
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly enantioselective copper(I)-catalyzed conjugate addition of 1,3-diynes to α,β-unsaturated trifluoromethyl ketones†
Amparo
Sanz-Marco
a,
Gonzalo
Blay
*a,
M. Carmen
Muñoz
b and
José R.
Pedro
*a
aDepartament de Química Orgànica, Facultat de Química, Universitat de València, C/Dr. Moliner 50, 46100-Burjassot, Spain. E-mail: jose.r.pedro@uv.es; gonzalo.blay@uv.es; Fax: +34 963544328; Tel: +34 962544336
bDepartament de Física Aplicada, Universitat Politècnica de València, Camí de Vera s/n, E-46022-València, Spain
First published on 23rd April 2015
Abstract
The conjugate diynylation of α,β-unsaturated trifluoromethyl ketones is carried out in the presence of a low catalytic load (2.5 mol%) of a copper(I)–MeOBIPHEP complex, triethylamine and a terminal 1,3-diyne. Pre-metalation of the terminal 1,3-diyne with stoichiometric or higher amounts of dialkylzinc reagent is not required. The corresponding internal diynes bearing a propargylic stereogenic center are obtained with good yields and excellent enantioselectivities.
The 1,3-diyne moiety is present in a large number of natural and artificial molecules.1 Naturally occurring diynes are found as metabolites in a variety of fungi, higher plants and marine organisms, and many of them have important biological and pharmacological properties ranging from antifungal to anticancer activities.2 Diynes and oligoynes are also of interest as probes of extended π-conjugation and can serve as active components in optoelectronic devices.3 Furthermore, conjugate diynes are intriguing building blocks due to the unique behavior of alkynes, especially in transition metal-catalyzed processes.4
In recent years the nucleophilic addition of terminal alkynes to prochiral electrophiles has emerged as one of the most efficient procedures for the synthesis of internal alkynes bearing a propargylic stereogenic center. Thus considerable success has been made in the enantioselective alkynylation of carbonyl compounds5 and imines,6 and, more recently, in the conjugate alkynylation of electrophilic double bonds.7 However, studies on enantioselective addition of 1,3-diynes are more limited, and all the examples reported in the literature involve the activation of terminal 1,3-diynes as diynylzinc reagents, which requires in most of the cases the use of stoichiometric or larger amounts of expensive dialkylzinc reagents. Thus, after the pioneering work of Carreira in 2003,8 on the use of 4 equivalents of Zn(OTf)2/N-methylephedrine to achieve the addition of a terminal 1,3-diyne to an aliphatic aldehyde, Trost9 reported in 2010 a catalytic enantioselective addition of 1,3-diynes to aldehydes using a dinuclear ProPhenol/zinc catalyst. Later on, Pu,10 Tykwinski11 and Wang12 developed their own versions of the asymmetric zinc-catalyzed addition of 1,3-diynes to aldehydes by using combinations of dialkylzinc with amino alcohol11,12 or binaphthol-type10 ligands. In 2011, Ma described the enantioselective addition of 1,3-diynylzinc reagents, generated in situ from Me2Zn and terminal diynes, to aromatic ketones in the presence of a Cu(II)–hydroxycamphor–sulfonamide complex and Me2Zn.13 On the other hand, the diynylation of different kinds of aldimines14 and fluorinated ketimines15 has been reported by the same group using terminal diynes, Me2Zn and binaphthol-type ligands. However, a procedure for the enantioselective addition of 1,3-diynes to electrophilic C–C double bonds, i.e. enones, has not been reported so far.
Following our research interest in the conjugate alkynylation reaction,7d,e,i,j we report in this communication our results on the enantioselective copper-catalyzed conjugate addition of terminal 1,3-diynes to enones, a reaction without any precedent in the literature. In our study, we have chosen α,β-unsaturated trifluoromethyl ketones (Scheme 1) as electrophiles because of the significance of fluorinated compounds in medicinal chemistry.15 Trifluoromethyl enones16,17 are a particularly challenging class of substrates for conjugate asymmetric transformations since the presence of the strongly electron-withdrawing trifluoromethyl group not only activates the alkene but also renders the ketone functionality highly reactive making the control over regioselectivity difficult. In fact, only two examples of alkynylation of trifluoromethyl enones have been reported so far, both taking place regioselectively on the carbonyl group.17a,18
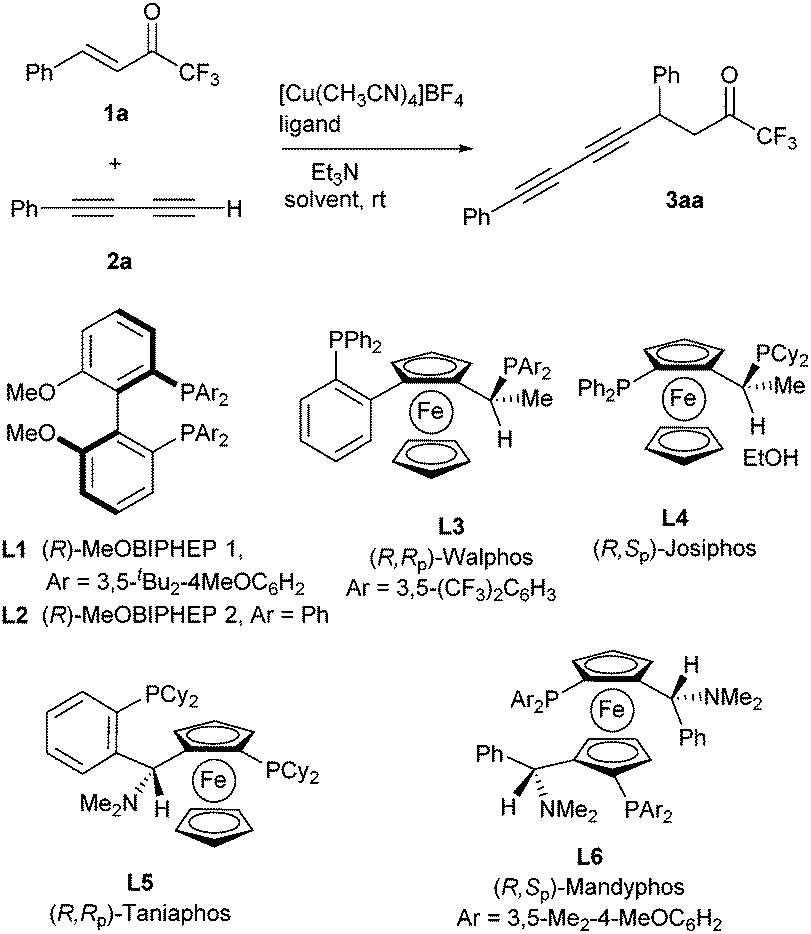 | ||
| Scheme 1 Conjugate diynylation of α,β-unsaturated trifluoromethyl ketones and ligands used in this study. | ||
In the onset of our investigation we studied the addition of phenyl-1,3-butadiyne (1a, R1 = Ph) to enone 2a (R2 = Ph) catalyzed by 20 mol% of [Cu(CH3CN)4]BF4 and (R)-MeOBIPHEP 2 (L1) in toluene (Scheme 1, Table 1, entry 1). Pleasantly, compound 3aa was obtained in 82% yield and 93% ee under these reaction conditions. No products arising from 1,2-addition to the carbonyl group were observed. Further studies with this catalytic system (Table 1, entries 2–6) showed that the catalyst load could be reduced to as low as 2.5 mol% and the amount of base decreased to sub-stoichiometric 0.1 equiv. without a significant effect on the reaction outcome. Furthermore, the amount of diyne was also reduced to only 1.3 equivalents with respect to enone. Other phosphine ligands L2–L6 were also tested (Table 1, entries 7–11). Surprisingly, from all the studied ligands, only biarylphosphine L1 provided a catalytic complex sufficiently active to promote the reaction with this low catalytic load. The use of copper(I) triflate (Table 1, entry 12) provided similar results to [Cu(CH3CN)4]BF4, whereas other solvents such as THF or CH2Cl2 decreased the reaction rate, compound 3aa being obtained in lower yields than in toluene, although still with good enantioselectivities (Table 1, entries 13 and 14). Diethyl ether performed better than THF, but with lower yield and ee than toluene (Table 1, entry 15). The use of other bases such as DIPEA did not improve the results obtained with Et3N (Table 1, entry 16).
| Entry | L | L-Cu (mol%) | 1a (equiv.) | Et3N (equiv.) | Solvent | t (h) | Yield (%) | eea (%) |
|---|---|---|---|---|---|---|---|---|
| a Determined by HPLC using chiral stationary phases. b CuOTf 0.5 Tol was used instead of [Cu(CH3CN)4]BF4. c DIPEA was used instead of Et3N. | ||||||||
| 1 | L1 | 20 | 7.4 | 1.0 | Toluene | 18 | 82 | 93 |
| 2 | L1 | 10 | 5 | 1.0 | Toluene | 18 | 70 | 93 |
| 3 | L1 | 5 | 3 | 1.0 | Toluene | 18 | 73 | 93 |
| 4 | L1 | 5 | 3 | 0.1 | Toluene | 18 | 75 | 93 |
| 5 | L1 | 2.5 | 1.3 | 0.1 | Toluene | 18 | 73 | 93 |
| 6 | L1 | 2.5 | 1.3 | 0 | Toluene | 18 | n.r. | — |
| 7 | L2 | 2.5 | 1.3 | 0.1 | Toluene | 18 | n.r. | — |
| 8 | L3 | 2.5 | 1.3 | 0.1 | Toluene | 18 | n.r. | — |
| 9 | L4 | 2.5 | 1.3 | 0.1 | Toluene | 18 | n.r. | — |
| 10 | L5 | 2.5 | 1.3 | 0.1 | Toluene | 18 | Trace | — |
| 11 | L6 | 2.5 | 1.3 | 0.1 | Toluene | 18 | Trace | — |
| 12b | L1 | 2.5 | 1.3 | 0.1 | Toluene | 18 | 65 | 93 |
| 13 | L1 | 2.5 | 1.3 | 0.1 | THF | 48 | 34 | 94 |
| 14 | L1 | 2.5 | 1.3 | 0.1 | CH2Cl2 | 48 | 17 | 90 |
| 15 | L1 | 2.5 | 1.3 | 0.1 | Et2O | 48 | 58 | 90 |
| 16 | L1 | 2.5 | 1.3 | 0.1c | Toluene | 18 | 65 | 91 |
Under the best reaction conditions available (Table 1, entry 5) we studied the scope of the reaction with various enones 1 and diynes 2.‡ First, we conducted the addition of diyne 2a with several trifluoromethyl enones bearing different substituents at the β position of the double bond. The results are gathered in Table 2. Good results were obtained with a variety of enones bearing a substituted aromatic ring at this position. Good enantiomeric excesses were obtained for enones bearing an aromatic ring substituted at either the ortho, meta or para positions (Table 2, entries 2–4). Aromatic rings bearing electron-withdrawing (Table 2, entries 5 and 6) or electron-donating (Table 2, entries 7 and 8) substituents were also tolerated yielding the expected products with enantiomeric excesses above 90%. Enone 1i bearing a bulky 2-naphthyl group also reacted under the optimized conditions to give the diynylated product 3ia with good yield and high ee. Remarkably, the reaction could also be carried out with enones, featuring aliphatic groups on the β-carbon, providing the corresponding products 3ja–3la with moderate yields but high enantiomeric excesses (84–88%, Table 2, entries 10–12), although a higher catalytic load (10 mol%) was required in these cases.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 1 | R1 | 2 | R2 | 3 | Yield (%) | eeb (%) |
| a 1 (0.14 mmol), 2 (1.3 equiv.), Et3N (0.1 equiv.), [Cu(CH3CN)4]BF4 (2.5 mol%), L6 (2.5 mol%), toluene, rt. b Determined by HPLC using chiral stationary phases. c Reaction carried out with 10 mol% of catalyst. d Reaction carried out with 0.6 mmol of 1a. | |||||||
| 1 | 1a | Ph | 2a | Ph | 3aa | 73 | 93 |
| 2 | 1b | 2-MeC6H4 | 2a | Ph | 3ba | 94 | 94 |
| 3 | 1c | 3-MeC6H4 | 2a | Ph | 3ca | 55 | 93 |
| 4 | 1d | 4-MeC6H4 | 2a | Ph | 3da | 56 | 92 |
| 5 | 1e | 2-BrC6H4 | 2a | Ph | 3ea | 76 | 94 |
| 6 | 1f | 4-BrC6H4 | 2a | Ph | 3fa | 55 | 92 |
| 7 | 1g | 2-MeOC6H4 | 2a | Ph | 3ga | 69 | 94 |
| 8 | 1h | 4-MeOC6H4 | 2a | Ph | 3ha | 41 | 92 |
| 9 | 1i | 2-Naphthyl | 2a | Ph | 3ia | 59 | 92 |
| 10c | 1j | PhCH2CH2 | 2a | Ph | 3ja | 50 | 84 |
| 11c | 1k | CH3(CH2)3 | 2a | Ph | 3ka | 53 | 87 |
| 12c | 1l | (CH3)2CHCH2 | 2a | Ph | 3la | 50 | 88 |
| 13 | 1a | Ph | 2b | 3-FC6H4 | 3ab | 50 | 90 |
| 14 | 1a | Ph | 2c | 4-FC6H4 | 3ac | 68 | 92 |
| 15 | 1a | Ph | 2d | 2-MeOC6H4 | 3ad | 89 | 92 |
| 16 | 1a | Ph | 2e | 4-MeOC6H4 | 3ae | 65 | 91 |
| 17 | 1a | Ph | 2f | 3-Thienyl | 3af | 72 | 94 |
| 18 | 1a | Ph | 2g | PhCH2CH2 | 3ag | 41 | 93 |
| 19 | 1b | 2-MeC6H4 | 2g | PhCH2CH2 | 3bg | 50 | 95 |
| 20 | 1a | Ph | 2h | 6-ClC4H8 | 3ah | 61 | 93 |
| 21 | 1a | Ph | 2i | TIPS | 3ai | 50 | 85 |
| 22d | 1a | Ph | 2a | Ph | 3aa | 62 | 92 |
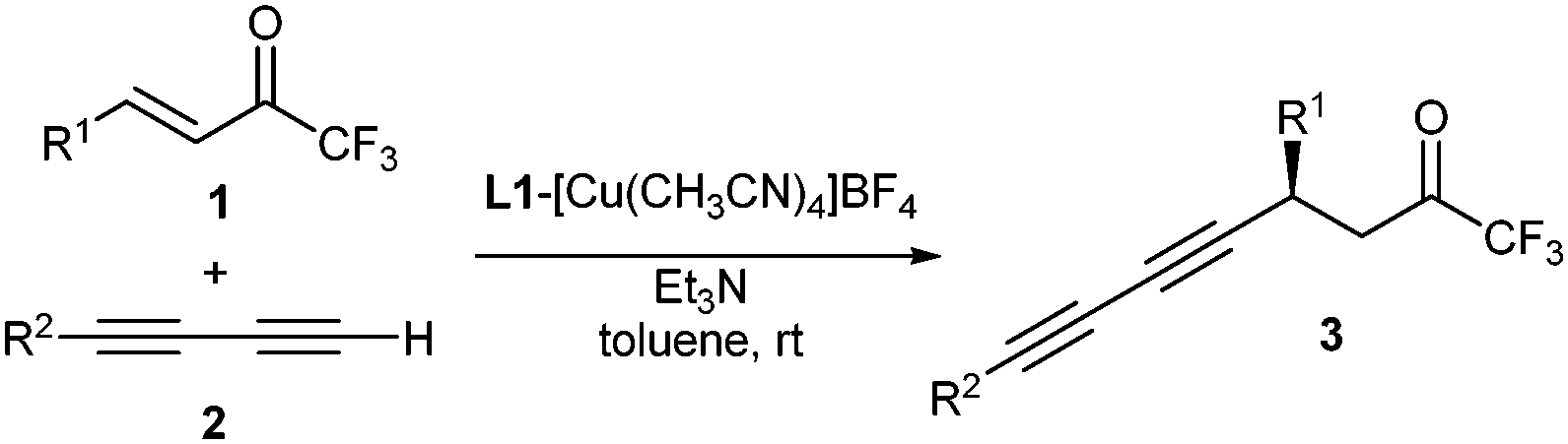
Next, we tested the diyne scope (Table 2, entries 13–21). Substituted phenyl-1,3-butadiynes bearing electron-donating (MeO) or electron-withdrawing (F) groups on the phenyl group reacted with compound 1a with variable yields but excellent enantioselectivities (Table 2, entries 13–16). The heterocyclic 3-thienyl-1,3-butadiyne (2f) reacted with 1a to give compound 3af with 72% yield and 94% ee (Table 2, entry 17). Next, we examined the reaction with aliphatic diynes, which reacted similarly to aromatic diynes. 6-Phenyl-1,3-hexadiyne reacted with enones 1a and 1b to give the corresponding chiral diynes 3ag and 3bg, respectively, with moderate yield but high enantioselectivity (Table 2, entries 18 and 19). 8-Chloro-1,3-octadiyne (2h) reacted in a similar way to give compound 3ah in 61% yield and 93% ee (Table 2, entry 20). These results are in contrast to those observed in the copper-catalyzed conjugate alkynylation of β-trifluoromethyl enones with terminal monoynes where aliphatic alkynes reacted with lower yields and enantioselectivities than aromatic and heteroaromatic ones.7d Finally, silyldiyne 2i, which is an equivalent of 1,3-butadiyne, could be reacted with enone 1a to give the diynylated product 3ai with 85% ee (Table 2, entry 21), showing the broad scope of the reaction regarding the diyne nucleophile. The reaction between enone 1a and diyne 2a was also carried out at a fourfold scale with a small decrease of yield but without a noticeable effect on the enantioselectivity (Table 2, entry 22).
The absolute stereochemistry of compound 3af (Table 2, entry 15) was elucidated by X-ray crystallographic analysis (see Fig. S2 in the ESI†),19 and for the rest of the products it was assigned on the assumption of a uniform reaction mechanism.
Some synthetic modifications of diynes 3 are presented in Scheme 2. Thus, full hydrogenation of both triple bonds in 3aa could be carried out over 10% Pd/C in ethyl acetate to give trifluoromethyl ketone 4 without any loss of optical purity. On the other hand, a chiral tetrahydrofuran 6 bearing a trifluoromethylated quaternary stereocenter could be obtained after diastereoselective addition of methylmagnesium chloride to compound 3aa followed by silver-catalyzed cyclization. We have also performed the desilylation of compound 3ai (70% yield) to give the chiral terminal diyne 7 upon treatment with TBAF and acetic acid in THF.
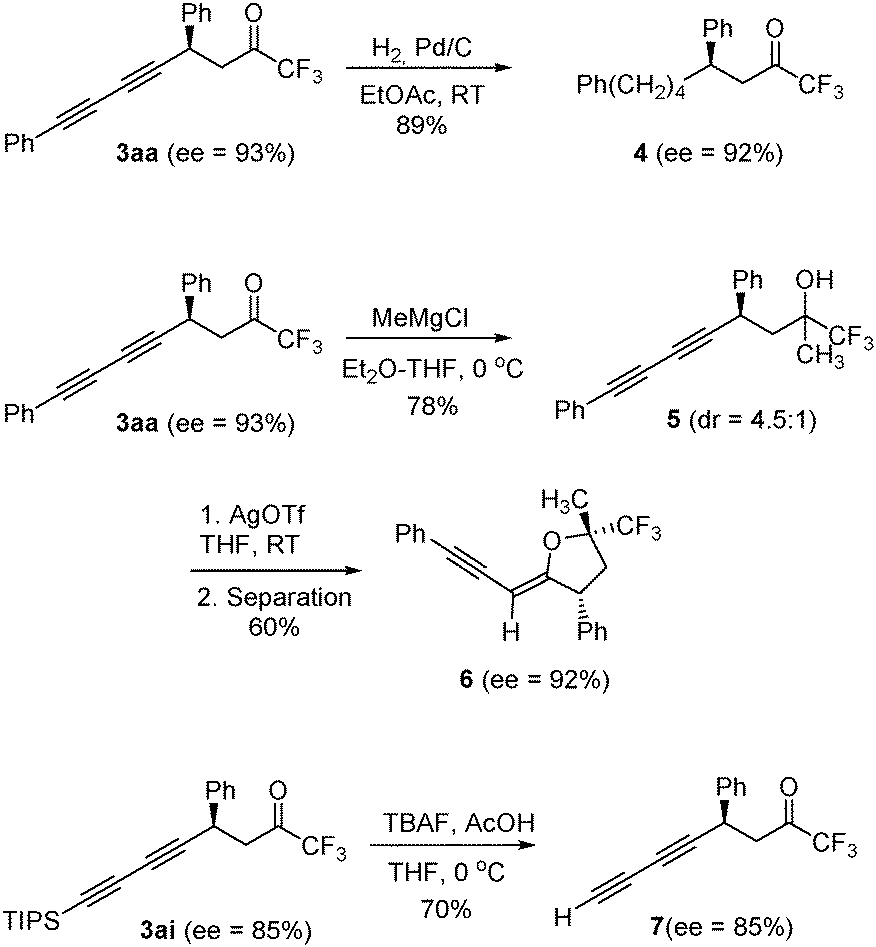 | ||
| Scheme 2 Examples of chemical transformations carried out on diynes 3. | ||
In summary, we have reported the first example of enantioselective conjugate diynylation of enones. The reaction requires only a small excess (1.3 equiv.) of a terminal diyne and is carried out in the presence of a low catalytic load of a copper(I)–biphosphine complex (0.025 equiv.) and an amine (0.1 equiv.) to provide the corresponding internal diynes bearing a propargylic stereogenic center with excellent enantioselectivities. The reaction is broad in scope for a wide range of trifluoromethyl ketones20 allowing variation of substituents on the enone β-carbon as well as on the diyne. It should be remarked that, unlike in other enantioselective diynylation reactions of carbonyl compounds and imines previously reported in the literature, pre-metalation of the terminal diyne with stoichiometric amounts of a dialkylzinc reagent is not required. Our results show that the transient diynyl-copper species formed from the terminal diyne and the copper(I) complex in the presence of an amine are sufficiently nucleophilic to react even with weak electrophiles. This may anticipate the possibility of other enantioselective diynylation reactions not requiring pre-metalation of terminal diynes with stoichiometric amounts of organometallic reagents in the future.
Financial support from the Ministerio de Economía y Competitividad (MINECO-Gobierno de España) and FEDER (EU) (CTQ2013-47494-P) and from Generalitat Valenciana (ISIC2012/001) is gratefully acknowledged. A. S.-M. thanks the MINECO for a predoctoral grant (FPI program). Access to NMR and MS facilities from the Servei Central de Suport a la Investigació Experimental (SCSIE)-UV is also acknowledged.
Notes and references
- Reviews: (a) A. L. K. Shi Shun, R. Rik and R. Tykwinski, Angew. Chem., Int. Ed., 2006, 45, 1034 CrossRef PubMed; (b) Modern Acetylene Chemistry, ed. P. J. Stang and F. Diederich, VCH, Weinheim, 1995 Search PubMed; (c) K. S. Sindhu and G. Anilkumar, RSC Adv., 2014, 4, 27867 RSC.
- (a) H.-J. Jung, B.-S. Min, J.-Y. Park, Y.-H. Kim, H.-K. Lee and K.-H. Bae, J. Nat. Prod., 2002, 65, 897 CrossRef CAS PubMed; (b) S. F. Mayer, A. Steinreiber, R. V. A. Orru and K. Faber, J. Org. Chem., 2002, 67, 9115 CrossRef CAS PubMed; (c) Y. Satoh, M. Satoh, K. Isobe, K. Mohri, Y. Yoshida and Y. Fujimoto, Chem. Pharm. Bull., 2007, 55, 561 CrossRef CAS; (d) N. P. McLaughlin, E. Butler, P. Evans, N. P. Brunton, A. Koidis and D. K. Rai, Tetrahedron, 2010, 66, 9681 CrossRef CAS PubMed; (e) D. Shin, J.-E. Yang, S. B. Lee and C. W. Nho, Bioorg. Med. Chem. Lett., 2010, 20, 7549 CrossRef CAS PubMed.
- (a) Review: M. B. Nielsen and F. Diederich, Chem. Rev., 2005, 105, 1837 CrossRef CAS PubMed; (b) W. B. Wan, S. C. Brand, J. J. Pak and M. M. Haley, Chem. – Eur. J., 2000, 6, 2044 CrossRef CAS; (c) K. West, C. Wang, A. S. Batsanov and M. R. Bryce, Org. Biomol. Chem., 2008, 6, 1934 RSC.
- (a) D.-G. Yu, F. de Azambuja, T. Gensch, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 9650 CrossRef CAS PubMed; (b) N.-K. Lee, S. Y. Yun, P. Mamidipalli, R. M. Salzman, D. Lee, T. Zhou and Y. Xia, J. Am. Chem. Soc., 2014, 136, 4363 CrossRef CAS PubMed; (c) S. Y. Yun, K.-P. Wang, N.-K. Lee, P. Mamidipalli and D. Lee, J. Am. Chem. Soc., 2013, 135, 4668 CrossRef CAS PubMed; (d) G. Huang, K. Xie, D. Lee and Y. Xia, Org. Lett., 2012, 14, 3850 CrossRef CAS PubMed; (e) S. Gupta, P. K. Agarwal, M. Saifuddin and B. Kundu, Tetrahedron Lett., 2011, 52, 5752 CrossRef CAS PubMed; (f) E. J. Cho, M. Kim and D. Lee, Org. Lett., 2006, 8, 5413 CrossRef CAS PubMed.
- B. M. Trost and A. Weiss, Adv. Synth. Catal., 2009, 351, 963 CrossRef CAS PubMed.
- G. Blay, A. Monleón and J. R. Pedro, Curr. Org. Chem., 2009, 13, 1498 CrossRef CAS.
- Cu catalysis: (a) T. F. Knopfel, P. Zarotti, T. Ichikawa and E. M. Carreira, J. Am. Chem. Soc., 2005, 27, 9682 CrossRef PubMed; (b) R. Yazaki, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 10275 CrossRef CAS PubMed; (c) R. Yazaki, N. Kumagai and M. Shibasaki, Org. Lett., 2011, 13, 952 CrossRef CAS PubMed; (d) A. Sanz-Marco, A. Garcia-Ortiz, G. Blay and J. R. Pedro, Chem. Commun., 2014, 50, 2275 RSC; (e) A. Sanz-Marco, A. Garcia-Ortiz, G. Blay, I. Fernandez and J. R. Pedro, Chem. – Eur. J., 2014, 20, 668 CrossRef CAS PubMed . Rh catalysis: ; (f) T. Nishimura, X.-X. Guo, N. Uchiyama, T. Katoh and T. Hayashi, J. Am. Chem. Soc., 2008, 130, 1576 CrossRef CAS PubMed; (g) T. Nishimura, T. Sawano and T. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 8057 CrossRef CAS PubMed; (h) E. Fillion and A. K. Zorzitto, J. Am. Chem. Soc., 2009, 131, 14608 CrossRef CAS PubMed . Zn catalysis: ; (i) G. Blay, L. Cardona, J. R. Pedro and A. Sanz-Marco, Chem. – Eur. J., 2012, 18, 12966 CrossRef CAS PubMed; (j) G. Blay, M. C. Muñoz, J. R. Pedro and A. Sanz-Marco, Adv. Synth. Catal., 2013, 555, 1071 CrossRef PubMed . For enantioselective addition of alkynyl organometallics see also: ; (k) S. Cui, S. D. Walker, J. C. S. Woo, C. J. Borths, H. Mukherjee, M. J. Chen and M. M. Faul, J. Am. Chem. Soc., 2010, 132, 436 CrossRef CAS PubMed; (l) Y.-S. Kwak and E. J. Corey, Org. Lett., 2004, 6, 3385 CrossRef CAS PubMed; (m) O. V. Larionov and E. Corey, Org. Lett., 2010, 12, 300 CrossRef CAS PubMed.
- S. Reber, T. F. Knöpfel and E. M. Carreira, Tetrahedron, 2003, 59, 6813 CrossRef CAS.
- B. M. Trost, V. S. Chan and D. Yamamoto, J. Am. Chem. Soc., 2010, 132, 5186 CrossRef CAS PubMed.
- M. Turlington, Y. Du, S. G. Ostrum, V. Santosh, K. Wren, T. Lin, M. Sabat and L. Pu, J. Am. Chem. Soc., 2011, 133, 11780 CrossRef CAS PubMed.
- E. R. Graham and R. R. Tykwinski, J. Org. Chem., 2011, 76, 6574 CrossRef CAS PubMed.
- B. Zheng, S.-N. Li, J.-Y. Mao, B. Wang, Q.-H. Bian, S.-Z. Liu, J.-C. Zhong, H.-C. Guo and M. Wang, Chem. – Eur. J., 2012, 18, 9208 CrossRef CAS PubMed.
- T.-L. Liu, H. Ma, F.-G. Zhang, Y. Zheng, J. Nie and J.-A. Ma, Chem. Commun., 2011, 47, 12873 RSC.
- T.-L. Liu, H.-X. Zhang, Y. Zheng, Q. Yao and J.-A. Ma, Chem. Commun., 2012, 48, 12234 RSC.
- (a) F.-G. Zhang, H. Ma, Y. Zheng and J.-A. Ma, Tetrahedron, 2012, 68, 7663 CrossRef CAS PubMed; (b) F.-G. Zhang, H. Ma, J. Nie, Y. Zheng, Q. Gao and J.-A. Ma, Adv. Synth. Catal., 2012, 354, 1422 CrossRef CAS PubMed; (c) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455 CrossRef CAS PubMed; (d) D. Cahard, X. Xu, S. Couve-Bonnaire and X. Pannecoucke, Chem. Soc. Rev., 2010, 39, 558 RSC; (e) K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305 CrossRef CAS; (f) J.-A. Ma and D. Cahard, Chem. Rev., 2008, 108, PR1 CrossRef CAS; (g) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- For recent examples involving asymmetric 1,4-addition reactions to trifluoromethyl enones see: (a) L. C. Morrill, S. M. Smith, A. M. Z. Slawin and A. D. Smith, J. Org. Chem., 2014, 79, 1640 CrossRef CAS PubMed; (b) P.-P. Yeh, D. S. B. Daniels, D. B. Cordes, A. M. Z. Slawin and A. D. Smith, Org. Lett., 2014, 16, 964 CrossRef CAS PubMed; (c) L. C. Morrill, J. Douglas, T. Lebl, A. M. Z. Slawin, D. J. Fox and A. D. Smith, Chem. Sci., 2013, 4, 4146 RSC; (d) Z.-K. Pei, Y. Zheng, J. Nie and J.-A. Ma, Tetrahedron Lett., 2010, 51, 4658 CrossRef CAS PubMed; (e) S. Sasaki, T. Yamauchi and K. Higashiyama, Tetrahedron Lett., 2010, 51, 2326 CrossRef CAS PubMed; (f) P. Li, Z. Chai, S.-L. Zhao, Y.-Q. Yang, H.-F. Wang, C.-W. Zheng, Y.-P. Cai, G. Zhao and S.-Z. Zhu, Chem. Commun., 2009, 7369 RSC; (g) C. Zheng, Y. Li, Y. Yang, H. Wang, H. Cui, J. Zhang and G. Zhao, Adv. Synth. Catal., 2009, 351, 1685 CrossRef CAS PubMed.
- For recent examples involving asymmetric 1,2-addition reactions to trifluoromethyl enones see: (a) G.-W. Zhang, W. Meng, H. Ma, J. Nie, W.-Q. Zhang and J.-A. Ma, Angew. Chem., Int. Ed., 2011, 50, 3538 CrossRef CAS PubMed; (b) Z.-J. Liu and J.-T. Liu, Chem. Commun., 2008, 5233 RSC; (c) X.-J. Wang, Y. Zhao and J. T. Liu, Org. Lett., 2007, 9, 1343 CrossRef CAS PubMed; (d) V. G. Nenajdenko, K. I. Smolko and E. S. Balenkova, Tetrahedron: Asymmetry, 2001, 12, 1259 CrossRef CAS.
- H. Wang, K.-F. Yang, L. Li, Y. Bai, Z.-J. Zheng, W. Q. Zhang, Z. W. Gao and L.-W. Xu, ChemCatChem, 2014, 6, 580 CrossRef CAS PubMed.
- CCDC 1046444.
- Other non-fluorinated enones such as 2-cyclohexenone, chalcone or 4-phenyl-3-buten-2-one did not react under the optimized conditions.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for new compounds, and X-ray data and ORTEP plot for compound 3af. CCDC 1046444. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc01676b |
| ‡ General procedure for the enantioselective conjugate diynylation reaction: [Cu(CH3CN)4]BF4 (1.1 mg, 0.0034 mmol) and (R)-L1 (4.1 mg, 0.0034 mmol) were added to a dried round bottom flask which was purged with nitrogen. Toluene (0.2 mL) was added via a syringe and the mixture was stirred for 1.5 h at room temperature under a nitrogen atmosphere. Then, a solution of α,β-unsaturated trifluoromethyl ketone 1 (0.144 mmol) in toluene (1.0 mL) was added via a syringe, followed by triethylamine (2 μL, 0.0144 mmol). The solution was stirred for 10 min at room temperature. Then a solution of 1,3-diyne 2 (0.188 mmol) in toluene (1.0 mL) was added via a syringe and the solution was stirred at room temperature until the reaction was complete (TLC). The reaction mixture was quenched with 20% aqueous NH4Cl (1.0 mL), extracted with CH2Cl2 (2 × 15 mL), washed with brine (15 mL), dried over MgSO4 and concentrated under reduced pressure. Purification by flash chromatography on silica gel eluting with hexane:ethyl acetate mixtures afforded compound 3. |
| This journal is © The Royal Society of Chemistry 2015 |