [Mn+2] = 0.80 mM the new plasmon band centered at ∼672 nm develops in less than 5 min, see Fig. 2.
 | ||
| Fig. 2 UV-vis spectra of aqueous solutions of AuNPs at (a) [Mn+2] = 0.20 mM, scans obtained every 2 min during 20 min; (b) [Mn+2] = 0.40 mM, scans at 3 min interval during 30 min, and (c) [Mn+2] = 0.81 mM, scans at 2.5 min interval during 25 min, scan 1 is due to gold nanoparticle solution in the absence of Mn+2 ions. | ||
Fig. 3(a) shows the comparative absorbance–time data for SPR band time evolution of AuNPs under three experimental conditions: (i) 10 mM AcH, in which no appreciable absorbance increase has been observed during 80 min; (ii) 10 mM AcH and 0.12 mM Mn+2, in which medium the nanoparticles seem to be stable also in, at least, 100 min, and (iii) 10 mM AcH containing 0.20 mM Mn+2, in which medium noticeable changes can be clearly observed. Fig. 3(b) shows the absorbance increase at 672 nm as a function of time for [Mn+2] equal to 0.81, 0.61, and 0.40 mM. At 0.81 mM, the blue colour of gold colloidal solutions develops in less than 5 min, whereas at half of this concentration, the colour change is much slower, being unappreciable even in 1 h, but the addition of 10 mM acetic acid strongly accelerates the process as it is shown in Fig. 3(c) by the corresponding TEM image that indicates complete aggregation in less than 40 min.
 | ||
| Fig. 3 Absorbance versus time for aqueous AuNPs in (a) (●) 10 mM AcH; (▼) 10 mM AcH and [Mn+2] = 0.12 mM, and (▲) 10 mM AcH and [Mn+2] = 0.20 mM; (b) (○) the uppermost curve [Mn+2] = 0.81 mM; (△) [Mn+2] = 0.61 mM, and (▽) the lowest curve [Mn+2] = 0.40 mM; and (c) the TEM photograph of AuNPs in 10 mM acetic acid and 0.40 mM Mn+2 (MnCl2) after 40 min of incubation. | ||
Data for absorbance (A) versus time (t) fit perfectly with the first-order integrated rate equation, and the observed rate constant, k0, of the self-assembly process was determined as a function of Mn+2 ion concentration. The results are listed in Table 1 and solid lines in Fig. 3(a) and (b) show the fit of the model to the experimental data.
| [AcH]a/mM | [Mn+2]/mM | λ max/nm | k 0/10−3 s−1 | ΔAb |
|---|---|---|---|---|
| a Acetic acid. b ΔA (=A∞ − A0). c In 100 min. | ||||
| — | 0.20 | 610 | Not measurable | ∼0 |
| 10 | — | 610 | Not measurable | ∼0 |
| 10 | 0.12 | 610 | Not measurable | 0.025c |
| 10 | 0.20 | 610 | 0.30 ± 0.01 | 0.186 |
| 10 | 0.40 | 610 | 3.60 ± 0.09 | 0.236 |
| 10 | 0.61 | 610 | 18.1 ± 0.2 | 0.317 |
| — | 0.40 | 672 | 0.180 ± 0.001 | 0.137 |
| — | 0.61 | 672 | 1.58 ± 0.01 | 0.300 |
| — | 0.80 | 672 | 5.60 ± 0.01 | 0.381 |
The analysis of these results indicates the negligible aggregation of gold nanoparticles in 10 mM AcH aqueous solutions in the absence or in the presence of 0.12 mM Mn+2 and in the timeframe of the observation. By contrast, working with double the [Mn+2] concentration, a new absorption band at longer wavelengths evolves with time due to nanoparticle assembly. The extension of aggregation increases with [Mn+2]; a fact which is further corroborated by the effect of [Mn+2] on the absorbance readings at 672 nm at infinite time, see the last column in Table 1.
An estimation of the variation of k0 with [Mn+2] leads to an exponential increase of the rate of the gold nanoparticle assembling process according to eqn (1), i.e. proportional to the fourth-degree of [Mn+2].
| k0 = k[Mn+2]4 | (1) |
This fact suggests a large efficiency of Mn+2 ions in the interaction with AuNPs to promote self-assembly. The rate constant of the assembly process was determined to be k = (1.31 ± 0.01) × 1011 M−4 s−1 in the presence of 10 mM acetic acid and k = (1.34 ± 0.04) × 1010 M−4 s−1 in the absence of acetic acid. The effect of acetic acid, in the sense of modulating the protonation of citrate, explains these results because lowering the net negative charge of NPs leads to a compression of the electrical double layer that surrounds AuNPs, which increases the self-assembly rate.
In aqueous solution the highest value determined for the rate constant of a diffusion-controlled process corresponds to the neutralization of H+ and OH−, kn = 1.5 × 1011 M−1 s−1, explained by Grothus’s mechanism.24 The value estimated in this study can be understood by considering the high number of charges of approaching NPs.
The pH of 10 mM acetic acid aqueous solution has been measured to be 3.30; aqueous solution of 0.85 nM AuNPs has a pH of 6.20, whereas the pH of 0.85 nM AuNPs in 10 mM acetic acid was measured to be 4.40. As the pKa of acetic acid is very close to the pK2 and lower than the pK3 of citric acid (pK1 = 3.1, pK2 = 4.8, and pK3 = 6.4), the addition of acetic acid may reduce the negative charge of citrate-capped gold NPs. The appearance of a red-shifted new band results from the coupling of the plasmon absorption of AuNPs assisted through self-assembly. For explaining the self-assembly process, two main factors can be considered. First, adsorption of Mn+2 reduces the negative charge of nanoparticles and, second, the carboxylate groups of citrate-capped neighbouring AuNPs are capable of complexation of Mn+2 ions with the concomitant crosslinked particle networks, Scheme 2.
 | ||
| Scheme 2 Schematic representation of interfacial interactions for Mn+2-mediated assembly of citrate-capped gold nanoparticles. | ||
We measured the ζ-potential of AuNPs as a function of the concentration of Mn+2 ions. The results are shown in Fig. 4, along with that determined by Murph et al.25 for gold nanoparticles slightly higher, 20 nm. Our results indicate that in 10 mM AcH, the addition of 0.12 mM Mn+2 ions increases the ζ-potential from −33 mV to −29.4 mV. In spite of this small variation of the surface charge of AuNPs, the rate of self-assembly due to the presence of bridging linkers, such as Cys, cap, or NOcap, is dramatically increased as we will show in the next sections. On the other hand, and contrary to that observed by Murph et al., higher concentrations of Mn+2 rapidly yield unstable AuNPs: the ζ-potential at [Mn+2] = 0.8 mM, measured just after adding Mn+2, was −20.5 mV, but increases with time (blue solution) to achieve ζ-potential values close to that accepted for neutral particles. This observation is in agreement with the kinetic results depicted either in Fig. 2(c) or 3(b) on the effect of 0.8 mM Mn+2, under which experimental conditions, the assembly of AuNPs is very significant in only 5 min.
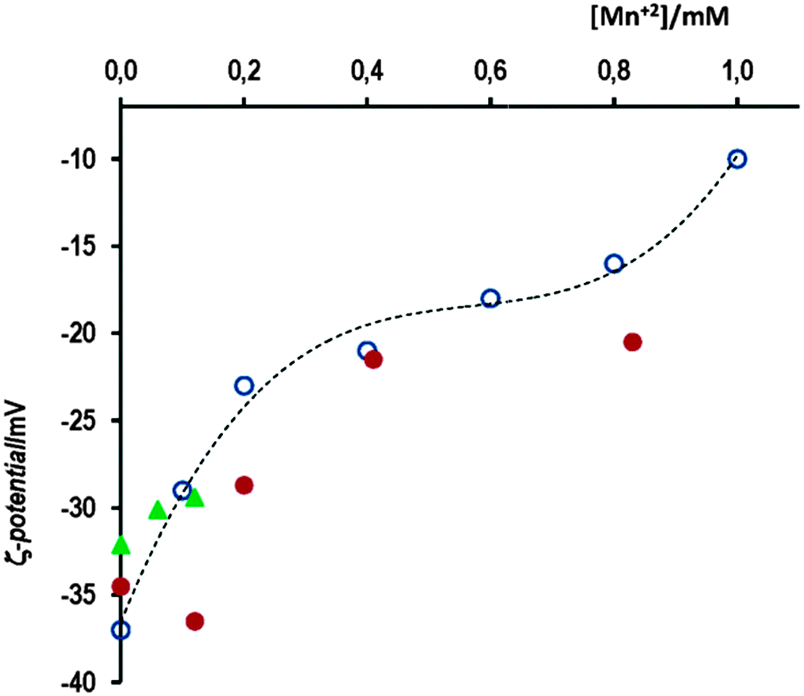 | ||
Fig. 4 Variation of the ζ-potential of AuNPs as a function of [Mn+2], ( ) in the presence of 10 mM AcH; ( ) in the presence of 10 mM AcH; ( ) no AcH, and ( ) no AcH, and ( ) Murph’s data for AuNPs of 20 nm size. ) Murph’s data for AuNPs of 20 nm size. | ||
The stability of gold nanoparticles in aqueous medium depends on the protecting capped shell of citrates that surround gold-nanoparticles by an electrical double layer. This layer contains the charges on the nanocrystal surface and the counterions that serve to protect the nanoparticles from aggregation. The presence of Mn+2 ions reduces the net charge of citrate-capped gold nanoparticles, due to the complexation of ions by the citrate-carboxylates. The effect increases with Mn+2 concentration and to the same extent the electrical double layer is compressed, which favours the interparticle interaction. As evident, when Mn+2 ions are added to a solution of citrate capped AuNPs, the rate of particle aggregation increases with an increase in Mn+2 concentration. The citrate anions have a dual role as stabilising agents for the gold core and as ligands to complex the manganese ions.
In the next sections we are going to demonstrate that stable Mn–AuNPs are improved sensors towards thiols.
2. Irreversible Cys-mediated assembly of gold nanoparticles
Thiol-containing amino acids, such as cysteine (Cys), were found to react with AuNPs through the strong covalent bonds between the –SH group of Cys and gold. Therefore, AuNP-based colorimetric detection has been used to detect Cys from the color change in the Cys-mediated assembly of AuNPs or gold nanorods.12,13,26 The main disadvantage of these detection methods was rather the long time required in the process.Fig. 5(a) presents the time evolution of UV-vis spectra of AuNP solutions in 10 mM acetic acid in the presence of 34 μM Cys. The increase in absorbance at long-wavelength (λmax ∼ 660 nm) is completed in less than 50 min, whereas a small decrease in the short-wavelength band (∼520 nm), typical for AuNP solution (the lowest red curve), also occurs. The spectral changes are accompanied by a red-to-blue color change that can be observed with the naked eye. The reaction spectra of the same sample in the absence of AcH recorded during 1 h hardly varies; in fact, the half-life time of the process can be estimated from the work of Zhang et al.12 to be as long as 4 h. By contrast, the Cys-mediated assembly of AuNPs is much more rapid in the presence of 0.12 mM Mn+2 ions, under which conditions only 5 min is enough to clearly observe the total time evolution of the SPR band.
 | ||
| Fig. 5 (a) UV-vis spectra of AuNPs in 10 mM AcH and 34 μM cysteine recorded as a function of time; the red scan corresponds to AuNPs only, scans 2 to 10 were obtained at 2.5 min interval; the dashed scan is after 50 min and (b) absorbance increase at 660 nm as a function of time for (◆) [Cys] = 34 μM, [AcH] = 5.5 mM; (●) [Cys] = 34 μM, [AcH] = 10 mM, see the spectra of (a), and (▼ [Cys] = 34 μM, [AcH] = 5.5 mM, [Mn+2] = 0.12 mM); in all experiments [AuNPs] = 0.85 nM. | ||
Fig. 5(b) displays comparative data for the absorbance increase at 660 nm as a function of time under different experimental conditions. Data for A–t fit the first-order integrated rate equation and values of the observed rate constant, k0, are listed in Table 2. As it can be seen, the rate constant of Cys-mediated AuNP aggregation in 10 mM AcH solution is more than 20-times faster than that measured in aqueous solutions of AuNPs. The effect of [Cys] is small and does not show first-order dependence; notice that Cys is introduced in excess, e.g. at 17 μM there are about 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules/gold nanoparticles. Under the same experimental conditions of [Cys] and [acetic acid], the addition of 0.12 mM Mn+2 ions multiplies by ∼6 times the rate of nanoparticle aggregation. When small amounts of AcH and Mn+2 ions are added to the aqueous colloidal gold solutions, concentrations of Cys as low as 34 nM can be detected and after incubation times as short as 40 min.
000 molecules/gold nanoparticles. Under the same experimental conditions of [Cys] and [acetic acid], the addition of 0.12 mM Mn+2 ions multiplies by ∼6 times the rate of nanoparticle aggregation. When small amounts of AcH and Mn+2 ions are added to the aqueous colloidal gold solutions, concentrations of Cys as low as 34 nM can be detected and after incubation times as short as 40 min.
| Cysteine (Cys)-mediated nanoparticle aggregation | |||||
|---|---|---|---|---|---|
| [Cys]/μM | [AcH]/mM | [Mn+2]/mM | k 0/10−3 s−1 | — | ΔA(660nm) |
| a Taken from ref. 12. b See Scheme 2 for their definition, the A–t data were fitted to A = A∞ + c1·exp(−k1t) + c2·exp(−k2t) with c1 and c2 being constants. c At [AuNPs] = 1.7 nM. | |||||
| 14a | — | — | 0.050 | — | — |
| 17 | 10 | — | 1.14 | — | 0.379 |
| 34 | 10 | — | 1.27 | — | 0.388 |
| 42.5 | 10 | — | 1.58 | — | 0.364 |
| 34 | 5.5 | 0.12 | 6.20 | — | 0.352 |
| 34 | 5.5 | — | 0.555 | — | 0.393 |
| 42.5 | 10 | 0.12 | 9.44 | — | 0.351 |
| 0.034 | 10 | 0.12 | 0.330 | — | 0.173 |
The effect of Cys addition to stable Mn–AuNPs in aqueous acetic acid solutions is double. On the one hand, the replacement of citrate by Cys reduces the surface charge of gold nanoparticles. On the other hand, each adsorbed Cys molecule leaves the zwitterionic amino acid group facing outward from the nanoparticle surface, which can act as bridging linkers between one nanoparticle and another due to the strong electrostatic and/or H-bonding interactions. Both effects acting in the same direction strongly increase the self-assembly rate.
Attempts to disassemble the Cys-mediated aggregates by the addition of 12 μL of 1.0 M NaOH, final pH 8.2, did not show any appreciable effect. In fact, the rate of the reversal of the SPR band of homocysteine (similar to Cys) at pH 11.35, which is accompanied by a change in the color of the solution from bluish to red purplish, is very slow, nearly 9 days are required.11
3. Reversible assembly of gold nanoparticles mediated by captopril- and S-nitrosocaptopril
The captopril molecule is a mercaptoproline derivative that contains the carboxylic group (pK1 = 3.52) of the proline moiety and the thiol group (pK2 = 10.00) of the propionyl residue. Thiols adsorb as their thiolate forms on gold surfaces.27A recent study shows that the pKa of sulfur-bound hydrogens of thiols on gold nanoparticles can be reduced by more than 10 pKa units; in fact, the SERS spectra indicate that the sulfur-bound hydrogen is completely deprotonated even at very low pH.28
Aqueous solutions of 0.85 nM citrate-capped gold nanoparticles have pH = 6.20. At this pH, the carboxylic group of cap is ionized, whereas the thiol group is not. Under these conditions, addition of cap to aqueous solutions of gold nanoparticles does not show any change in the SPR band, at least in the time scale of these experiments. This fact does not mean that cap molecules are not adsorbed onto the gold surface. Changes in the SPR band are observed when the interaction between particles takes place, which is not possible between the highly negative charged particles. Addition of 10 mM acetic acid induces cap-mediated assembly of AuNPs, and a new plasmon band centered at 620 nm develops.
Fig. 6(a) shows the corresponding time evolution spectra, in which the dashed spectrum was taken after 2 h of incubation; at this moment, the addition of 12 μL of 1 M NaOH (total vol. 1 mL, final pH 8.2) yields the dotted spectrum (follow the arrow) that matches with spectrum 1 recorded in the absence of cap. This fact evidences the complete reversal of the assembly cap-mediated process. The TEM image of this solution (after adding NaOH) resembles that shown in Fig. 1(c), i.e. no evidence of aggregation was observed; by contrast, the transmission electron microscope image taken after 60 min of incubation of AuNPs with cap in 5.5 mM acetic acid medium indicates the complete aggregation of both spherical and triangular gold nanoparticles, as it can be seen in Fig. 6(c).
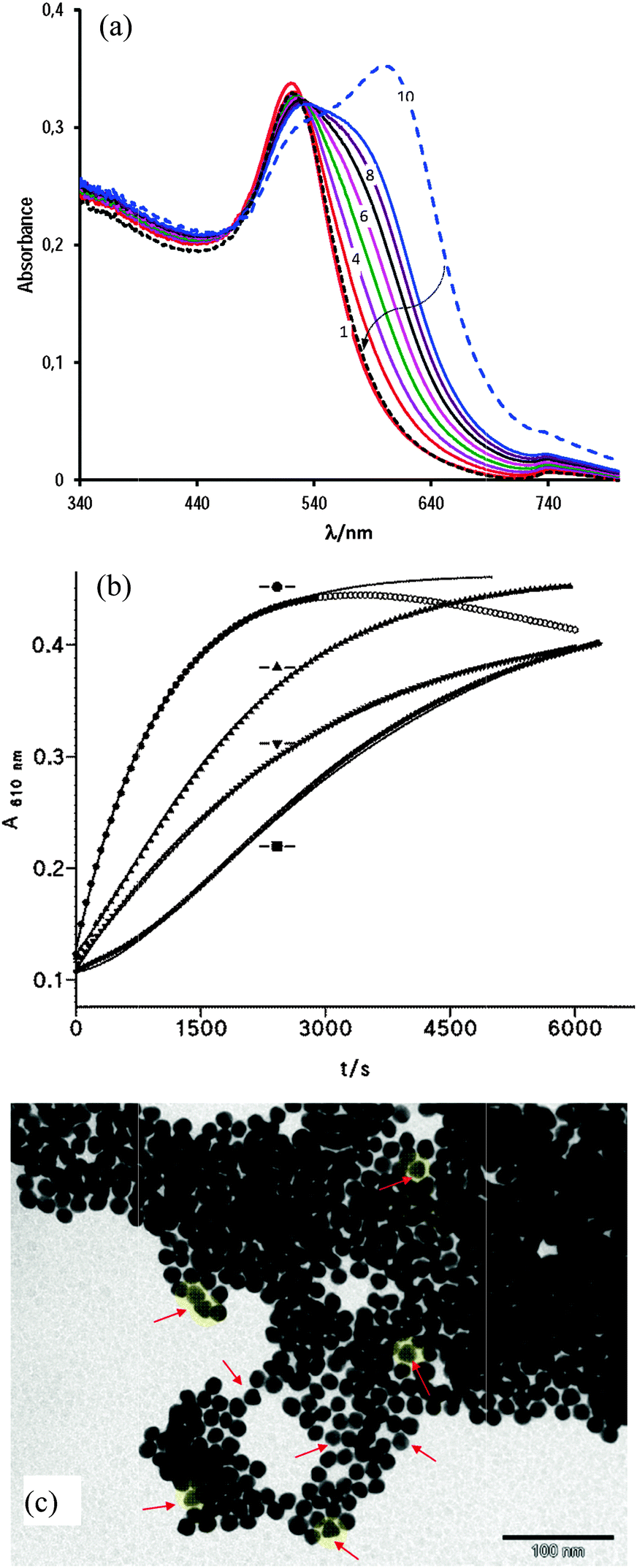 | ||
| Fig. 6 (a) Absorption spectrum of AuNPs in 10 mM AcH at [cap] = 0.75 mM every 6 min; (---) after 120 min; and (⋯) at the end of the reaction after adding 12 μL of 1 M NaOH; (b) increase in absorbance as a function of time for [cap] = 0.75 mM, [Mn+2] = 0.12 mM and [AcH] equal to (●) 10 mM: the uppermost curve is fitted to the first-order integrated rate equation, open circles not included – or to (▲) 5.5 mM – solid line fits the double exponential, and (▼) [cap] = 1.24 mM; [Mn+2] = 0.12 mM; solid line fits the first-order integrated rate equation; (■) [cap] = 0.75 mM; [AcH] = 10 mM: the lowest curve, solid line fits the double exponential; (c) the TEM image of AuNPs in the presence of [cap] = 1.1 mM and 10 mM acetic acid after 60 min. | ||
The presence of small amounts of Mn+2 ions, as low as 0.12 mM, facilitates the crosslinking process between gold nanoparticles, but the coexistence of both additives, i.e. Mn+2 ions in mild acid medium ([Mn+2] = 0.12 mM in 10 mM acetic acid) yields the most favorable conditions for visible interactions of cap-mediated gold nanoparticle assembly.
As for Cys, captopril molecules replace citrate from the gold surface due to the formation of strong covalent bonds S–Au. This fact disrupts the negatively charged citrate shell of AuNPs and decreases the gold nanoparticle stability. The cap molecule bulkier than Cys makes the cap adsorption onto the gold surface much slower than that with Cys, and more [cap] is required to observe the interaction between AuNPs; in fact the kinetic profiles of cap-mediated nanoparticle aggregation show a induction period under some experimental conditions. On the other hand, the carboxyl group of adsorbed cap molecules remains exposed outward the NP surface and favors nanoparticle interactions via hydrogen-bonding. By deprotonating these carboxyl groups, e.g. by adding NaOH, the H-bonds are broken as well and, consequently, the process is reverted.
The plot of the increase in absorbance (A) with time (t) under different experimental conditions, showing the comparative effect of additives, is depicted in Fig. 6(b). We must indicate that the last additive to the sample was the Mn+2 ions and/or acetic acid; therefore, the possible binding of cap molecules to the AuNP-surface was allowed prior to the start of aggregation. High concentrations of either Mn+2 or acid result in A–t profiles that fit the first-order integrated rate equation, whereas at low acid concentrations or in the absence of Mn+2 the kinetic profiles show an induction period.29 These profiles can be interpreted as consecutive processes due to (1) binding of cap to gold nanoparticles with a rate constant k1 and (2) aggregation of nanoparticles due to hydrogen bonding interactions between the surface carboxyl groups of cap of the adjacent nanoparticles of rate constant k2, see Scheme 3. As H-bonding interactions are weaker than electrostatic interactions, the cap concentrations required to observe nanoparticle assembly are higher than the cysteine concentration; in addition, the observed rate constant of the assembly process is higher in the case of Cys than the corresponding values of cap, compare k0 and k2 in Table 2.
 | ||
| Scheme 3 Schematic representation of cap adsorption onto Mn–AuNPs, color change after aggregation, and H-bonding between the adjacent nanoparticles. | ||
From the uppermost curve in Fig. 6(b), one can see that cap-capped gold nanoparticles are unstable (absorbance decreases at ca. t > 3000 s) and coalesce. The solution changes from blue to colorless and the aggregates stay at the bottom of the cuvette; but upon adding NaOH to final pH 8.2 (12 μL of 1 M NaOH) the aggregates redissolve instantaneously and the solution becomes red, like well-dispersed citrate-capped AuNPs.
A similar response to the addition of NaOH was observed in the disassembly of AuNP aggregates mediated by glutathione; nevertheless, the degree of SPR band reversal is not completed. In a similar manner, upon addition of citrate to the GSH–AuNP assembly after certain reaction time, the spectral evolution of the SPR band can be prevented from further progression, either frozen or slowly reverted depending on the citrate concentration.30 Kim et al. demonstrated that Pb+2 also mediated aggregation of gold nanoparticles capped with 11-mercaptoundecanoic acid, and addition of EDTA partially reverses the response due to Pb+2 chelation.31 In the same sense, the SPR band of AuNP aggregation mediated by PEN or NAP is pH-dependent and the assembly–disassembly process is demonstrated to be fully reversible when the pH was cycled between 7 and 3.16
Finally, we analyze the interaction of S-nitrosocaptopril, NOcap, with colloidal gold solutions. The only structural difference between cap and NOcap is the replacement of –SH in cap by –SNO in NOcap, which allow us to obtain information on sulfur–gold interaction.
In aqueous acid medium of sodium nitrite, captopril is readily converted to S-nitrosocaptopril. The UV-vis spectrum of NOcap aqueous solutions shows the characteristic band at 332 nm (ε = 1020 M−1 cm−1) and a much less intense absorption band centered at 547 nm (ε = 20 M−1 cm−1).21
S-Nitrosothiols (RSNOs) release NO in the presence of AuNPs because of the affinity between gold and thiols, which catalysed and tuned the NO deliver process because of the affinity between gold and thiols, as shown in an interesting study by Hervés et al.32 The available Au-surface determines the released amount of NO. By employing a sensitive NO electrode, the stoichiometry of the reaction was estimated and the results indicate that the amount of NO released is not quantitative at low concentrations of AuNPs. The rate equation is of zero-order with respect to [RSNO] and first-order for [AuNPs], and the reactivities of the different RSNOs analysed, which include the nitrosothiols of PA, N-acetylpenicillamine, and GSH, towards the Au-surface are quite similar with rate constants of the order of ∼5 × 10−2 s−1.
Fig. 7(a) shows the reaction spectra of nanoparticle assembly mediated by S-nitrosocaptopril. The band centered at 332 nm is due to NOcap, characteristic of RSNOs. The SPR band of aqueous citrate-capped gold nanoparticles centered at 520 nm shifts to 620 nm in the presence of NOcap, i.e., the reaction spectra within the 440–800 nm interval of NOcap-mediated assembly is not different from those of AuNP aggregation by cap, as expected, because cap molecules are immobilized on the gold surface by strong S–Au bonds. The green dashed spectrum, which matches that at zero time, was recorded once the reaction was completed (after ∼60 min) and just after adding 12 μL of 1 M NaOH to the reaction sample. The result evidences the reversal of the gold nanoparticle aggregation process.
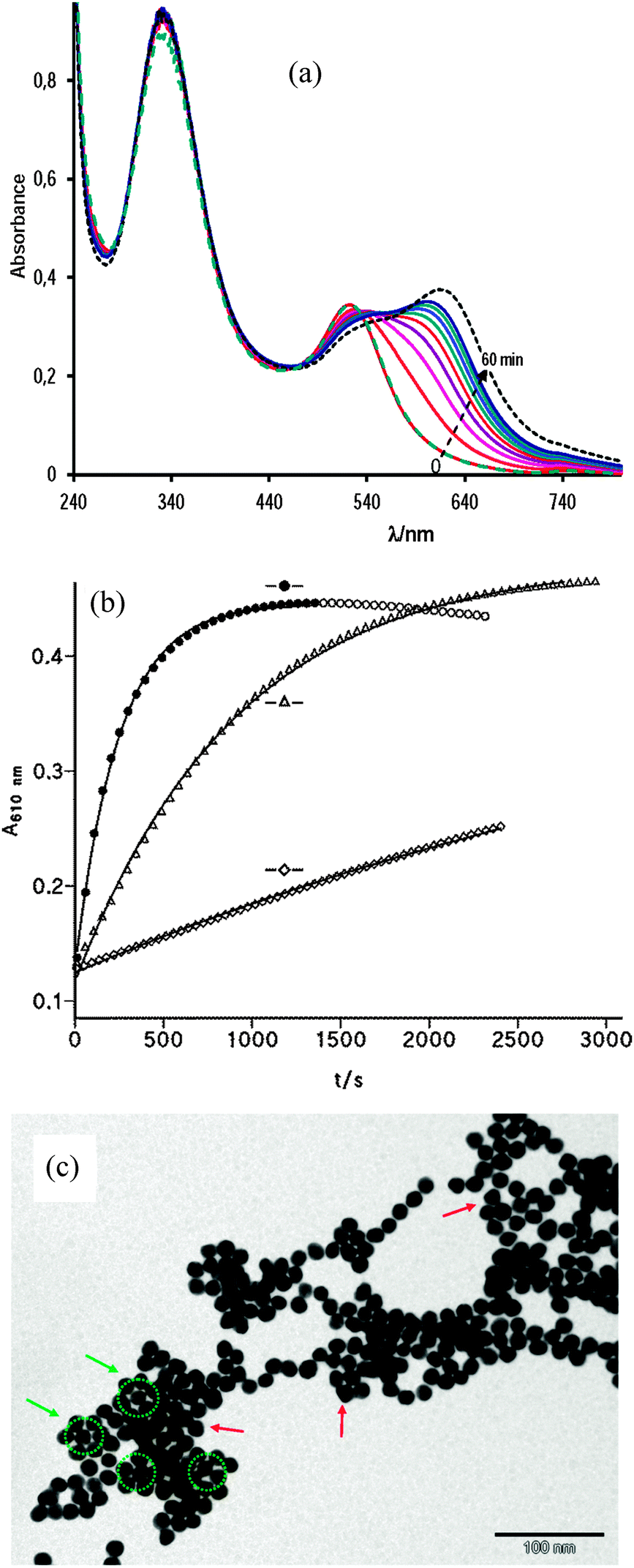 | ||
| Fig. 7 (a) UV-vis absorption spectra of 0.85 nM AuNPs in 10 mM acetic acid and [NOcap] = 0.75 mM recorded at 6 min interval; (---) after 120 min; and (⋯) at the end of the reaction after adding 12 μL of 1 M NaOH (b) increase in absorbance of AuNPs with time in the presence of [NOcap] = 0.755 mM at (●) [AcH] = 10 mM, and [Mn+2] = 0.12 mM, open circles not included in the fitting process; (△) [AcH] = 10 mM, and (⋄) [Mn+2] = 0.12 mM; and (c) the TEM image of AuNPs incubated for 40 min with 0.75 mM NOcap in 10 mM acetic acid and 0.12 mM Mn+2. | ||
Fig. 7(b) shows the effect of acetic acid and Mn+2 ion concentration on the rate of AuNP assembly mediated by NOcap. The presence of 0.12 mM Mn+2 accelerates the reaction; the catalytic effect is much more marked in 10 mM acetic acid, but the coexistence of both additives enables the reaction to reach the end in a few minutes. In each case the absorbance–time profiles fit the first-order integrated rate equation. The values of the observed rate constant, k0, are listed in Table 2 along with the corresponding experimental conditions. The comparison of k0 with the equivalent rate constant, k2, for cap-mediated aggregation evidences the accelerating effect of NOcap.
The NOcap-mediated assembly of AuNPs shows good first-order kinetics under all the experimental conditions studied; in other words, the binding of NOcap molecules to gold NPs is much faster than the nanoparticle aggregation, contrarily to that observed with cap, that is, gold nanoparticles stabilized by NOcap seem to reach the equilibrium faster (step 1 in Scheme 3) than those stabilized with cap. The only difference between cap and NOcap is the –S–H bond in cap against –S–NO in NOcap. Literature results show that decomposition of S-nitrosothiols occurs through thermal homolysis of the RS–NO bond.33 The RS–NO homolytic dissociation energies (around 20–30 kcal mol−1) are remarkably lower than heterolytic cleavage by an average of 29 kcal.34 The gold thiolate (S–Au) bond energy is about 40 kcal mol−1,35i.e. greater than that of the RS–NO bond. Hence, the RS–NO bond can be easily cleaved in the presence of AuNPs that favor the S–Au bond formation. Therefore, citrate capped gold nanoparticles enhance the NO release rate from S-nitrosothiol based systems, being modulated by affecting the size and concentration of gold nanoparticles.33,36
On the other hand, RS–H bond energy is about 87 kcal mol−1,35i.e. much greater than that of the RS–Au bond. Consequently, the thiol must adsorb onto the gold surface in the thiolate form,27 whose concentration at acidic pH is quite small, a fact that explains the slow reaction rate of cap adsorption against NOcap.
The TEM analysis of the samples confirmed that at neutral and basic pH the particles are well-dispersed and randomly distributed on the TEM grid, while in 10 mM acetic acid or in the presence of a small amount of manganese ions the particles tend to coalesce as it is shown in Fig. 7(c) for NOcap-mediated assembly. Notice the abundance of symmetrical arrangements of spherical and triangular gold nanoparticles.
In summary, aqueous solutions of 0.85 nM AuNPs and 10 mM acetic acid have a pH around 4.40; but, as the thiol group of Cys or cap binds to the gold surface, the releasing protons reduce the pH to around 3.5. At this pH, the amine group of Cys is protonated and the carboxyl group is ionized, while the carboxyl groups of either cap or NOcap are partially protonated. This important fact evidences the existence of electrostatic interactions in Cys-modified gold nanoparticles, and H-bonding interactions in cap (or NOcap) mediated gold nanoparticle aggregation. Therefore, as NOcap – or cysteine or captopril – is being exchanged by citrate-capped gold NPs, the pH of the solution decreases and the particles tend to aggregate, leading to plasmon coupling, which results in broadening and red-shift of the maximum absorption, the maximum observed wavelength being at around 672 nm in the Mn+2-mediated aggregation; 660 nm with Cys and 620 nm with cap or NOcap serve as average values because the many factors that condition λmax make the precise shape of the new bands to vary even from sample to sample. Actually it is well-established that the origin of nanoparticle aggregation in these systems is the strong interaction of gold–sulfur or due to a strong reduction in the zeta potential at high concentrations of manganese ions. Consequently, we assume that Cys, cap or NOcap is immobilized on the gold surface via the thiolate group. Therefore, the stability of Cys – or cap or NOcap – modified AuNPs will be determined by the interactions of amine and/or carboxylic groups exposed to the exterior surface of NPs.
Conclusions
The presence of small amounts of Mn+2 ions in mild acid medium has proven to dramatically accelerate the assembly of thiol-capped gold nanoparticles obtained through ligand exchange in aqueous solution. Two mechanisms are proposed to explain the nanoparticle assembly in this study. The assembly via electrostatic interactions between zwitterionic amino acids is operative when using cysteine as the ligand; the resulting process is irreversible upon increasing pH to basic, and the kinetics show first-order behaviour with rate constants strongly dependent on the concentration of both the acid and manganese ions. The assembly via hydrogen bonding is proposed when the ligand is captopril or S-nitrosocaptopril. In contrast to that observed with Cys, the process is fully reversible when the pH is increased to basic levels, a fact that proves the stronger electrostatic interactions than H-bonding. The aggregation rate was found to be significantly higher for NOcap than for cap, showing the important role that the specific nature of the ligand molecule plays, which, in the present case, can be explained taking into account the bond energies of both the broken and formed bonds. The kinetics of the assembly process show first-order behaviour with NOcap under all tested conditions, while with cap, consecutive reactions appear to occur at low concentrations either of acid or cap.Acknowledgements
We are indebted to Dr Sarah Fiol and Dr Juan Antelo of the University of Santiago de Compostela (Instituto de Investigaciones Tecnológicas) for their assistance in the measurement of ζ-potentials.Notes and references
- M.-E. Stewart, C. R. Anderton, L. B. Thompson, J. Maria, S. K. Gray, J. A. Rogers and R. G. Nuzzo, Chem. Rev., 2008, 108, 494–521 CrossRef CAS PubMed.
- C. M. Cobley, J. Chen, E. C. Cho, L. V. Wang and Y. Xia, Chem. Soc. Rev., 2011, 40, 44–56 RSC.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- H. Jans and Q. Huo, Chem. Soc. Rev., 2012, 41, 2849–2866 RSC.
- S. Zeng, D. Baillargeat, H.-P. Ho and K.-T. Yong, Chem. Soc. Rev., 2014, 43, 3426–3452 RSC.
- V. Myroshnychenko, J. Rodríguez-Fernández, I. Pastoriza-Santos, M. A. Funston, C. Novo, P. Mulvaney, L. M. Liz-Marzán and F. J. García de Abajo, Chem. Soc. Rev., 2008, 37, 1792–1805 RSC.
- J. Turkevich, P. C. Stevenson and J. A. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plench, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
- D. C. Hone, A. H. Haines and D. A. Russell, Langmuir, 2003, 19, 7141–7144 CrossRef CAS.
- K. J. Fahnestock, M. Manesse, H. A. McIlwee, C. I. Schauer, R. Boukherroub and S. Szunerits, Analyst, 2009, 134, 881–886 RSC.
- I.-I. S. Lim, W. Ip, E. Crew, P. M. Njoki, D. Mott, C.-J. Zhong, Y. Pan and Z. Zhou, Langmuir, 2007, 23, 826–833 CrossRef CAS PubMed.
- F. X. Zhang, I. Han, I. B. Israel, J. G. Daras, M. M. Maye, N. K. Ly and C.-J. Zhong, Analyst, 2002, 127, 462–465 RSC.
- L. Li and B. Li, Analyst, 2009, 134, 1361–1365 RSC.
- B. Hu, X. Cao and P. Zhang, New J. Chem., 2013, 37, 3853–3856 RSC.
- S. Basu, S. Panigrahi, S. Praharaj, S. K. Ghosh, S. Pande, S. Jana and T. Pal, New J. Chem., 2006, 30, 1333–1339 RSC.
- P. Taladriz-Blanco, N. J. Buurma, L. Rodríguez-Lorenzo, J. Pérez-Juste, L. M. Liz-Marzán and P. Hervés, J. Mater. Chem., 2011, 21, 16880–16887 RSC.
- H. M. Zakaria, A. Shah, M. Konieczny, J. A. Hoffmann, A. J. Nijdam and M. E. Reeves, Langmuir, 2013, 29, 7661–7673 CrossRef CAS PubMed.
- Z. Zhong, S. Patskovskyy, P. Bouvrette, J. Luong and A. Gedanken, J. Phys. Chem. B, 2004, 108, 4046–4052 CrossRef CAS.
- R. Stewart, The proton applications to organic chemistry, Academic Press, London, 1985 Search PubMed.
- S. G. Tajc, B. S. Tolbert, R. Basavappa and B. L. Miller, J. Am. Chem. Soc., 2004, 126, 10508–10509 CrossRef CAS PubMed.
- A. Sexto and E. Iglesias, Org. Biomol. Chem., 2011, 9, 7207–7216 CAS.
- J. Loscalzo, D. Smick, N. Andon and J. S. Cooke, J. Pharmacol. Exp. Ther., 1989, 249, 726–729 CAS.
- Addition of 0.074 mM acetic acid to a 18.7 nm colloidal gold solution at pH 3 (adjusted by dropwise addition of HCl) does not cause the aggregation of gold after incubation as long as 1000 min, according to the data from ref. 17.
- K. J. Laidler, Chemical Kinetics, Harper Collins Publishers, 3rd edn, ch. 6, 1987 Search PubMed.
- E. H. Murph, S. Jacobs, J. Liu, T. C. C. Hu, M. Siegfired, S. M. Serkiz and J. Hudson, J. Nanopart. Res., 2012, 14, 658–669 CrossRef.
- P. K. Sudeep, S. T. S. Joseph and K. G. Thomas, J. Am. Chem. Soc., 2005, 127, 6516–6517 CrossRef CAS PubMed.
- J. E. Mattiesen, D. Jose, C. M. Sorensen and K. I. Klabunde, J. Am. Chem. Soc., 2012, 134, 9376–9379 CrossRef PubMed , and references therein.
- S. M. Ansar, G. S. Perera, D. Jiang, R. A. Holler and D. Zhang, J. Phys. Chem. C, 2013, 117, 8793–8798 CAS.
- J. H. Espenson, Chemical Kinetics and Reaction Mechanisms, McGraw-Hill, 2nd edn, ch. 4, 1995 Search PubMed.
- I.-I. S. Lim, D. Mott, W. Ip, P. M. Njoki, Y. Pan, S. Zhou and C.-J. Zhong, Langmuir, 2008, 24, 8857–8863 CrossRef CAS PubMed.
- Y. Kim, R. C. Johnson and J. T. Hupp, Nano Lett., 2001, 1, 165–167 CrossRef CAS.
- P. Taladriz-Blanco, V. Pastoriza-Santos, J. Pérez-Juste and P. Hervés, Langmuir, 2013, 29, 8061–8069 CrossRef CAS PubMed.
- M. D. Bartberger, J. M. Mannion, S. C. Powel, J. S. Stamler, K. N. Houk and E. J. Toone, J. Am. Chem. Soc., 2001, 123, 8868–8869 CrossRef CAS PubMed.
- J.-M. Lu, J. M. Wittbrodt, K. Wang, Z. Wen, H. B. Schlegel, P. G. Wang and J.-P. Cheng, J. Am. Chem. Soc., 2001, 123, 2903–2904 CrossRef CAS PubMed.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS PubMed.
- S. Priya, T. Kaviyarasan and S. Berchmans, Analyst, 2012, 137, 1541–1543 RSC.
| This journal is © the Owner Societies 2015 |