
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Binding or aggregation? Hazards of interpretation in studies of molecular recognition by porphyrins in water†
Charles M.
Renney
ab,
Gaku
Fukuhara
b,
Yoshihisa
Inoue
b and
Anthony P.
Davis
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: Anthony.Davis@bristol.ac.uk; Fax: +44-117-9298611; Tel: +44-117-9546334
bDepartment of Applied Chemistry, Osaka University, Suita 565-0871, Japan
First published on 6th May 2015
Abstract
Reports have suggested that polar porphyrins such as tetraphenylporphine tetrasulfonate (TPPS) can serve as carbohydrate receptors in water. Here we find that TPPS shows changes in UV-visible absorption when treated with glucose, but that these are best explained by altered aggregation states and not by formation of a closely-bound complex.
Biomimetic carbohydrate recognition presents a major challenge to supramolecular chemists.1 The problem is significant because saccharide recognition is a central natural phenomenon which plays roles in many biological processes.2 At the same time, theory suggests that binding carbohydrates in water should be unusually difficult. Polar interactions must compete with solvation, while the hydrophobic surfaces in saccharides are relatively small. In accordance, natural carbohydrate recognition is generally weak.3 The affinities (Ka) of lectins (carbohydrate-binding proteins) for monosaccharides are typically in the range 102–104 M−1, much smaller than most other biomolecular interactions.4
In general, research on synthetic carbohydrate receptors has confirmed the scale of the challenge. For example our own efforts to bind glucose in water, using macrocyclic “synthetic lectins”, have achieved binding constants of just 60–90 M−1.5 However, the literature contains some counter-examples in which simple systems perform surprisingly well. In particular a range of porphyrins with polar substituents have been studied in water (usually with small amounts of added methanol) by UV-visible or fluorescence spectroscopy, and found to bind monosaccharides with remarkable affinities.6 Porphyrins 1 and 2 are just two of many which have been investigated. Tetraphenylporphine tetrasulfonate (TPPS, 1) was reported to bind glucose 3 and galactose 4 with Ka = 120 and 135 M−1 respectively (water–methanol, 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5),6c while the tetraphosphonate 2 gave corresponding values of 17600 and 19700 M−1.6b
:5),6c while the tetraphosphonate 2 gave corresponding values of 17600 and 19700 M−1.6b
The apparent success of these systems raised interesting questions concerning the geometry and driving force for binding, which were not fully answered in the original reports. We therefore undertook a further study of the interaction between commercially available TPPS 1 and glucose 3. Herein we report the results, which highlight the difficulty of exploiting porphyrin units in water-soluble receptors. Although porphyrins provide rigid scaffolds and extended hydrophobic surfaces, their tendency to aggregate can lead to complex behaviour which is readily misinterpreted. Unfortunately we can find no clear evidence that discrete, closely-associated complexes form between 1 and glucose under the conditions of these experiments.
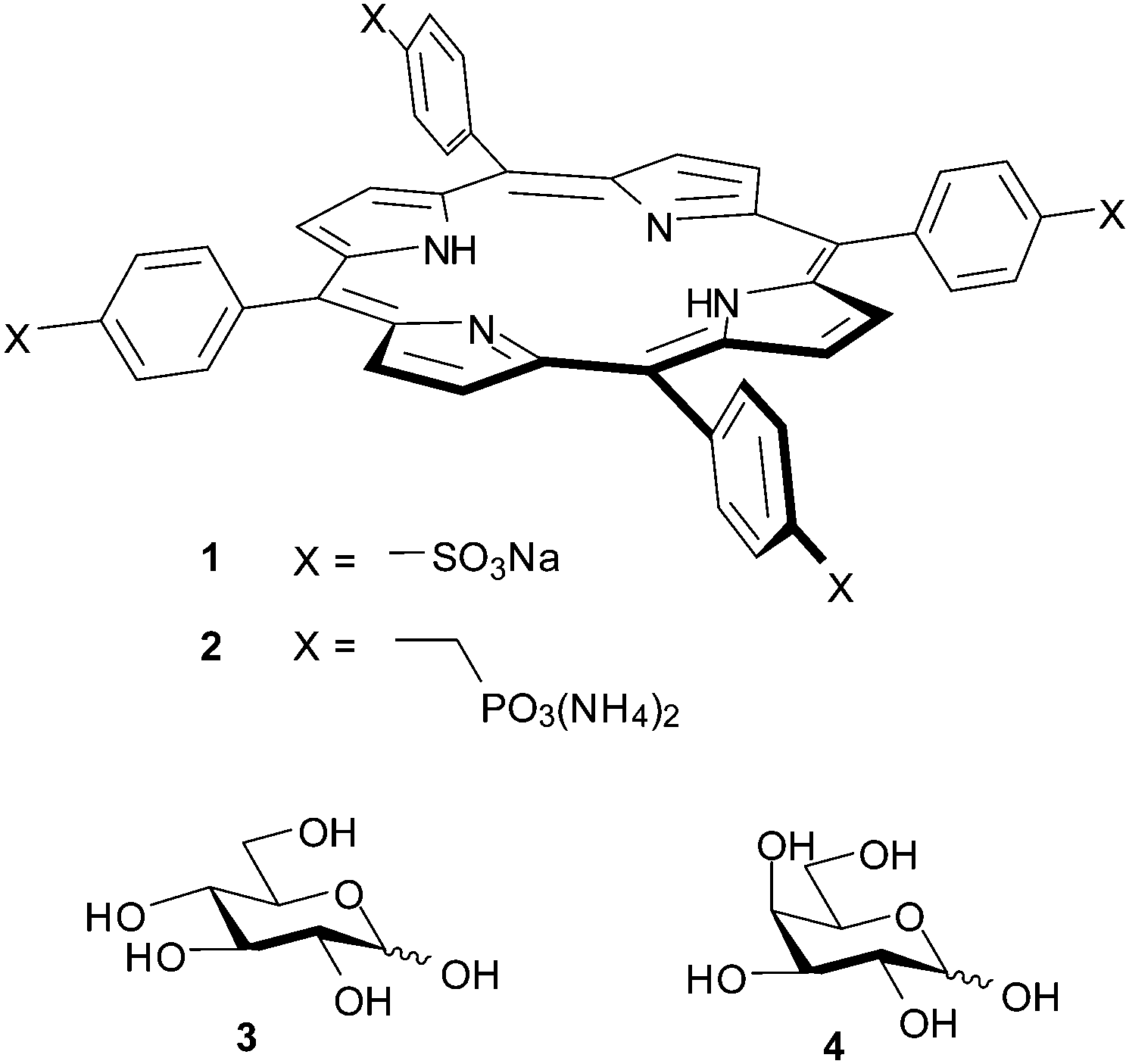
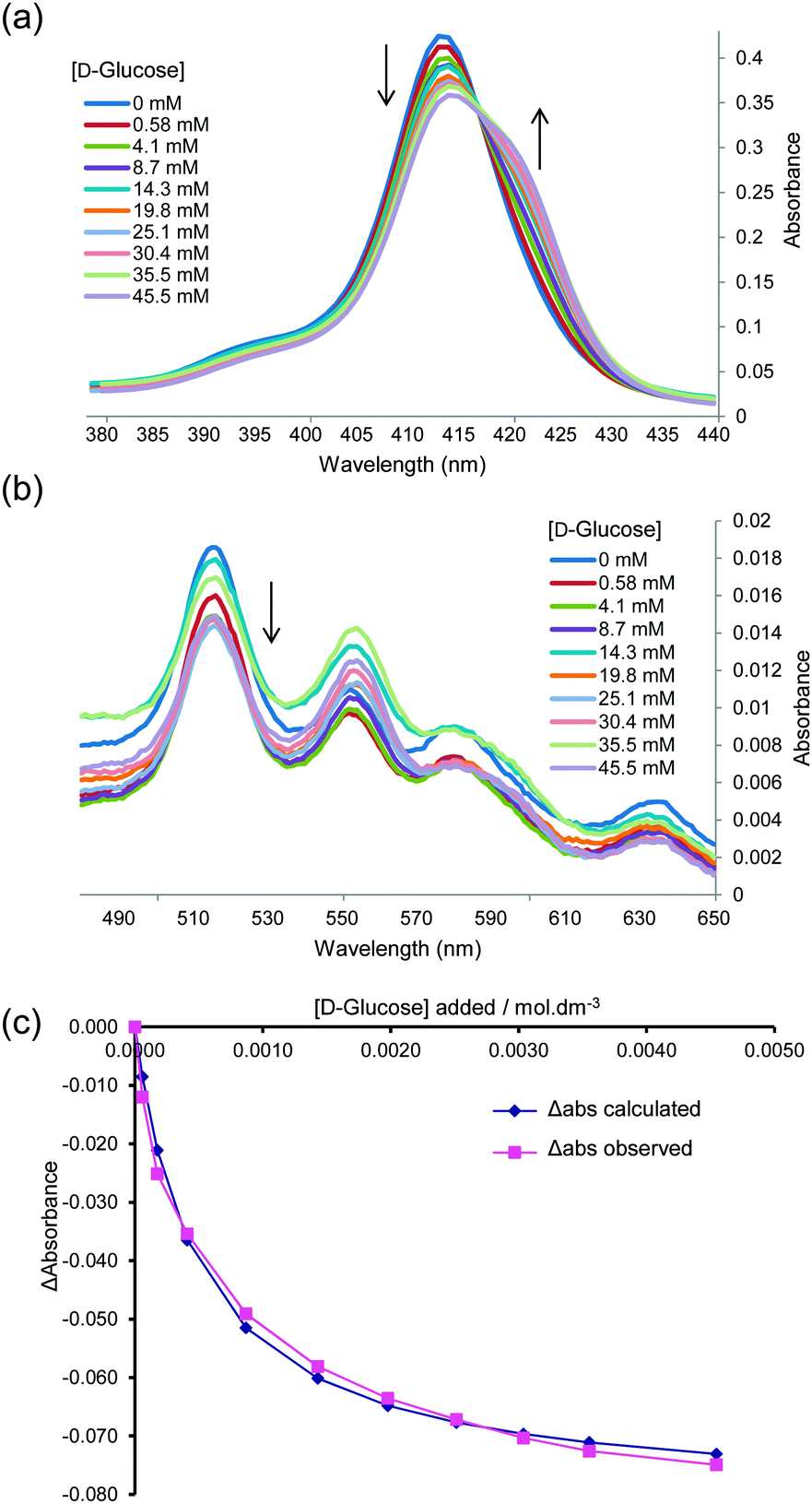
We began by adding glucose to TPPS 1 (6 μM) in water–methanol 95:5, and following the interaction by UV-visible spectroscopy (as performed in the original work). Significant changes were observed in both the Soret and Q absorption bands of 1, as reported previously6c and as illustrated in Fig. 1a and b. Analysis of the data according to a 1:1 binding model could give tolerably good fits, as illustrated in Fig. 1c. The Ka value obtained from this analysis was 2000 M−1, which is significantly higher than that reported previously.6c To check for aggregation of 1 we performed a dilution study following the intensity of the Soret band in the concentration range 3–60 μM, again following the earlier work. As shown in Fig. 2, no significant deviation was observed from the Beer–Lambert law, suggesting that 1 was monomeric at these concentrations.
 | ||
| Fig. 1 UV-visible titration of D-glucose into TPPS 1 (6.15 μM in water–MeOH 95:5). (a) and (b) Absorption in the Soret and Q bands respectively. Arrows indicate decrease or increase of absorbance as glucose is added. (c) Experimental and calculated values obtained by fitting the absorbance data at 413 nm to a 1:1 binding model. Apparent Ka = 2000 M−1. Similar curves may be obtained from experiments in 100% water; see for example ESI,† Fig. S1–S3. | ||
 | ||
| Fig. 2 The absorbance of TPPS 1 in water–methanol 95:5 at 412 nm plotted against concentration. For spectra see Fig. S4 (ESI†). | ||
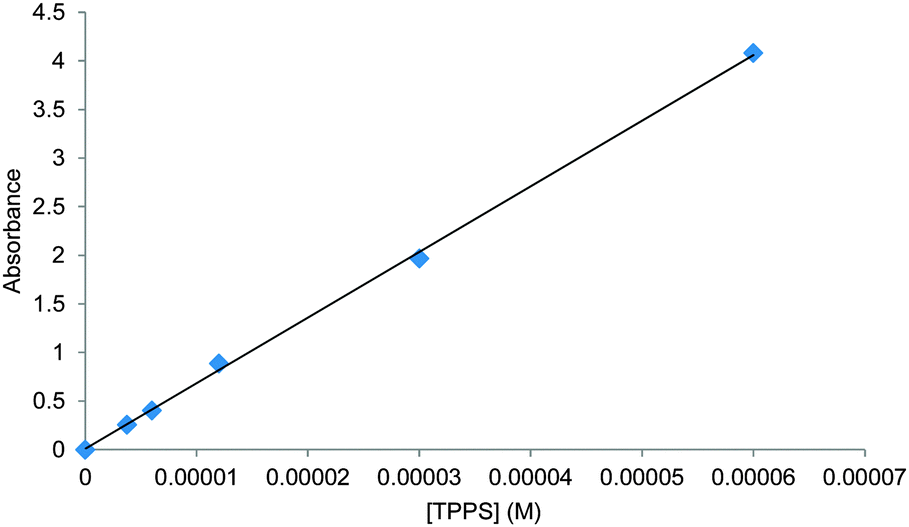
Although these results could be viewed as promising, the initial experiments gave some cause for concern. While some titrations gave good fits to 1:1 binding, others did not. The change in observed absorption varied considerably between experiments and, where analysis seemed possible, the derived binding constants varied considerably. We therefore sought confirmation by other techniques. We were especially interested to apply 1H NMR, as chemical shift changes can often be related directly to contacts between interacting molecules. In the present case, the porphyrin ring current is known to induce dramatic changes to the chemical shifts of neighbouring protons.7 If the glucose is tightly associated with TPPS in any reasonable geometry, some effects should be expected. Accordingly, a 1H NMR titration experiment was performed in which TPPS 1 (up to 10 mM) was added to glucose (2 mM) in D2O. As shown in Fig. 3a, the signals due to glucose were almost entirely unaffected by the added porphyrin. The signals due to the TPPS moved significantly, but the changes matched those for a dilution study in the absence of glucose (see Fig. S5, ESI†) and are presumably due to aggregation. The reverse experiment in which glucose was added to TPPS again showed no sign of interaction between the species (Fig. 3b). Other methods were also investigated. Isothermal titration calorimetry (ITC) of glucose into 1 (0.5 mM) revealed almost no enthalpy change and no evidence for saturation as expected for 1:1 binding (see Fig. S6, ESI†). Attempts to detect induced CD on addition of glucose to 1 were also unsuccessful (Fig. S7, ESI†).
 | ||
| Fig. 3 1H NMR spectra of mixtures of TPPS 1 and glucose. (a) Addition of TPPS to glucose (2 mM) in D2O. Signals due to glucose (3–4 ppm) are unchanged (b) addition of glucose to TPPS (2 mM) in D2O. Signals due to TPPS (>7.5 ppm) are unchanged. | ||
Faced with these contradictory results, we considered whether some other explanation might account for the spectra in Fig. 1. In particular we realised we should revisit the question of aggregation. Porphyrins are known to associate strongly in aqueous solution, even when furnished with powerful water-solubilising substituents. TPPS itself is much studied8 and the literature suggests that, depending on conditions, aggregation can occur even at μM concentrations.8a,e We therefore decided to supplement the UV-visible dilution study (Fig. 2) with fluorescence spectroscopy. Fluorescence emission spectra were obtained of 1 in water–methanol 95:5 at concentrations between 6 and 0.05 μM. As shown in Fig. 4, clear changes were observed. As the sample was diluted, emission at 605 nm increased relative to other wavelengths. Fluorescence lifetime measurements confirmed that this was due to the appearance of a new species (see Table S1, ESI†). It thus seemed that the UV-visible study was misleading, and that aggregation was occurring even at these very low concentrations.
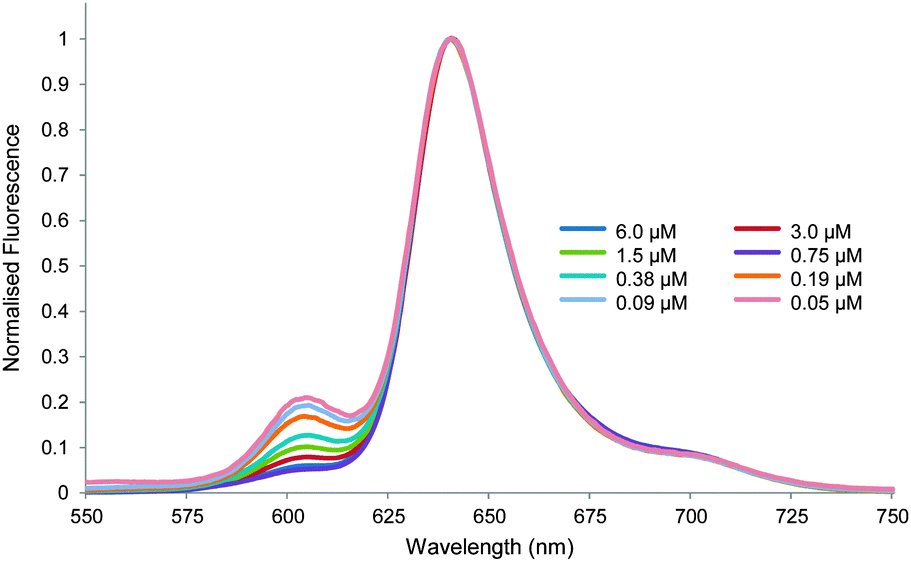 | ||
| Fig. 4 Normalised fluorescence emission spectra for the dilution of TPPS 1 in water–methanol 95:5. Excitation wavelength = 426 nm. | ||
A key observation made during these studies was that the UV-visible absorption spectrum of 1 varied with sample age. For example, the 413 nm Soret absorption of 1 (6 μM) in water, prepared by dilution from a 1 mM stock solution, decayed gradually by ∼3% over a period of 24 hours (see Fig. 5 and Fig. S8, S9). Though unexpected to us, this phenomenon is not new. Others have noted that the UV-visible spectra of 1 depend on sample history, and equilibrate over slow timescales, due to gradual changes in aggregation state.8c,e In the present case dilution of the stock solution disturbs the equilibrium position, and the redistribution between different types of aggregate clearly takes several hours. We also observed that the rate and scale of absorption changes could be affected by additives, including carbohydrates. Thus addition of glucose (600 μM) to the above solution caused a reduction in the Soret absorption of 7% over 24 hours, while glycerol (600 μM) produced >8% decay over 12 hours (Fig. 5). Both fluorescence lifetime and dynamic light scattering experiments confirmed that these changes were due to slow redistribution between aggregation states. The former technique detected two species emitting at 605 nm with lifetimes of ∼2 and ∼10 ns respectively. In all three solutions, the abundance of the 2 ns species increased substantially (from ∼50% to ∼80%) over the 24 hour period (see Table S2, ESI†). Dynamic light scattering on the solutions of 1 and 1 + glucose revealed the presence of both large (∼3000 nm) and small (1–2 nm) aggregates immediately after sample preparation. The large particles became smaller and less predominant over a 16 hour period (see Fig. S10, S11 and Table S3, ESI†).
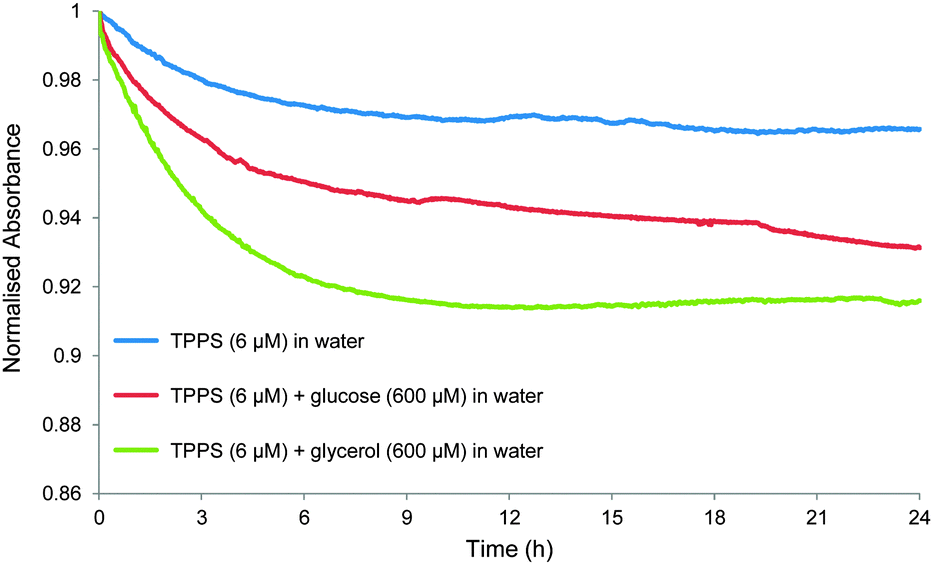 | ||
| Fig. 5 Changes in normalised absorbance at 413 nm over time for TPPS 1 in water (6 μM) (blue line); TPPS (6 μM) + glucose (600 μM) in water (red line); and TPPS (6 μM) + glycerol (600 μM) in water (green line). | ||
These results suggest an explanation for the data in Fig. 1. At the start of the experiment, dilution of the stock solution of 1 gives a metastable solution in which much of the porphyrin is collected in large assemblies. As the titration proceeds, the aggregation state of 1 in the cuvette slowly changes, mediated in part by the added glucose. Depending on the time taken between additions a curve results which, by coincidence, may approximate to a 1:1 binding isotherm. Consistent results are possible if the operator works in a systematic manner, but any variation in procedure leads to different curve shapes and/or apparent binding constants (as observed by us). The exact role of the glucose is unclear, and may involve some degree of binding to specific aggregates. However, the changes in the absorption spectra do not reflect the formation of discrete 1:1 complexes. Moreover the effect is not specific, as glycerol seems to behave similarly.
In conclusion, we have re-examined the porphyrin 1 as a receptor for carbohydrates in aqueous solution, and have found no evidence for the formation of 1:1 complexes. Although addition of glucose may induce changes in the UV-visible spectra of 1, these seem to be associated with complex and kinetically slow adjustments to the aggregation state of the porphyrin. Changes can occur even when nothing is added to the TPPS, and other additives (e.g. glycerol) can also serve as promoters. Titration experiments may yield curves which seem consistent with 1:1 binding, but this impression is illusory. While these conclusions apply only to TPPS 1, it seems likely that other simple porphyrins will behave similarly. Reports of carbohydrate recognition by porphyrins in aqueous solution should therefore be viewed with caution, unless it is quite clear that changes in aggregation state cannot be responsible for the observations.
We thank the Japanese Society for the Promotion of Science for a Summer Programme grant, and the Bristol Chemical Synthesis Centre for Doctoral Training, funded by EPSRC (EP/G036764/1), Novartis and the University of Bristol, for a PhD studentship (both to C.M.R.).
Notes and references
- For reviews, see: (a) A. P. Davis and R. S. Wareham, Angew. Chem., Int. Ed., 1999, 38, 2978–2996 CrossRef; (b) A. P. Davis, Org. Biomol. Chem., 2009, 7, 3629–3638 RSC; (c) S. Jin, Y. F. Cheng, S. Reid, M. Y. Li and B. H. Wang, Med. Res. Rev., 2010, 30, 171–257 CAS; (d) M. Mazik, Chem. Soc. Rev., 2009, 38, 935–956 RSC; (e) Y. Nakagawa and Y. Ito, Trends Glycosci. Glycotechnol., 2012, 24, 1–12 CrossRef CAS . For recent examples, see: ; (f) C. Nativi, O. Francesconi, G. Gabrielli, I. De Simone, B. Turchetti, T. Mello, L. D. C. Mannelli, C. Ghelardini, P. Buzzini and S. Roelens, Chem. – Eur. J., 2012, 18, 5064–5072 CrossRef CAS PubMed; (g) G. Fukuhara and Y. Inoue, J. Am. Chem. Soc., 2011, 133, 768–770 CrossRef CAS PubMed; (h) A. Pal, M. Berube and D. G. Hall, Angew. Chem., Int. Ed., 2010, 49, 1492–1495 CrossRef CAS PubMed; (i) M. Rauschenberg, S. Bomke, U. Karst and B. J. Ravoo, Angew. Chem., Int. Ed., 2010, 49, 7340–7435 CrossRef CAS PubMed; (j) O. Francesconi, C. Nativi, G. Gabrielli, M. Gentili, M. Palchetti, B. Bonora and S. Roelens, Chem. – Eur. J., 2013, 19, 11742–11752 CrossRef CAS PubMed; (k) S. Rieth, M. R. Miner, C. M. Chang, B. Hurlocker and A. B. Braunschweig, Chem. Sci., 2013, 4, 357–367 RSC.
- See for example: (a) The Sugar Code – Fundamentals of Glycoscience, ed. H.-J. Gabius, Wiley-Blackwell, Weinheim, 2009 Search PubMed; (b) B. Wang and G.-J. Boons, Carbohydrate Recognition: Biological Problems, Methods and Applications, Wiley, Hoboken, 2011 Search PubMed; (c) D. Solís, N. V. Bovin, A. P. Davis, J. Jiménez-Barbero, A. Romero, R. Roy, K. Smetana Jr. and H. J. Gabius, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 186 CrossRef PubMed.
- M. Ambrosi, N. R. Cameron and B. G. Davis, Org. Biomol. Chem., 2005, 3, 1593–1608 CAS.
- E. J. Toone, Curr. Opin. Struct. Biol., 1994, 4, 719–728 CrossRef CAS.
- N. P. Barwell, M. P. Crump and A. P. Davis, Angew. Chem., Int. Ed., 2009, 48, 7673–7676 CrossRef CAS PubMed; C. Ke, H. Destecroix, M. P. Crump and A. P. Davis, Nat. Chem., 2012, 4, 718–723 CrossRef PubMed; H. Destecroix, C. Renney, T. J. Mooibroek, T. S. Carter, P. F. N. Stewart, M. P. Crump and A. P. Davis, Angew. Chem., Int. Ed., 2015, 54, 2057–2061 CrossRef PubMed.
- (a) O. Rusin and V. Král, Chem. Commun., 1999, 2367–2368 RSC; (b) V. Král, O. Rusin, J. Charvatova, P. Anzenbacher and J. Fogl, Tetrahedron Lett., 2000, 41, 10147–10151 CrossRef; (c) J. Charvatova, O. Rusin, V. Král, K. Volka and P. Matejka, Sens. Actuators, B, 2001, 76, 366–372 CrossRef CAS; (d) V. Král, O. Rusin and F. P. Schmidtchen, Org. Lett., 2001, 3, 873–876 CrossRef PubMed; (e) O. Rusin, M. Hub and V. Král, Mater. Sci. Eng., C, 2001, 18, 135–140 CrossRef; (f) O. Rusin and V. Král, Sens. Actuators, B, 2001, 76, 331–335 CrossRef CAS; (g) O. Rusin, K. Lang and V. Král, Chem. – Eur. J., 2002, 8, 655–663 CrossRef CAS; (h) J. Kralova, J. Koivukorpi, Z. Kejik, P. Pouckova, E. Sievanen, E. Kolehmainen and V. Král, Org. Biomol. Chem., 2008, 6, 1548–1552 RSC.
- R. J. Abraham, G. R. Bedford, D. McNeillie and B. Wright, Org. Magn. Reson., 1980, 14, 418–425 CrossRef CAS PubMed.
- (a) N. C. Maiti, M. Ravikanth, S. Mazumdar and N. Periasamy, J. Phys. Chem., 1995, 99, 17192–17197 CrossRef CAS; (b) A. S. R. Koti and N. Periasamy, J. Mater. Chem., 2002, 12, 2312–2317 RSC; (c) M. A. Castriciano, A. Romeo, V. Villari, N. Angelini, N. Micali and L. M. Scolaro, J. Phys. Chem. B, 2005, 109, 12086–12092 CrossRef CAS PubMed; (d) B. A. Friesen, K. R. A. Nishida, J. L. McHale and U. Mazur, J. Phys. Chem. C, 2009, 113, 1709–1718 CrossRef CAS; (e) J. V. Hollingsworth, A. J. Richard, M. G. H. Vicente and P. S. Russo, Biomacromolecules, 2012, 13, 60–72 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplementary figures showing spectra, ITC and DLS data, tables showing fluorescence lifetime and DLS data. See DOI: 10.1039/c5cc02768c |
| This journal is © The Royal Society of Chemistry 2015 |