
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unique Group 1 cations stabilised by homoleptic neutral phosphine coordination†
Marina Carravetta,
Maria Concistre,
William Levason,
Gillian Reid* and
Wenjian Zhang
Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK. E-mail: G.Reid@soton.ac.uk
First published on 8th May 2015
Abstract
Homoleptic coordination of the neutral diphosphines Me2P(CH2)2PMe2 and o-C6H4(PMe2)2 to the hard Li+ and Na+ cations is achieved using Li[Al{OC(CF3)3}4] and Na[B{3,5-(CF3)2-C6H3}4] as ‘naked’ cation sources. Crystallographic, solid state and solution multinuclear NMR studies confirm distorted octahedral coordination solely via three chelating diphosphines in these unique species.
Neutral phosphine ligands, PR3 (R = alkyl, aryl), are ubiquitous in transition metal chemistry, owing to their capacity to tune the electronic and steric properties, and hence the reactivity, of the complexes, and to the strong σ-donor properties of the soft phosphine donor functions. This has led to wide utilisation of phosphine co-ligands in many transition metal reagents and catalysts.1 Phosphine complexes of many p-block acceptors have also developed substantially in recent years.2 However, complexes involving coordination of neutral phosphine ligands towards the strongly electropositive s-block elements, particularly the Group 1 cations, have remained extremely elusive, and there are no reported examples with exclusively PR3 coordination. This is no doubt in part due to the high affinity of the alkali metal and alkaline earth cations for hard, electronegative Lewis bases such as water, alkoxide, amide etc., their high lability, as well as the high lattice energies often associated with many Group 1 and 2 salts, which severely limit their solubilities in non-competitive organic media. Thus, to-date there has been only one reported example of a neutral phosphine co-ligand coordinated to an alkali metal cation, the organometallic silylamide dimer [Li{N(Ar)CC(R)Si(R)2NAr}(μ-Me2PCH2CH2PMe2)]2 (mean d(Li−P) = 2.650(3) Å),3 and two structurally authenticated species with PR3 coordination to alkaline earth ions; [BeCl2(κ1-Ph2PCH2PPh2)2]4 and the dinuclear [Be2Cl2(μ-Cl)2(PCy3)2],5 both containing distorted tetrahedral Be(II).
A small number of anionic ligands bearing phosphine functions have been coordinated to s-block cations, including [Mg{C6H3-2,6-(CH2PMe2)2}2], in which the Mg–P bonds are also stabilised by the anionic pincer ligand framework,6 [{Li(2-PPh2-C6H4)}2(OEt2)2] (d(Li–P) = 2.69–2.75 Å),7 hindered alkoxy–phosphine complexes, including [Li(μ-OCtBu2CH2PR2)]2 (R = Me or Ph) and [Li(μ-OCtBu2CH2PPh2)2Li(OCtBu2)] (d(Li–P) = 2.50–2.65 Å),8 [Na(H2Al{P(SiMe3)2}2)(dme)2],9 [Li(solvent)x{Ph2B(CH2PiPr2)2}] (solvent = thf, x = 2; Et2O, x = 1) (d(Li–P) = 2.596(3), 2.608(3) Å),10 as well as a small number of Li+ complexes with (phosphinomethyl)aluminate ligands.11 The negative charge on the anionic ligands in these species brings a significant electrostatic component to the bonding, and contrasts the covalent metal–phosphine bonding present in the d- and p-block acceptor ions.
In recent work we reported12 that complexes of Na+ with polyamines and aza macrocycles, including the [Na(Me3-tacn)2]+ sandwich cation and the distorted five-coordinate [Na(thf)(Me4-cyclam)]+ cation (Me3-tacn = 1,4,7-trimethyl-1,4,7-triazacyclononane, Me4-cyclam = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane), could be prepared readily by using Na[BArF]·2thf ([BArF]− = [B{3,5-(CF3)2-C6H3}4]−). This [BArF]− salt13 has good solubility in weak-donor solvents such as CH2Cl2 and toluene. Very recently we extended this chemistry by reporting the homoleptic octathia coordination to Na+ in the macrocyclic complex, [Na([24]aneS8)][BArF] ([24]aneS8 = 1,4,7,10,13,16,19,22-octathiacyclotetracosane), containing distorted dodecahedral coordination.14 To develop this chemistry further we sought to establish whether it would be possible to induce coordination of softer, neutral phosphine ligands towards Group 1 cations without the additional stability offered by the macrocyclic frameworks employed in the aza and thioether chemistry. To achieve this we have used both the [BArF]−15 and [Al{OC(CF3)3}4]− weakly coordinating anions.16–19
We describe here the first series of Group 1 cations coordinated only to neutral phosphine ligands, in the form of distorted octahedral Li+ and Na+ cations containing tris-diphosphine coordination.
Reaction of Li[Al{OC(CF3)3}4]19 with three mol. equiv. of Me2PCH2CH2PMe2 (dmpe) or o-C6H4(PMe2)2 (diphos) in anhydrous toluene gives [Li(dmpe)3][Al{OC(CF3)3}4] and [Li(diphos)3][Al{OC(CF3)3}4], respectively as white solids in very good yield.‡ The corresponding [Na(dmpe)3][BArF] and [Na(diphos)3][BArF] salts were obtained similarly from a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diphosphine: Na[BArF] ratio. Attempts to prepare the analogous K+ complexes by reaction of the diphosphine with K[BArF] in a 3:1 molar ratio failed, while the weaker donor and sterically bulkier o-C6H4(PPh2)2, and the diarsine, o-C6H4(AsMe2)2 (the direct analogue of diphos), did not coordinate to Li+ or Na+ under similar conditions.
:1 diphosphine: Na[BArF] ratio. Attempts to prepare the analogous K+ complexes by reaction of the diphosphine with K[BArF] in a 3:1 molar ratio failed, while the weaker donor and sterically bulkier o-C6H4(PPh2)2, and the diarsine, o-C6H4(AsMe2)2 (the direct analogue of diphos), did not coordinate to Li+ or Na+ under similar conditions.
The coordination environments present in the new complexes were established unambiguously from X-ray crystallographic studies on three examples. The structure§ of [Na(dmpe)3][BArF] contains discrete Na+ cations coordinated to three chelating dmpe ligands, in a distorted octahedral environment (Fig. 1), with discrete [BArF]− anions providing charge balance. The Na–P bond distances lie in the range 2.95–3.03 Å, suggesting relatively weak coordination, and the P–Na–P angles within the five-membered chelate rings are very acute (69.8–73.4°). A similar structure is present in [Na(diphos)3][BArF],§ with coordination at Na+ through six P-donor atoms from three chelating diphos ligands, with d(Na–P) = 2.92–3.07 Å (Fig. 2). As in the dmpe complex, these are considerably longer than the sum of the ionic radius for Na (1.02 Å) and the covalent radius for P (1.06 Å). They compare with [Na(H2Al{P(SiMe3)2}2)(dme)2] (d(Na–P) = 3.052(1), 3.092(1) Å).9 The P–Na–P angles within the chelate rings are even more acute, ca. 65°, reflecting the smaller bite angle associated with the rigid o-phenylene diphosphine cf. the dimethylene-linked dmpe. The large [BArF]− anions remain discrete, but interleave between the cations (Fig. S1, ESI†).
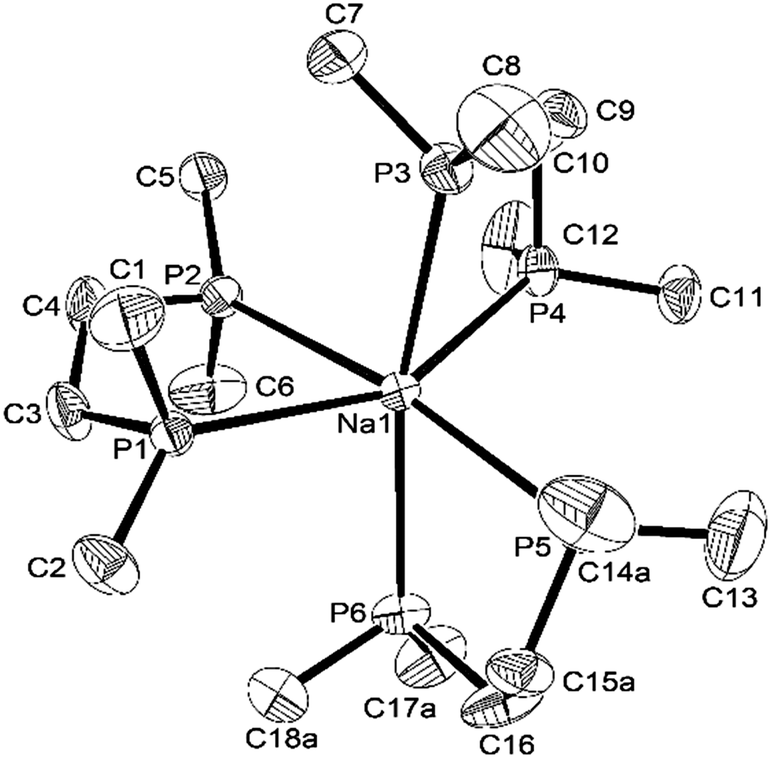 | ||
| Fig. 1 View of the structure of the [Na(dmpe)3]+ cation with numbering scheme adopted. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Na1–P1 3.0287(8), Na1–P2 3.0035(8), Na1–P3 2.9621(8), Na1–P4 2.9459(8), Na1–P5 2.9718(8), Na1–P6 2.9960(8), P1–Na1–P2 69.801(18), P3–Na1–P4 73.40(2), P5–Na1–P6 71.48(2). | ||
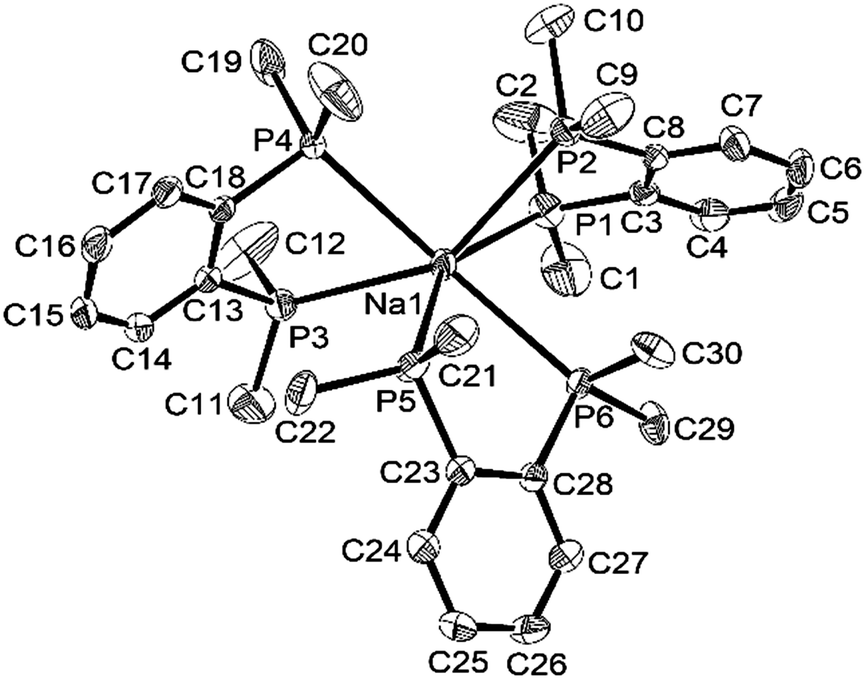 | ||
| Fig. 2 View of the structure of the [Na(diphos)3]+ cation with numbering scheme adopted. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. Selected bond lengths (Å) and angles (o): Na1–P1 2.9216(12), Na1–P2 3.0677(12), Na1–P3 3.0087(12), Na1–P4 2.9937(12), Na1–P5 2.9992(12), Na1–P6 3.0400(12), P1–Na1–P2 65.36(3), P3–Na1–P4 65.49(3), P5–Na1–P6 65.57(3). | ||
The structure§ of the lithium–diphosphine complex, [Li(diphos)3][Al{OC(CF3)3}4] was also determined from a small, weakly diffracting crystal. While the weak diffraction data mean that detailed geometric comparisons require caution, the presence of three chelating neutral diphos ligands at Li+, giving homoleptic P6-coordination, is unequivocal (Fig. 3). The aluminate anion provides charge balance, but does not interact with the cation. The Li–P bond distances are considerably shorter (by ca. 0.4 Å) than d(Na–P) in these systems, while the P–Li–P angles within the chelate rings are correspondingly larger (ca. 75°), as expected due to the smaller ionic radius.
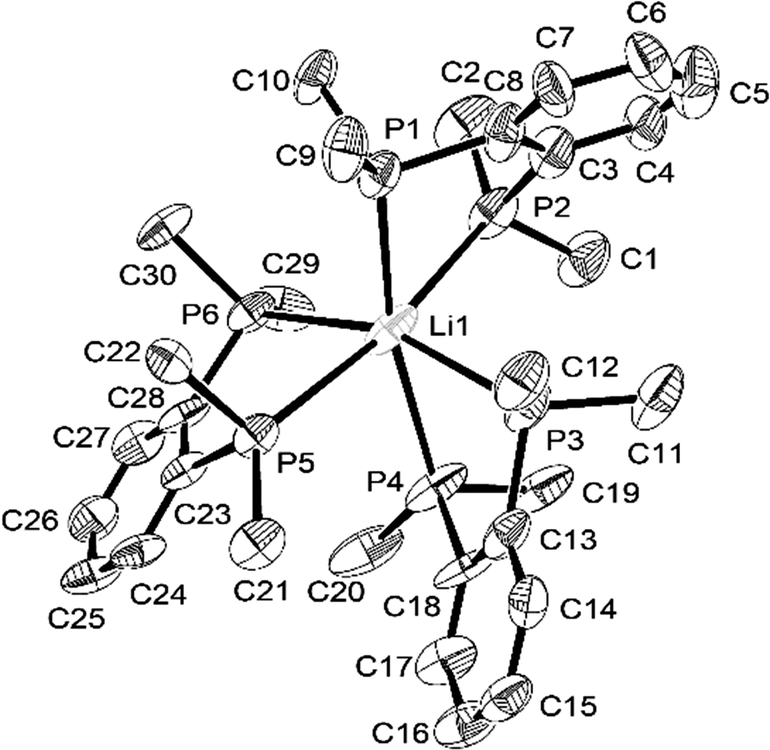 | ||
| Fig. 3 View of the structure of the [Li(diphos)3]+ cation with numbering scheme adopted. Ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. | ||
To investigate the properties of these unusual complexes further, we obtained the MAS NMR spectroscopic data (31P, 23Na and 7Li) from the powdered solids. The NMR data are summarised in Table 1. The spectra for [Li(dmpe)3]+ are shown in Fig. 4 (the other spectra are provided as ESI,† Fig. S2–S4).¶ The 31P NMR data from direct excitation (Fig. 4(a)) exhibits two peaks, the main one at −54.5 ppm is attributed to the six equivalent P-donor atoms in the complex cation; the minor peak at −48.5 ppm is consistent with the chemical shift for ‘free’ dmpe in solution (−48 ppm).20 This is further confirmed by 31P cross-polarization (CP) MAS21 data (Fig. 4(b)), where the second peak is absent, in accord with the highly mobile nature of ‘free’ dmpe.
| Complex | δ31P/ppm | δ7Li/ppm | δ23Na/ppmc | |||
|---|---|---|---|---|---|---|
| Solid | Solnb | Solid | Solnb | Solid | Solnb | |
| a Small amounts of uncomplexed ligand (literature δ31P = −48 (dmpe)20 and −55 (diphos)20) were also observed in the solid state spectra in some of the samples, arising from some sample degradation during spectral acquisition. b Li complexes recorded in d8-toluene solution (298 K); Na complexes recorded in CD2Cl2 solution (298 K). c δ23Na measured for Na[BArF] = −35.5 (s) ppm. | ||||||
| [Li(diphos)3][Al{OC(CF3)3}4] | −59.2 | −53.0 | +0.4 | −0.1 | — | — |
| [Li(dmpe)3][Al{OC(CF3)3}4] | −54.5 | −45.9 | −0.7 | −1.3 | — | — |
| [Na(diphos)3][BArF] | −61.5, −59.0 (1:1) |
−50.3 | — | — | +3.8 | +5.5 |
| [Na(dmpe)3][BArF] | −57.4 | −49.7 | — | — | +8.9 | +2.5 |
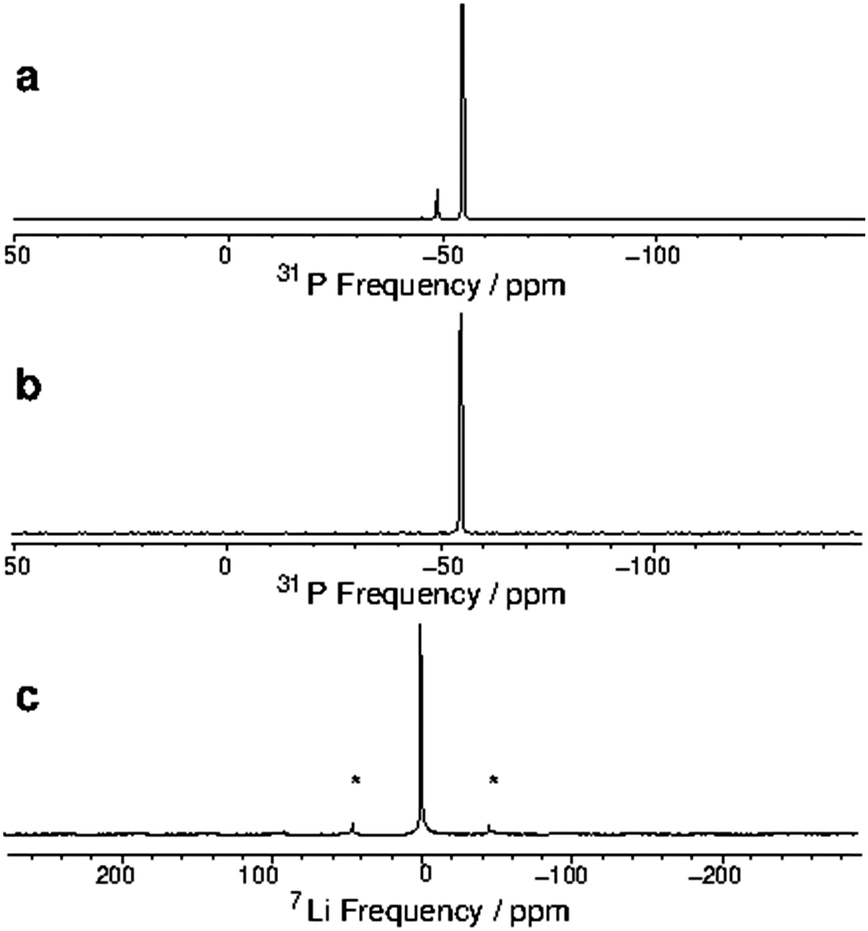 | ||
| Fig. 4 Solid state MAS NMR measurements at 9.4 T and 7.1 kHz spinning frequency on [Li(dmpe)3][Al{OC(CF3)3}4]. Spinning sidebands are labelled with asterisks. (a) Direct acquisition 31P NMR spectrum. The spectrum is an average of 8 scans with inter-pulse delay of 60 s. The main peak at −54.5 ppm is due to the complex, while the small peak at −48.5 ppm corresponds to ‘free’ dmpe (due to a small amount of decomposition of the complex during data acquisition). (b) 31P CP MAS NMR spectrum, where the peak from the very mobile reagent (‘free’ dmpe) is absent. The spectrum is an average of 16 scans with inter-pulse delay of 10 s. (c) Direct excitation 7Li NMR spectrum. The spectrum is an average of 32 scans with inter-pulse delay of 15 s. The signal is at −0.7 ppm. | ||
The chemical shift differences between ‘free’ and coordinated diphosphine (Δ) are small and negative for all four complexes; (L = dmpe: Δ = −6 ppm for Li+; −9.4 ppm for Na+; L = diphos: Δ = −5 ppm for Li+; ca. −6 ppm for Na+−). These contrast with the large, positive Δ values typically observed in transition metal phosphine complexes which contain five-membered chelate rings.22 No 7Li–31P/23Na–31P couplings are evident in the spectra, presumably due to the small magnitude of the J values, which fall within the line width.
Fig. 4(c) shows the 7Li NMR data for [Li(dmpe)3][Al{OC(CF3)3}4], showing a single peak at −0.7 ppm.
Solution 1H and 31P{1H} NMR spectra (d8-toluene or CD2Cl2) on the four compounds also show very small coordination shifts, although in these spectra the resonances are closer to the respective ‘free’ ligand. These, as well as the 7Li and 23Na solution spectra, are essentially unchanged upon cooling to 183 K (it seems likely that the low temperature-limiting spectrum is not reached at the freezing point of the solvent). These observations may indicate that in solution the complexes are partially dissociated, leading to chemical shifts closer to the free ligand values. Sharp singlets are observed by 7Li and 23Na NMR spectroscopy, with chemical shifts similar to those from the solid state spectra (Table 1).
These results demonstrate that unusual homoleptic neutral phosphine complexes of the Group 1 cations can be readily accessed in (non-polar) organic media through the use of the strong σ-donating bidentate ligands with ‘naked’ metal cation sources.
We thank the EPSRC for support through a Programme Grant (EP/I033394/1). The SCFED Project (http://www.scfed.net) is a multidisciplinary collaboration of British universities investigating the fundamental and applied aspects of supercritical fluids. MCa also thanks the Royal Society for a University Research Fellowship.
Notes and references
- See for example Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon Press, Oxford, 1987 Search PubMed; Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2004 Search PubMed; P. W. N. M. van Leeuwen, Homogeneous Catalysis: Understanding the Art, Kluwer Academic Publishers, Dordrecht, 2004 Search PubMed; R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, 2005 Search PubMed.
- J. Burt, W. Levason and G. Reid, Coord. Chem. Rev., 2014, 260, 65 CrossRef CAS PubMed.
- R. J. Bowen, M. A. Fernandes, P. B. Hitchcock, M. F. Lappert and M. Lay, J. Chem. Soc., Dalton Trans., 2002, 3253 RSC.
- G. Frenking, N. Holzmann, B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2010, 636, 1772 CrossRef CAS PubMed.
- H. Braunschweig and K. Gruss, Z. Naturforsch., 2011, B66, 55 CrossRef PubMed.
- A. Pape, M. Lutz and G. Müller, Angew. Chem., Int. Ed., 1994, 33, 2281 CrossRef PubMed.
- S. Harder, L. Brandsma, J. A. Kanters, A. Duisenberg and J. H. van Lenthe, J. Organomet. Chem., 1991, 420, 143 CrossRef CAS.
- L. M. Engelhardt, J. M. Harrowfield, M. F. Lappert, I. A. MacKinnon, B. H. Newton, C. L. Raston, B. W. Skelton and A. H. White, J. Chem. Soc., Chem. Commun., 1986, 846 RSC.
- C. von Hänisch and B. Rolli, Z. Anorg. Allg. Chem., 2004, 630, 1987 CrossRef PubMed.
- J. C. Thomas and J. C. Peters, Inorg. Chem., 2003, 42, 5055 CrossRef CAS PubMed.
- H. H. Karsch, A. Appelt and G. Müller, Organometallics, 1985, 4, 1624 CrossRef CAS; H. H. Karsch, A. Appelt and G. Müller, J. Chem. Soc., Chem. Commun., 1984, 1415 RSC.
- M. Everett, A. Jolleys, W. Levason, D. Pugh and G. Reid, Chem. Commun., 2014, 50, 5843 RSC.
- M. Brookhart, B. Grant and A. F. Volpe Jr., Organometallics, 1992, 11, 3920 CrossRef CAS.
- M. J. D. Champion, J. M. Dyke, W. Levason, M. E. Light, D. Pugh, H. Bhakhoa, L. Rhyman, P. Ramasami and G. Reid, Inorg. Chem., 2015, 54, 2497 CrossRef CAS PubMed.
- S. Strauss, Chem. Rev., 1993, 93, 927 CrossRef CAS.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066 CrossRef CAS PubMed.
- T. S. Cameron, A. Decken, I. Dionne, M. Fang, I. Krossing and J. Passmore, Chem. – Eur. J., 2002, 8, 3386 CrossRef CAS.
- I. Krossing and L. van Wullen, Chem. – Eur. J., 2002, 8, 700 CrossRef CAS.
- I. Raabe, K. Wagner, K. Guttsche, M. Wang, M. Grätzel, G. Santiso-Quiñones and I. Krossing, Chem. – Eur. J., 2009, 15, 1966 CrossRef CAS PubMed.
- R. J. Burt, J. Chatt, W. Hussain and G. J. Leigh, J. Organomet. Chem., 1979, 182, 203 CrossRef CAS; E. P. Kyba, S. T. Liu and R. L. Harris, Organometallics, 1983, 2, 1877 CrossRef.
- G. Metz, X. Wu and S. O. Smith, J. Magn. Reson., Ser. A, 1994, 110, 219 CrossRef CAS.
- P. Garrou, Chem. Rev., 1981, 81, 229 CrossRef CAS.
- G. M. Sheldrick, SHELXS-97, Program for crystal structure solution, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for crystal structure refinement, University of Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: A packing diagram for [Na(diphos)3][BArF] and MAS NMR data (7Li, 23Na and 31P) for the phosphine complexes reported. CCDC 1044099–1044101. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc03184b |
| ‡ Synthetic procedure. Schlenk techniques and a glove-box were used for all manipulations, which were conducted under anhydrous and anaerobic conditions. [Li(diphos)3][Al{OC(CF3)3}4]: Li[Al{OC(CF3)3}4] (121 mg, 0.125 mmol) in 3 mL toluene was added o-C6H4(PMe2)2 (74 mg, 0.375 mmol) with stirring. After 5 min, the volatiles were removed in vacuo. The white solid residue was washed with pentane and dried in vacuo. Yield: 181 mg 92%. Colourless single crystals were grown by slow evaporation from a toluene solution under N2. Anal, required for C46H48AlF36LiO4P6 (1568.5): C, 35.2%; H, 3.0%. Found: C, 35.2 %; H, 3.0 %. 1H NMR (CD2Cl2, 298 K): 1.35 (s, [36H], Me), 7.37–7.43 (m, [6H], o-C6H4), 7.47–7.53 (m, [6H], o-C6H4) ppm. [Na(diphos)3][B{3,5-(CF3)2-C6H3}4]: method as above, using Na[B{3,5-(CF3)2-C6H3}4] (110 mg, 0.125 mmol) o-C6H4(PMe2) (74 mg, 0.375 mmol). White solid. Yield: 160 mg, 86%. Colourless single crystals were grown by slow evaporation from a toluene solution under N2. Anal, required for C62H60BF24NaP6 (1480.7): C, 50.3; H, 4.0%. Found: C, 50.4; H, 4.0%. 1H NMR (CD2Cl2, 298 K): 1.20 (s, [36H], Me), 7.34–7.37 (m, [6H], o-C6H4), 7.42–7.48 (m, [10H], overlapping o-C6H4 and [BArF]− H4), 7.62 (s, [8H], [BArF]− H2/6) ppm. [Li(dmpe)3][Al{OC(CF3)3}4]: Li[Al{OC(CF3)3}4] (121 mg, 0.125 mmol) in 5 ml toluene was added dmpe (58 mg, 0.375 mmol) with stirring. After 5 min, the volatiles were removed in vacuo. The white solid residue was washed with anhydrous pentane and dried in vacuo. Yield: 164 mg, 91%. Anal, required for C34H48AlF36LiO4P6 (1424.4): C, 28.6; H, 3.3%. Found: C, 28.7; H, 3.0%. 1H NMR (CD2Cl2, 298 K): 1.30 (br s, [36H], Me), 1.85 (br s, [12H], CH2) ppm. [Na(dmpe)3][B{3,5-(CF3)2-C6H3}4]: As above, using Na[B{3,5-(CF3)2-C6H3}4] (110 mg, 0.125 mmol) and dmpe (58 mg, 0.375 mmol). White solid. Yield: 143 mg, 85%. Colourless single crystals were grown by slow evaporation from a toluene solution under N2. Anal, required for C50H60BF24NaP6 (1336.6): C, 44.9; H, 4.5%. Found: C, 44.8; H, 4.5%. 1H NMR (CD2Cl2, 298 K): 1.01 (br s, [36H], Me), 1.45 (br s, [12H], CH2), 7.46 (s, [4H], [BArF]− H4), 7.62 (s, [8H], [BArF]− H2/6) ppm. |
| § Crystal data for [Na(dmpe)3][BArF]: C50H60BF24NaP6, MW = 1336.60, monoclinic, P21/c (no. 14), a = 18.4514(10), b = 13.2631(10), c = 26.133(2) Å, β = 90.826(3)°, U = 6394.6(8) Å3, Z = 4, μ = 0.277 mm−1, T = 100 K, 35720 total reflections, 12513 unique reflections, Rint = 0.059, R1 (I > 2σ(I)) = 0.068, R1 (all data) = 0.105, wR2 (I > 2σ(I)) = 0.169, wR2 (all data) = 0.197. Crystal data for [Na(diphos)3][BArF]: C62H60BF24NaP6, MW = 1480.72, triclinic, P Crystal data for [Li(diphos)3][Al{OC(CF3)3}4]: C46H48AlF36LiO4P6, MW = 1568.58, orthorhombic, Pna21 (no. 33), a = 15.092(2), b = 27.737(5), c = 15.405(3) Å, U = 6449.1(19) Å3, Z = 4, μ = 0.325 mm−1, 38 |
| ¶ Solid state NMR experiments. All measurements were performed on a Bruker 9.4 T magnet with a Chemagnetics Infinity console using a double-resonance 4 mm APEX probe. Solid powdered samples were transferred into 4 mm zirconium oxide thin wall rotors within the glovebox, using special end caps with o-rings to exclude air. Magic angle spinning (MAS) conditions have been applied with a spinning speed of 7.1 kHz at room temperature, using N2 gas flow for bearing and drive. The chemical shift scales were referenced by setting at 0 ppm the signals of LiCl 1 M, NaCl 1 M and 85% H3PO4, respectively for 7Li, 23Na and 31P. Spectra were recorded with direct excitation using a 90° pulse followed by acquisition, without proton decoupling. For 31P NMR, additional measurements were also performed with ramped CP methods with 3 ms contact time. |
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2),
(no. 2), | This journal is © The Royal Society of Chemistry 2015 |