
Electron small polarons and their transport in bismuth vanadate: a first principles study
Kyoung E.
Kweon
a,
Gyeong S.
Hwang
*a,
Jinhan
Kim
b,
Sungjin
Kim
b and
SeongMin
Kim
b
aDepartment of Chemical Engineering, University of Texas at Austin, Austin, Texas 78712, USA. E-mail: gshwang@che.utexas.edu
bCAS Center, Samsung Advanced Institute of Technology (SAIT), Yongin 449-712, South Korea
First published on 3rd November 2014
Abstract
Relatively low electron mobility has been thought to be a key factor that limits the overall photocatalytic performance of BiVO4, but the behavior of electrons has not been fully elucidated. We examine electron localization and transport in BiVO4 using hybrid density functional theory calculations. An excess electron is found to remain largely localized on one V atom. The predicted hopping barrier for the small polaron is 0.35 eV (with inclusion of 15% Hartree–Fock exchange), and tends to increase almost linearly with lattice constant associated with pressure and/or temperature changes. We also examine the interaction between polarons, and discuss the possible concentration-dependence of electron mobility in BiVO4.
1. Introduction
Bismuth vanadate (BiVO4) is one of the most promising photoanode candidates for water splitting.1–11 Its band gap Eg is sufficiently small (≈2.4 eV) to allow the absorption of visible light as compared to only ultraviolet light absorption by titanium oxide (TiO2, Eg ≈ 3.0–3.2 eV), which is the most widely used. However, in practice, the solar-to-hydrogen (STH) conversion efficiency of BiVO4 is far lower than the maximum theoretical value.12 Previous experimental and theoretical studies have identified that the overall photoelectrochemical performance of BiVO4 is largely limited by poor electron transport,8,9 which facilitates the recombination of photogenerated electron–hole pairs. To overcome the drawback, many efforts have been made to find a robust and effective way to enhance the photoinduced charge separation, including doping4,7–9 and heterostructure formation.10,11 Although electron mobility appears to be a major bottleneck that we have to solve for realizing the desired efficiency of BiVO4-based photoanodes, so far the nature of an excess electron and its transport properties in BiVO4 have not been well studied.In oxide semiconductors, excess charge carriers (electrons and holes) often remain localized.13,14 The charge localization is commonly accompanied by lattice distortions, which results in a so-called polaron.13–15 Density functional theory (DFT) is perhaps the most successful computational method for the study of semiconductor and oxide materials; however, in practice, the results rely on approximations for the exchange–correlation potential which accounts for the many-body electron–electron interaction. In particular, the microscopic description of localized charge states tends to depend strongly on the exchange–correlation (xc) energy functional employed. The (semi)local functionals of conventional DFT may fail to predict charge localization due to their inherent self-interaction error (which erroneously favors charge delocalization).16,17 The self-interaction effects can be effectively canceled by the partial inclusion of non-local Hartree–Fock (HF) exchange;16–18 the HF/DFT hybrid approach has been shown to be capable of predicting charge localization and transport in many metal oxide semiconductors.19–21
In this work, we investigate the behavior of excess electrons in BiVO4 using hybrid DFT calculations. We first identify the formation and structure of a small electron polaron, and evaluate its relative stability with respect to the completely delocalized (nonpolaronic) state by varying the fraction α of HF exchange. The α dependence of the polaron formation energy is also analyzed by decoupling it in terms of electronic energy gain and strain energy loss. Next, we show a variation in the predicted barrier for adiabatic electron hopping with α and discuss the underlying reasons. Finally, the possible effects of electron concentration and hydrostatic pressure on the mobility of small electron polarons are discussed.
2. Computational methods
Spin-polarized hybrid DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP 5.2.2).22 The projector augmented wave (PAW) method with a planewave basis set (Ecut = 450 eV) was used to describe the interaction between the core and valence electrons.23 We employed a PBE (Perdew–Burke–Ernzerhof)24-based hybrid functional in which a fraction α of the PBE exchange is replaced with HF exchange. Here, the slowly decaying long-range part of the HF interaction was excluded following the Heyd–Scuseria–Ernzerhof (HSE) screened approach.25 As such, the xc energy is given by| EHSExc = αEHF,SRx(μ) + (1 − α)EPBE,SRx(μ) + EPBE,LRx(μ) + EPBEc, |
Bulk BiVO4 was modeled using a supercell consisting of 36 BiVO4 formula units with periodic boundary conditions in all three directions; for a charged system, a homogeneous countercharge was added to maintain overall charge neutrality. The atoms in the supercell were fully relaxed until the residual forces on all the constituent atoms become smaller than 0.02 eV Å−1. The Brillouin zone was sampled using a single k-point (Γ). For electron-doped BiVO4, we considered both localized (polaronic) and delocalized (nonpolaronic) electron states; to create a polaronic state, we initially applied a small perturbation around a selected VO4 tetrahedron to break the lattice symmetry prior to structural relaxation, while a nonpolaronic state was obtained by relaxing the structure without such initial perturbation.
3. Results and discussion
Fig. 1(a) shows a schematic illustration of the tetragonal (scheelite) structure of BiVO4 which consists of corner-sharing BiO8 dodecahedra and VO4 tetrahedra. As shown in the orbital-resolved density of states (DOS) in Fig. 1(a), the low-lying empty conduction bands are mainly composed of V 3d states with minor contributions from the Bi 6p and O 2p states. Note also the d orbital splitting into t2 and e states by the tetrahedral crystal field; additionally, the compression in the z-direction causes a splitting in the e states27,28 while leaving the dz2 levels at the bottom of the conduction band (CB).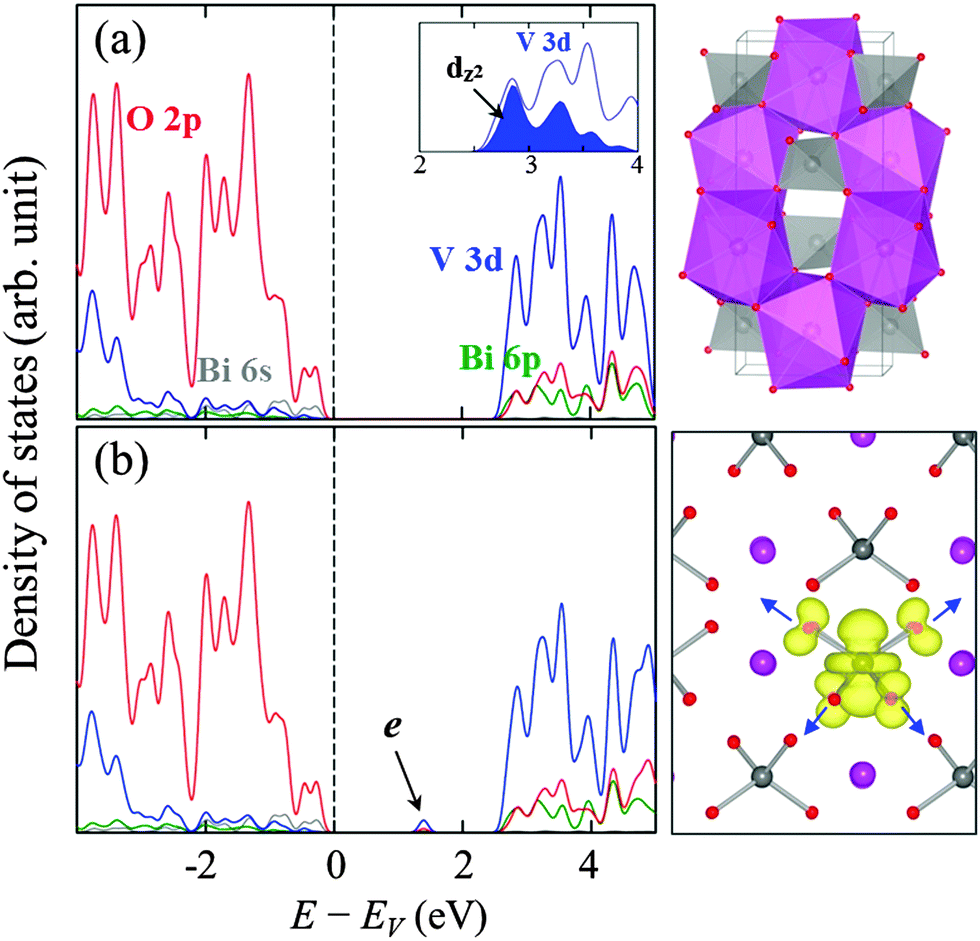 | ||
| Fig. 1 (left panels) Orbital projected density of states (DOS) for (a) pristine and (b) electron-doped tetragonal BiVO4; the grey, green, blue, and red lines represent the Bi 6s, Bi 6p, V 3d, and O 2p states, respectively, and the energy zero is set at the Fermi level (EF) which is indicated by the vertical dashed line. In addition, the V dz2 states (shaded in blue) are shown in the inset of (a), and the electron polaron state is indicated by an arrow in (b). (upper-right panel) Crystal structure representation of scheelite BiVO4 with indication of BiO8 dodecahedra (in purple) and VO4 tetrahedra (in gray). (lower-right panel) Band-decomposed charge density of the localized electron state (with an isosurface value of 0.02 e Å−3); purple, silver, and red balls represent Bi, V, and O atoms, respectively, and the arrows indicate elongation of the V4+–O bonds. | ||
As shown in Fig. 1(b), adding an electron to the BiVO4 matrix creates a deep localized state within the band gap, which is evidently due to local trapping of the excess electron; most of the excess charge tends to reside on a V atom by preferentially filling up the empty dz2 states, and only a small fraction is on the surrounding four O atoms. The electron self-trapping is accompanied by significant local lattice distortions around the reduced V4+ site, forming a small polaron. It is worth noting that noticeable elongation of V4+–O bonds in the electron-doped case (with respect to V5+–O bonds in undoped BiVO4) can be largely attributed to the filling of antibonding V 3d–O 2p states.
We also evaluated the relative stability of the polaronic state with respect to the completely delocalized (nonpolaronic) state (in which the excess electron spreads over entire V atoms and the Fermi level is shifted above the bottom of the CB, not shown here). The nonpolaronic state with no local lattice distortion has the nearly same configuration as the undoped structure, as expected.29 As summarized in Fig. 2(a), the total energy difference between the polaronic and nonpolaronic states, referred to as the polaron formation energy (Ep),30 varies with the fraction α of Hartree–Fock exchange added to the PBE-GGA functional; a positive Ep value indicates that the polaronic state is energetically more favorable than the non-polaronic state. The pure PBE functional (α = 0%) tends to erroneously favor electron delocalization, which is due to the incomplete cancellation of the Coulomb self-interaction.16,17 The Ep is predicted to be 40 meV at α = 5%, and increases monotonically with increasing α; note that adding the HF exchange partially cancels the unphysical self-interaction in the Hartree approximation, which in turn makes the localized state energetically favorable.16
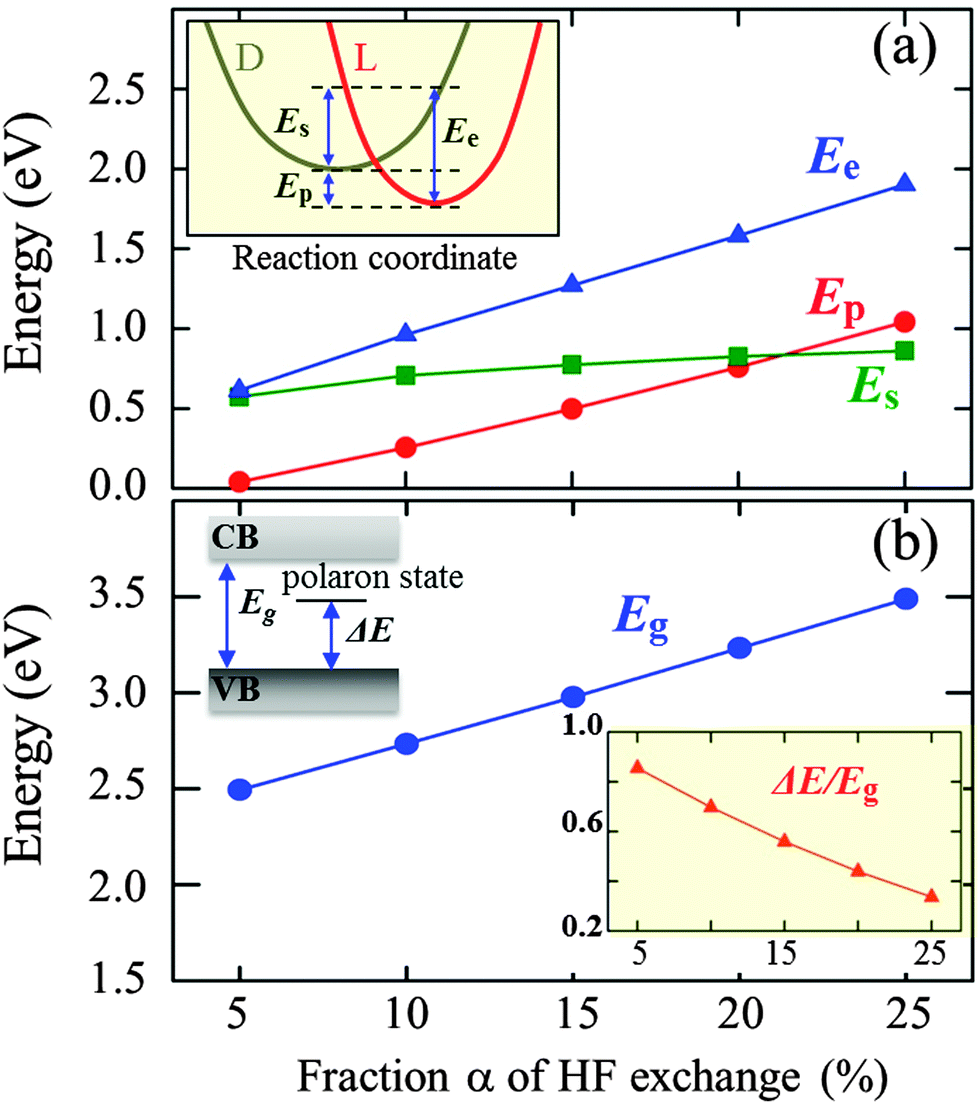 | ||
| Fig. 2 Results from hybrid DFT as a function of the fraction α of Hartree–Fock (HF) exchange. (a) Polaron formation (Ep), electronic (Ee), and lattice strain (Es) energies. The inset shows a graphical interpretation of Ep, Ee, and Es, and D and L represent the delocalized and localized states, respectively. (b) Band gap (Eg) and localized polaron state location from the valence band (ΔE), as illustrated in the upper left corner. The relative polaron state location with respect to the band gap (ΔE/Eg) is also shown in the inset graph. | ||
We would better understand the α dependence of Ep by decoupling it in terms of the electronic energy gain (Ee) due to lattice polarization and the strain energy loss (Es) caused by local polaronic lattice distortions,30,31 as illustrated in the inset of Fig. 2(a). For each case, Es is obtained from the total energy difference between the undistorted lattice and the distorted lattice due to polaron formation but without the excess electron,31 and Ee is estimated by adding Es and Ep. As shown in Fig. 2(a), while both Ee and Es increase with α, the former increases more rapidly than the latter; apparently, the large increase in Ee is primarily attributed to the increased Coulomb interaction energy as a result of the increase of charge localization with inclusion of HF exchange.16,17 A similar trend can be seen from DFT + U calculations;30,32 a larger value of Hubbard U (analogous to α in hybrid DFT) leads to more localization of charge and in turn a greater Ep. Our calculations also show that the band gap (Eg) and the position of the localized polaron state (ΔE/Eg) are a strong function of α [Fig. 2(b)]. The Eg is predicted to be 2.5 eV at α = 5%, and almost linearly increases with increasing α, which is overestimated in comparison to the experimental value1 of 2.34 eV while the pure PBE functional somewhat underestimates the band gap.33 The relative position of the polaron state within the gap is found to be shifted closer to the valance band as α is increased, implying enhancement in the stability of the polaron which is also well demonstrated by the increase in Ep.
A small polaron may undergo thermally-activated hopping from site to site.13 The activation energy (Ea) for polaron hopping can be estimated by calculating the variation of potential energy as the polaronic lattice configuration is displaced from the initial to the final geometry; if the initial and final states are identical, the transition state may occur midway between the two local minima. In Fig. 3, we present the predicted Ea for electron hopping along the 〈101〉 (or 〈011〉) direction as a function of the HF exchange fraction α; another probable path in the 〈100〉 (or 〈010〉) direction was considered, but it turns out to be energetically less favorable (not reported here). Ea is found to monotonically increase from 0.10 eV to 0.46 eV as α increases from 5% to 25%. In addition, Ea decreases upon decreasing the supercell size, and the magnitude of the decrease becomes larger with decreasing α. These results can be explained based on the theory for small polaron transport, as described in the following.
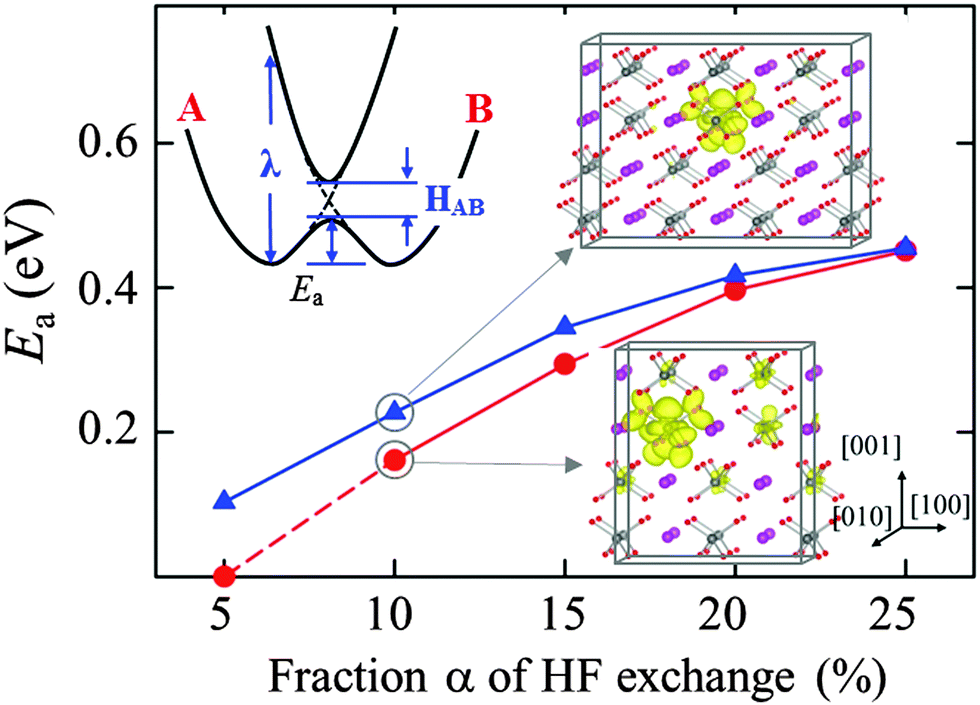 | ||
| Fig. 3 Predicted activation energies (Ea) for electron polaron hopping as a function of the fraction α of Hartree–Fock (HF) exchange for two different supercell sizes as depicted. The schematic diagram in the upper left corner illustrates the Marcus–Emin–Holstein–Austin–Mott theory for small polaron transfer between two adjacent sites; as detailed in the text, the Ea is determined by the reorganization energy (λ) which represents the energy needed to distort the initial-state structure to the final-state structure without electron transfer occurring and the electronic coupling between the initial and final states (HAB). In this work, the initial (A) and final (B) states are V4+–V5+ and V5+–V4+, respectively. In the supercell diagrams, the band-decomposed charge densities of corresponding polaron states (with an isosurface value of 0.005 e Å−3) are also shown to demonstrate the supercell-size dependence of electron localization; purple, silver, and red balls represent Bi, V, and O atoms, respectively. | ||
As illustrated in the inset of Fig. 3, for adiabatic electron transfer that follows the thermal hopping mechanism, the activation barrier can be estimated in terms of the reorganization energy (λ) and the electronic coupling between the initial and final states (HAB),34i.e., Ea = λ/4 − [|HAB| + λ/2 − (λ2/4 + |HAB|2)1/2]. According to the Marcus–Emin–Holstein–Austin–Mott theories,35,36 the initial and final states correspond to minima on their respective potential energy surfaces A and B which are assumed to be parabolic in shape. The steepness of the potential well is characterized by λ (which represents the energy needed to distort the initial-state structure to the final-state structure without electron transfer occurring). The height of the crossing point between the two potential energy surfaces is the diabatic activation energy, and the hopping barrier is reduced by the electronic interaction HAB between the initial and final states while the localized electron undergoes migration in an adiabatic way.
The reorganization energy λ is directly related to the polaron formation energy Ep which tends to almost linearly increase with α.34,36 Moreover, the more localized electron at a larger α may result in a smaller electronic coupling HAB. Hence, it is expected that Ea will increase sublinearly as α is increased. In addition, a smaller supercell (which gives a shorter distance between the polaron and its periodic images) may result in an increase in HAB, thereby leading to a further reduction in Ea. These hypotheses are well demonstrated and supported by our calculation results in Fig. 3.
In addition, for α < 20%, the substantial change in Ea with supercell size may suggest a possible dependence of electron mobility on its concentration in highly electron-doped BiVO4, due to the overlap between the diffusing electron wavefunctions. If the polaron–polaron separation is sufficiently small, the polaron wavefunctions can overlap to become stabilized.32 As shown in the inset of Fig. 3, indeed, the excess electron appears to spread to a noticeable extent over neighboring V atoms particularly along the 〈101〉 and/or 〈100〉 directions, as the supercell size is decreased; for the small and large supercells, the distances between the polaron and its periodic images in the 〈100〉 direction are 10.3 Å and 15.4 Å, respectively. Clearly, the increased wavefunction overlap is largely responsible for the reduction of Ea from 0.35 eV (large supercell) to 0.29 eV (small supercell) at α = 15%; the concentration effect can be overestimated or underestimated, depending on the choice of α, such that its careful selection is necessary. A similar reduction in Ea due to the overlap of polaron orbitals has been also suggested for hole hopping in Li2O2 and Li2CO3.32 The orbital overlapping will diminish as the polaron concentration decreases, and appears to be unimportant when the polarons are separated by more than several atomic distances if they are strongly localized. Nonetheless, our theoretical study, warranting further experimental validations, highlights the possibility that the Ea of electron hopping could be concentration dependent in highly electron-doped BiVO4.
Although an optimal α that gives an accurate description of electron localization and transport would hardly be determined, we could choose a reasonable value through comparison to experimental observations. In view of this, the good agreement between our predicted Ea (≈0.35 eV) at α = 15% for the low electron concentration case (large supercell) and the experimentally estimated value37 of 0.29 eV in 0.3% W-doped BiVO4 may suggest that α = 15% would offer a reasonable description of the behavior of small electron polarons in BiVO4. Perhaps, we could also validate the selection of α via comparison to the spectroscopic characterization of electronic states. As shown in the inset of Fig. 2(a), the deep polaron level below the conduction band can be also detected using various optical measurements such as infrared and core-level X-ray spectroscopy and electron spin resonance spectroscopy;31,38 therefore, we would like to call for experimentalists to investigate the formation and nature of localized polaronic states in electron-doped BiVO4.
Finally, we examined the effect of hydrostatic pressure on the barrier Ea for electron hopping by varying the supercell volume (or lattice constant). Fig. 4 presents predicted Ea values at α = 15% as a function of the supercell volume normalized with respect to the equilibrium volume of V0 = 2768 Å3 (=15.40 × 15.40 × 11.67 Å3). Ea increases with volume (or lattice constant); that is, Ea increases (decreases) under tension (compression). The Ea variation of a few tens of meV is not trivial considering that electron mobility is exponentially dependent on Ea (μ = μ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ea/kBT), where μ0 is the mobility prefactor13). The inset shows the calculated value of Ep, following the same trend as Ea; this indicates that polaron formation is facile under tensile conditions, which can be well understood given that the energy cost for the lattice distortions associated with a polaron is relatively less in comparison to the compressive case. The direct correlation between Ep and Ea may suggest that the increase of Ea with pressure is mainly attributed to an increase in λ, with a minor contribution from HAB. We think that the extent of HAB would be also affected to a certain degree by the changes in lattice constant, as the degree of electron localization is likely to vary with volume (see ρe variation in Fig. 4).
exp(−Ea/kBT), where μ0 is the mobility prefactor13). The inset shows the calculated value of Ep, following the same trend as Ea; this indicates that polaron formation is facile under tensile conditions, which can be well understood given that the energy cost for the lattice distortions associated with a polaron is relatively less in comparison to the compressive case. The direct correlation between Ep and Ea may suggest that the increase of Ea with pressure is mainly attributed to an increase in λ, with a minor contribution from HAB. We think that the extent of HAB would be also affected to a certain degree by the changes in lattice constant, as the degree of electron localization is likely to vary with volume (see ρe variation in Fig. 4).
 | ||
| Fig. 4 Predicted activation energies (Ea) for electron polaron hopping as a function of normalized supercell with respect to its equilibrium value (V0), i.e., ΔV = 100 × (V − V0)/V0; in the hybrid DFT calculations, 15% of HF exchange was included. The variations of the polaron formation energy (Ep) and the degree of electron localization around the VO4 tetrahedron (ρe) are also shown in the inset. | ||
The pressure dependence of Ea may raise a question regarding the temperature effect, as there is a direct relationship between temperature and volume, i.e., the volume (or lattice constant) increases with temperature. The measured volumetric thermal expansion coefficients for BiVO4 are around 8 × 10−6 °C−1 in the monoclinic phase and 50 × 10−6 °C−1 in the tetragonal phase.39 Taking those values, the volume expansion of BiVO4 is estimated to be 0.2% (1.5%) as the temperature is increased by 100 °C in the monoclinic (tetragonal) phase, corresponding to an increase in Ea by about 2 (20) meV. This suggests that the temperature effect could be important if the sample temperature varies significantly.
4. Conclusions
We examined the behavior of excess electrons in BiVO4 using hybrid functional calculations with the varying fractions α of HF exchange. Our calculation clearly shows that an excess electron remains largely localized on one V atom, accompanied by significant local lattice distortions around the reduced V4+ site. At α = 15%, the small polaron state is predicted to be about 0.5 eV more favorable than the completely delocalized state; the polaron formation energy Ep increases monotonically with increasing α, as a result of the larger increase in the electronic energy gain Ee relative to the strain energy loss Es. The polaron hopping barrier Ea is found to increase monotonically from 0.10 eV to 0.46 eV as α increases from 5% to 25%; the predicted values are consistent with the recently reported experimental estimates (≈0.3 eV in 0.3% W-doped BiVO4). Our study also demonstrates the possibility of overlap between the diffusing electron wavefunctions, leading to the concentration dependence of electron mobility in highly electron-doped BiVO4 (where the polaron–polaron separation is sufficiently small). Moreover, the hopping barrier Ea tends to increase with lattice constant; however, under typical operating conditions for pressure and temperature, the variation of Ea appears to be small, but not negligible. The improved understanding could assist in identifying effective ways to enhance the electron mobility and thus photocatalytic performance of BiVO4.Acknowledgements
This work was partially supported by the Welch Foundation (F-1535), the National Science Foundation (DMR-1122603), and the Samsung Advanced Institute of Technology (SAIT). We also acknowledge the Texas Advanced Computing Center for the use of their computing resources.References
- S. Tokunaga, H. Kato and A. Kudo, Chem. Mater., 2001, 13, 4624 CrossRef CAS.
- J. Yu and A. Kudo, Adv. Funct. Mater., 2006, 16, 2163 CrossRef CAS.
- H. Luo, A. H. Mueller, T. M. McCleskey, A. K. Burrell, E. Bauer and Q. X. Jia, J. Phys. Chem. C, 2008, 112, 6099 CAS.
- H. S. Park, K. E. Kweon, H. Ye, E. Paek, G. S. Hwang and A. J. Bard, J. Phys. Chem. C, 2011, 115, 17870 CAS.
- Y. Park, K. J. McDonald and K. Choi, Chem. Soc. Rev., 2013, 42, 2321 RSC.
- H. S. Park, K. C. Leonard and A. J. Bard, J. Phys. Chem. C, 2013, 117, 12093 CAS.
- F. F. Abdi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. van de Krol, Nat. Commun., 2013, 4, 1 Search PubMed.
- F. F. Abdi, N. Firet and R. van de Krol, ChemCatChem, 2013, 5, 490 CrossRef CAS.
- J. A. Seabold, K. Zhu and N. R. Neale, Phys. Chem. Chem. Phys., 2014, 16, 1121 RSC.
- S. J. Hong, S. Lee, J. S. Jang and J. S. Lee, Energy Environ. Sci., 2011, 4, 1781 CAS.
- J. Su, L. Guo, N. Bao and C. A. Grimes, Nano Lett., 2011, 11, 1928 CrossRef CAS PubMed.
- Z. Chen, T. F. Jaramillo, T. G. Deutsch, A. Kleiman-Shwarsctein, A. J. Forman, N. Gaillard, R. Garland, K. Takanabe, C. Heske, M. Sunkara, E. W. McFarland, K. Domen, E. L. Miller, J. A. Turner and H. N. Dinh, J. Mater. Res., 2010, 25, 3 CrossRef CAS.
- A. J. Bosman and H. J. van Daal, Adv. Phys., 1970, 19, 1 CrossRef CAS.
- A. L. Shluger and A. M. Stoneham, J. Phys.: Condens. Matter, 1993, 5, 3049 CrossRef CAS.
- D. Emin, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13691 CrossRef CAS.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Science, 2008, 321, 792 CrossRef CAS PubMed.
- P. Mori-Sánchez, A. J. Cohen and W. Yang, Phys. Rev. Lett., 2008, 100, 146401 CrossRef.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029 CrossRef CAS PubMed.
- D. M. Ramo, A. L. Shluger, J. L. Gavartin and G. Bersuker, Phys. Rev. Lett., 2007, 99, 155504 CrossRef.
- J. B. Varley, A. Janotti, C. Franchini and C. G. Van de Walle, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 081109 CrossRef.
- T. Maxisch, F. Zhou and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 104301 CrossRef.
- G. Kresse and J. Furthmuller, VASP: The Guide, Vienna University of Technology, Vienna, Austria, 2001 Search PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS PubMed.
- L. Schimka, J. Harl and G. Kresse, J. Chem. Phys., 2011, 134, 024116 CrossRef PubMed.
- M. Itoh and T. Aoki, J. Phys.: Condens. Matter, 2010, 22, 045503 CrossRef PubMed.
- V. V. Laguta, A. Vedda, D. Di Martino, M. Martini, M. Niki, E. Mihóková, J. Rosa and Y. Usuki, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 235108 CrossRef.
- K. E. Kweon and G. S. Hwang, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 205202 CrossRef.
- M. Setvin, C. Franchini, X. Hao, M. Schmid, A. Janotti, M. Kaltak, C. G. Van de Walle, G. Kresse, U. Diebold, 2014, arXiv:1401.7817 [cond-mat.str-el].
- A. Janotti, C. Franchini, J. B. Varley, G. Kresse and C. G. Van de Walle, Phys. Status Solidi RRL, 2013, 7, 199 CrossRef CAS.
- J. M. Garcia-Lastra, J. S. G. Myrdal, R. Christensen, K. S. Thygesen and T. Vegge, J. Phys. Chem. C, 2013, 117, 5568 CAS.
- K. E. Kweon and G. S. Hwang, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 165209 CrossRef.
- J. Blumberger and K. P. Mckenna, Phys. Chem. Chem. Phys., 2013, 15, 2184 RSC.
- D. Emin and T. Holstein, Ann. Phys., 1969, 53, 439 Search PubMed; I. G. Austin and N. F. Mott, Adv. Phys., 2001, 50, 757 CrossRef.
- N. A. Deskins and M. Dupuis, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 195212 CrossRef.
- A. J. E. Rettie, H. C. Lee, L. G. Marshall, J. Lin, C. Capan, J. Lindermuth, J. S. McCloy, J. Zhou, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2013, 135, 11389 CrossRef CAS PubMed.
- D. C. Cronemeyer, Phys. Rev., 1959, 113, 1222 CrossRef CAS.
- R. M. Hazen and J. W. E. Mariathasan, Science, 1982, 216, 991 CAS.
| This journal is © the Owner Societies 2015 |