
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of radiolabelled aryl azides from diazonium salts: experimental and computational results permit the identification of the preferred mechanism†
Sameer M.
Joshi
a,
Abel
de Cózar
bcde,
Vanessa
Gómez-Vallejo
f,
Jacek
Koziorowski
g,
Jordi
Llop
*a and
Fernando P.
Cossío
*bde
aRadiochemistry and Nuclear Imaging, CIC biomaGUNE, Paseo Miramón 182, Parque Tecnológico de San Sebastián, 20009, San Sebastián/Donostia, Spain. E-mail: jllop@cicbiomagune.es
bDepartamento de Química Orgánica I, Facultad de Química, Universidad del País Vasco/Euskal Herriko Unibertsitatea (UPV/EHU), 20018, San Sebastián/Donostia, Spain. E-mail: fp.cossio@ehu.es
cIkerbasque, Basque Foundation for Science, 48018, Bilbao, Spain
dCentro de Innovación en Química Avanzada (ORFEO-CINQA), Spain
eDonostia International Physics Center (DIPC), 20018, San Sebastián/Donostia, Spain
fRadiochemistry Platform, CIC biomaGUNE, Paseo Miramón 182, Parque Tecnológico de San Sebastián, 20009, San Sebastián/Donostia, Spain
gDepartment of Radiation Physics and Department of Medical and Health Sciences, Linköping University, Linköping, Sweden
First published on 17th April 2015
Abstract
Experimental and computational studies on the formation of aryl azides from the corresponding diazonium salts support a stepwise mechanism via acyclic zwitterionic intermediates. The low energy barriers associated with both transition structures are compatible with very fast and efficient processes, thus making this method suitable for the chemical synthesis of radiolabelled aryl azides.
The relevance of organic azides, first synthesized by Grieß in 1864,1 ranges from the preparation of heterocycles, peptides2 and pharmaceuticals,3 to the synthesis of anilines and nitrenes.4 The most prominent fields are currently Huisgen 1,3-dipolar azide–alkyne cycloadditions5 and different variants of the Staudinger ligation.6 Among organic azides, aryl azides have found industrial and biological application7 in different fields due to their relatively high stability, and are important intermediates in organic chemistry.
Several approaches can be used for the preparation of aryl azides, including the reaction of diazonium salts with hydrazine,8O-benzylhydroxylamine hydrochloride9 or azide ions.10 Despite the latter reaction having been widely exploited for decades, its mechanism is still unclear and has been the subject of controversy.
In principle, at least three possible mechanisms can be predicted for this reaction. The first one consists of an Sn2Ar process similar to that observed for solvolysis reactions of diazonium salts,11 as indicated in eqn (1):
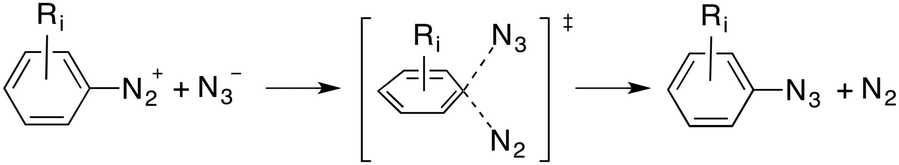 | (1) |
A second plausible mechanism involves a thermal (3+2) cycloaddition to form a 1H-pentazole cycloadduct12 that, in turn, can yield the product via a second retro-(3+2) reaction:
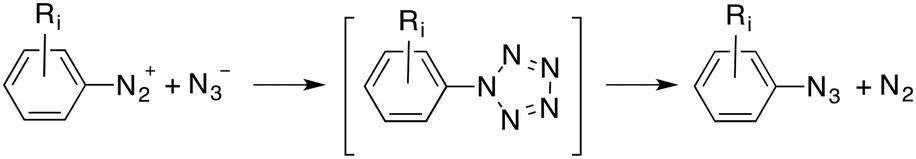 | (2) |
Finally, an addition–elimination process via an acyclic intermediate can be also considered, according to eqn (3):
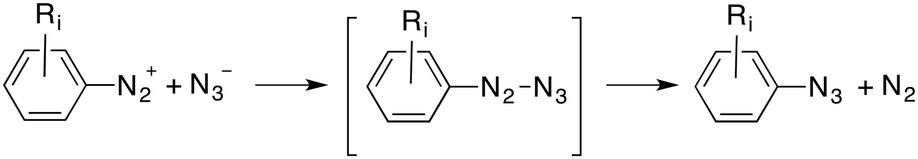 | (3) |
Nitrogen-13 (13N) is a positron emitter with a half-life of 9.97 minutes, and can be efficiently produced by proton irradiation of natural oxygen via the 16O(p,α)13N nuclear reaction. When water is irradiated with 8–16 MeV protons, a mixture of [13N]NO3−, [13N]NO2− and [13N]NH4+ is obtained, [13N]NO3− being the major species (ca. 85% of total radioactivity). [13N]NO3− can be quantitatively reduced to [13N]NO2− by passing [13N]NO3− over cadmium, and in our hands this labeling agent has proved useful for the synthesis of [13N]nitrosamines,14 [13N]nitrosothiols15 and [13N]azo derivatives.16
In continuation of our work, and with the ultimate goal of synthesizing 13N-labeled polysubstituted triazoles, we decided to approach the preparation of 13N-labeled phenyl azides by adapting a recently reported methodology,17 based on the reaction of an aromatic amine with NaNO2 and hydrazine hydrate (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:5) in the presence of acetic acid. According to the authors, one equivalent of sodium nitrite reacts with the aromatic amine to yield the corresponding diazonium salt; simultaneously, the reaction of another equivalent of nitrite with hydrazine hydrate generates in situ the azide ion, resulting in the formation of the aryl azide. When transitioning to radioactive conditions the reaction mechanism may have an impact on the radiochemical yield. If the reaction proceeds via the mechanism shown in eqn (1), the radiolabelling information contained in the azide anion should be completely transferred to the corresponding aryl azide; however, if the radiolabelled diazonium salt were reacted with non-radioactive azide ion, labeled aryl azide would never be obtained. Similar reasoning can be applied to mechanisms shown in eqn (2) and (3); hence the position of the label is paramount to prevent the formation of [13N]N2 with the consequent decrease in the labeling efficiency. With the aim of optimizing radiochemical yields, we envisaged a unique opportunity to further explore the mechanism of this reaction.
:2:5) in the presence of acetic acid. According to the authors, one equivalent of sodium nitrite reacts with the aromatic amine to yield the corresponding diazonium salt; simultaneously, the reaction of another equivalent of nitrite with hydrazine hydrate generates in situ the azide ion, resulting in the formation of the aryl azide. When transitioning to radioactive conditions the reaction mechanism may have an impact on the radiochemical yield. If the reaction proceeds via the mechanism shown in eqn (1), the radiolabelling information contained in the azide anion should be completely transferred to the corresponding aryl azide; however, if the radiolabelled diazonium salt were reacted with non-radioactive azide ion, labeled aryl azide would never be obtained. Similar reasoning can be applied to mechanisms shown in eqn (2) and (3); hence the position of the label is paramount to prevent the formation of [13N]N2 with the consequent decrease in the labeling efficiency. With the aim of optimizing radiochemical yields, we envisaged a unique opportunity to further explore the mechanism of this reaction.
The synthetic process for the preparation of 13N-labeled aryl azides was approached using two experimental settings (Scheme 1). In the first approach, denoted as A in Scheme 1, aniline was first reacted with sodium nitrite in the presence of hydrochloric acid, to yield the non-labeled diazonium salt (1). In a different vial, hydrazine hydrate was reacted with [13N]NO2− in the presence of acetic acid, to yield the 13N-labeled azide ion (Scheme 1A). Both solutions were finally mixed in a capped vial to enable the formation of 13N-labeled phenyl azide. In the second approach, denoted as B in Scheme 1, aniline was first reacted with [13N]NO2− in the presence of hydrochloric acid, to yield the 13N-labeled diazonium salt ([13N]1). In a different vial, hydrazine hydrate was reacted with sodium nitrite in the presence of acetic acid, to yield the non-labeled azide ion (Scheme 1B). Again, both solutions were finally mixed in a capped vial to enable the formation of 13N-labeled phenyl azide.
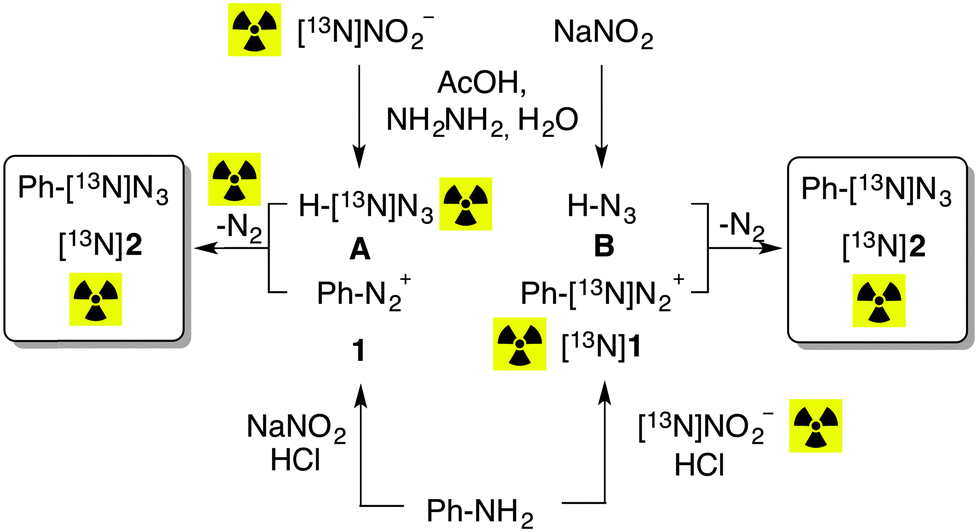 | ||
| Scheme 1 The two alternative strategies A and B followed to synthesize 13N-labeled phenyl azide ([13N]2). | ||
After termination of the reaction, the amount of radioactivity was measured with a dose calibrator (A1), the vials were flushed with nitrogen to remove radioactive gases, the amount of radioactivity was measured again (A2), and the reaction mixture was analyzed by HPLC using a radiometric detector in series with a UV detector. Identification of the 13N-labeled azide was confirmed by co-elution with the reference standard (see ESI† for further details).
Both synthetic strategies led to the formation of 13N-labeled phenyl azide (2). However, the amount of radioactive gas generated during the reaction, determined as the difference between A1 and A2, and referred to the total amount of 13N-labeled azide (the latter calculated as the product A2 × AUC, where AUC is the area under the peak for phenyl azide as measured in the radiometric detector and expressed as percentage with respect to all integrated peaks in the chromatogram) was 100.3 ± 1.7% and 4.0 ± 1.1% for strategies A and B, respectively. Analysis of the flushed gas by radio-GC-MS showed the presence of a single radioactive peak, which was identified as [13N]N2, while the amount of labeled azide obtained in 1B was twice the amount obtained in A. These results confirm that approximately half of the radioactivity is lost as [13N]N2 when route A is followed, while the information of the radiolabel is almost quantitatively transferred to the azide under route B (Scheme 1).
The experimental data completely discard the reaction mechanism based on SN2Ar (eqn (1)) and cleavage of the C–heteroatom bond, which would lead to a complete radioactivity loss (as [13N]N2) when route B is followed. On the other hand, they strongly suggest that the formation of the intermediate ring (eqn (2)) is not taking place; in such a case, [13N]N2 would be detected in a significant amount (ca. 100% with respect to the final amount of labelled aryl azide) when route B was used.
In view of these results, we performed DFT18 calculations on the parent PhN2+(1) + N3− → PhN3(2) + N2 reaction in order to obtain evidence concerning the most plausible reaction mechanism and get a better understanding of the experimental data. A M06-2X19 (PCM)20/def2-TZVPP21 study of the reactants in aqueous solution revealed the presence of a local minimum associated with a weak complex formed denoted as RC in Fig. 1.
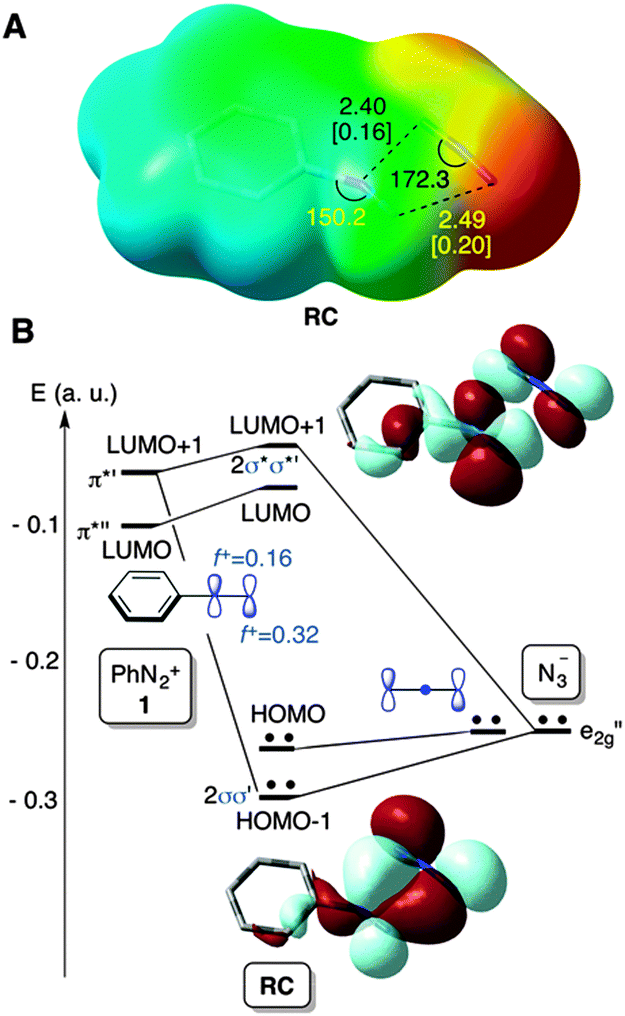 | ||
| Fig. 1 (A) Electrostatic potential and chief geometric features of complex RC associated with the interaction between azide anion and diazonium cation 1. Bond distances and angles are given in Å and deg., respectively. Numbers in square brackets are the corresponding bond indices. (B) Selected Kohn–Sham molecular orbitals of RC. Descriptors f+ on the nitrogen atoms correspond to the local electrophilic Fukui indices. | ||
This stationary point on the potential energy surface (PES) consists of a charge transfer complex, in which both ionic reactants are in close contact, with a calculated charge transfer of 0.5 a.u. The new N–N bond distances are ca. 2.4–2.5 Å (Fig. 1A), the respective Wiberg bond indices22 being ca. 0.2. This weak bonding pattern stems from a two-electron interaction between one of the e2g′′ MOs of the azide anion and the in-plane π*′ LUMO+1 of 1 (Fig. 1B). The occupied MOs π*′′ of 1 and e1g of N3− lead to a destabilizing four-electron interaction (not shown), thus resulting in a very weak bonding pattern between both the reactants at RC. Actually, this stationary point is not stabilized with respect to the separate reactants at 298 K (Fig. 2).
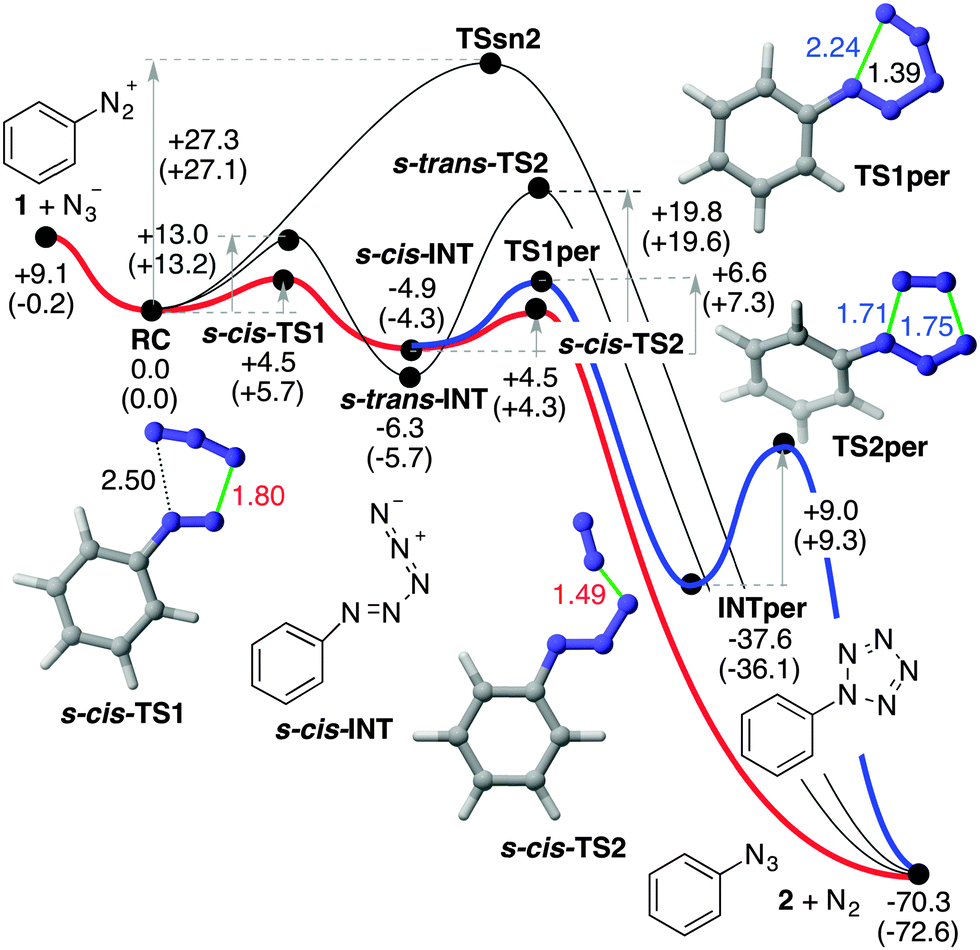 | ||
| Fig. 2 M06-2X(PCM)/def2-TZVPP reaction profiles associated with the reaction between diazonium cation 1 and azide anion to yield phenyl azide 2 and dinitrogen. Numbers close to reactants, intermediates and products indicate the relative energies in kcal mol−1. Numbers close to the arrows indicate the respective activation energies, in kcal mol−1. Numbers in parentheses indicate the respective Gibbs energies, computed at 298 K, in kcal mol−1. The lowest energy reaction paths are highlighted in red and blue. | ||
From these reactants we characterized the saddle point TSSN2 (Fig. 2) with the computed activation energy of ca. 27 kcal mol−1. The geometric features of this transition structure are quite similar to those obtained for solvolysis reactions of aromatic diazonium salts.11 In our case, however, there is an additional interaction between the diazonium and azide moieties (Fig. 1). This remarkable barrier and our experimental results permit us to discard the SN2Ar mechanism for this particular reaction.
The frontier MOs of the reactants at RC are also indicated in Fig. 1B and correspond to the in-plane MOs e2g′′ and π*′′. These computational data are compatible with a high electrophilicity associated with the terminal nitrogen of the diazonium moiety of 1, with a local electrophilic Fukui index23f+ of 0.32 a.u. (Fig. 1B). The interaction between the terminal nitrogen atoms of both reactants according to the mechanism reported in eqn (3) leads to saddle points s-cis- and s-trans-TS1 (Fig. 2). The former transition structure was calculated to be ca. 7 kcal mol−1 less energetic than the latter (Fig. 2). The chief geometric features of s-cis-TS1 closely resemble those expected for an asynchronous transition structure associated with a (3+2) cycloaddition.24 However, all our attempts to connect s-cis-TS1 directly with 2-phenyl-2H-pentazole INTper were unfruitful. Instead, this saddle point led to the zwitterionic intermediate s-cis-INT, from which we located a transition structure TS1per. This latter saddle point led to 2-phenyl-2H-pentazole INTper (Fig. 2), associated with the hypothetical (3+2) cycloaddition. From this local minimum we found the saddle point TS2per leading to phenyl azide 2. Although this latter transition structure associated with a retro-(3+2) cycloaddition is compatible with the reaction scheme gathered in eqn (2), it is important to note that INTper does not stem from RC but from s-cis-INT, which constitutes the key intermediate of the less energetic reaction profiles. In addition, our calculations indicate that the formation of INTper occurs with an activation barrier that is ca. 2 kcal mol−1 higher than that associated with the formation of phenyl azide 2.
Intrinsic Reaction Coordinate25 (IRC) scans from both s-cis- and s-trans-TS1 led to the corresponding zwitterionic intermediates s-cis- and s-trans-INT (see ESI†). The relative stabilities of these polar intermediates were found to be the opposite ones with respect to the corresponding transition structures. Therefore, the preferred route to yield azidobenzene 2 and dinitrogen occurs vias-cis-TS2 in which the cleavage of the PhN3(−)–N2(+) delocalized bond is produced (Fig. 2). This low barrier is associated with the formation of dinitrogen and azidobenzene 2, two neutral stabilized species.
In order to confirm the preference for the mechanism outlined in eqn (3) we carried out Car–Parrinello26 Molecular Dynamics (CPMD)27 within the DFT framework, using the BLYP gradient-corrected functional28 and ultrasoft Vanderbilt pseudopotentials.29 These simulations were carried out at different temperatures with a 1 fs time step for the integration of the equations of motion. Our CPMD results for the entire PhN2+(1) + N3− → PhN3(2) + N2 reaction confirmed that the reactants at internuclear distances similar to those found in complex RC form s-cis-INT zwitterion in less than 100 fs at 100 K (Fig. 3). This intermediate is stable at this temperature within a time span of at least 1.2 ns. When the system was heated at ca. 300 K, the system reached s-cis-TS2 in ca. 300 fs to yield the reaction products,30 thus confirming the stepwise nature of the reaction via open intermediates of type INT. These results are in agreement with our experimental results and provide a rationale for the loss of radioactivity observed when radiolabelled azide anion was used following method A (Scheme 1).
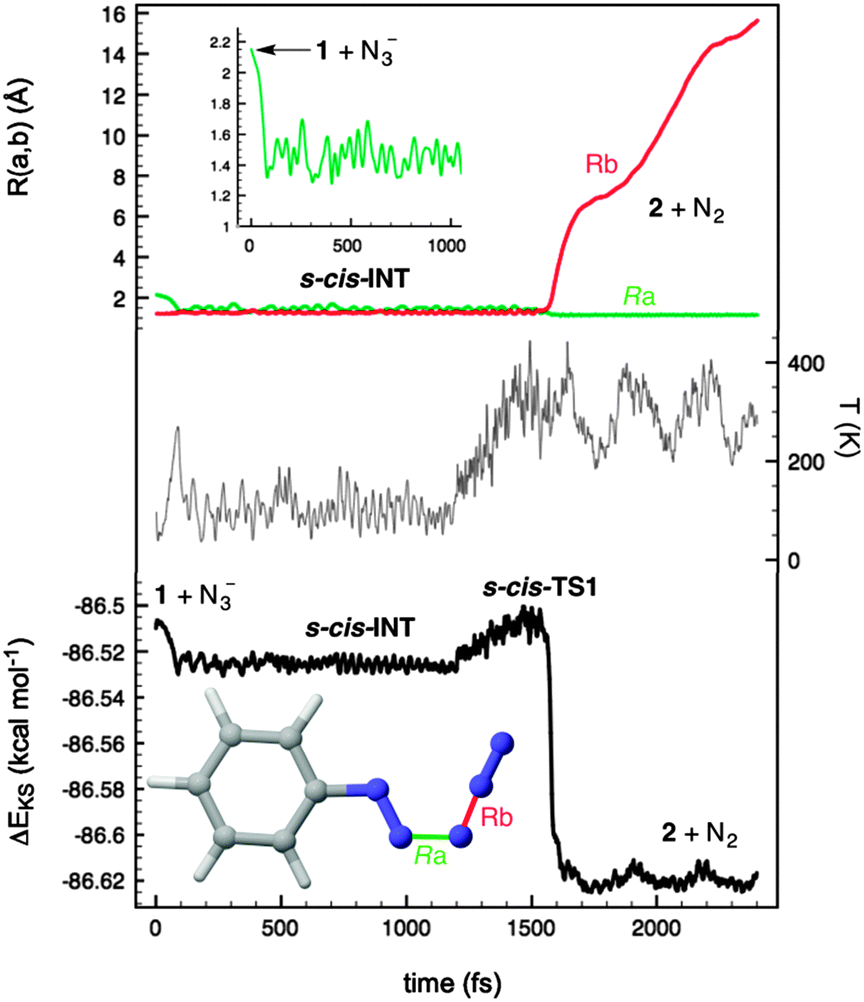 | ||
| Fig. 3 Car–Parrinello Molecular Dynamics (CPMD) plots of the reaction between the diazonium cation 1 and the azide anion to yield phenyl azide 2 and dinitrogen. | ||
Kinetic simulations carried out using reaction paths highlighted in red and blue in Fig. 2 indicate that ca. 99% of 2 stems from s-cis-TS2, whereas ca. 1% of the reaction product is formed viaINTper (see the ESI† for additional details). These results are in good agreement with the release of [13N]N2 obtained in our experimental studies following the synthetic strategy shown in Scheme 1, method A.
In conclusion, we have demonstrated using experimental and computational data that the formation of aryl azides from the corresponding diazonium salts occurs via a stepwise mechanism via acyclic zwitterionic intermediates. The use of the short-lived positron emitter nitrogen-13 for the elucidation of reaction mechanisms is unprecedented; hence, the work reported here can inspire future applications of this radionuclide beyond the preparation of radiolabelled compounds for imaging studies.
We acknowledge financial support from the RADIOMI project (EU FP7-PEOPLE-2012-ITN-RADIOMI), the Ministerio de Economía y Competitividad (MINECO) of Spain and FEDER (project CTQ2013-45415-P), the University of the Basque Country (UPV/EHU, UFI11/22 QOSYC), and the Basque Government (GV/EJ, grant IT-324-07). A. de C. and F. P. C. thank the SGI/IZO-SGIker (UPV/EHU) and the DIPC for generous allocation of computational resources.
Notes and references
- (a) P. Grieß, Philos. Trans. R. Soc. London, 1864, 13, 377 Search PubMed; (b) P. Grieß, Justus Liebigs Ann. Chem., 1865, 135, 131 CrossRef PubMed.
- (a) Y. S. Klausner and M. Bodanshky, Synthesis, 1974, 549–559 CrossRef CAS; (b) S.-Y. Han and Y.-A. Kim, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS PubMed.
- T. S. Lin and W. H. T. Prusoff, J. Med. Chem., 1978, 21, 109–112 CrossRef CAS.
- H. M. S. Kumar, B. V. S. Reddy, S. Anjaneyulu and J. S. Yadav, Tetrahedron Lett., 1999, 40, 8305–8306 CrossRef CAS.
- G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278–308 CrossRef CAS PubMed.
- C. I. Schilling, N. Jung, M. Biskup, U. Schepers and S. Bräse, Chem. Soc. Rev., 2011, 40, 4840–4871 RSC.
- (a) S. X. Cai, D. J. Glenn, K. R. Gee, M. D. Yan, R. E. Cotter, N. L. Reddy, E. Weber and J. F. W. Keana, Bioconjugate Chem., 1993, 4, 545–548 CrossRef CAS; (b) S. X. Cai, D. J. Glenn, M. Kanskar, M. Wybourne and N. J. F. W. Keana, Chem. Mater., 1994, 6, 1822–1829 CrossRef CAS; (c) E. W. Meijer, S. Nijhuis and F. C. B. M. Van Vroonhoven, J. Am. Chem. Soc., 1988, 110, 7209–7210 CrossRef CAS.
- (a) E. Noelting and O. Michel, Ber. Dtsch. Chem. Ges., 1893, 26, 86–87 CrossRef PubMed; (b) E. Noelting and O. Michel, Ber. Dtsch. Chem. Ges., 1893, 26, 88–92 CrossRef PubMed.
- E. Noelting, E. Grandmougin and O. Michel, Ber. Dtsch. Chem. Ges., 1892, 25, 3328–3342 CrossRef PubMed.
- (a) J. C. Kauer and R. A. Carboni, J. Am. Chem. Soc., 1967, 89, 2633–2637 CrossRef CAS; (b) M. Takahashi and D. Suga, Synthesis, 1998, 986–990 CrossRef CAS PubMed.
- (a) A. García-Martínez, S. de la Moya-Cerero, J. Osío-Barcina, F. Moreno-Jiménez and B. Lora-Maroto, Eur. J. Org. Chem., 2013, 6098–6107 CrossRef PubMed; (b) B. R. Ussing and D. A. Singleton, J. Am. Chem. Soc., 2005, 127, 2888–2899 CrossRef CAS PubMed; (c) Z. Wu and R. Glaser, J. Am. Chem. Soc., 2004, 126, 10632–10639 CrossRef CAS PubMed; (d) I. M. Cuccovia, M. A. da Silva, H. M. C. Ferraz Jr., J. R. Pliego, J. M. Riveros and H. J. Chaimovich, J. Chem. Soc., Perkin Trans. 2, 2000, 1896–1907 RSC.
- For analogous (3+2) cycloadditions involving arsa-diazonium salts see: M. Kuprat, A. Schultz and A. Villinger, Angew. Chem., Int. Ed., 2013, 52, 7126–7130 CrossRef CAS PubMed.
- R. N. Butler, A. Fox, S. Collier and L. A. Burke, J. Chem. Soc., Perkin Trans. 2, 1998, 2243–2247 RSC.
- V. Gómez-Vallejo, K. Kato, M. Hanyu, K. Minegishi, J. I. Borrell and J. Llop, Bioorg. Med. Chem. Lett., 2009, 19, 1913 CrossRef PubMed.
- (a) J. Llop, V. Gómez-Vallejo, M. Bosque, G. Quincoces and I. Peñuelas, Appl. Radiat. Isot., 2009, 67, 95 CrossRef CAS PubMed; (b) V. Gómez-Vallejo, K. Kato, I. Oliden, J. Calvo, Z. Baz, J. I. Borrell and J. Llop, Tetrahedron Lett., 2010, 51, 2990 CrossRef PubMed.
- (a) V. Gomez-Vallejo, J. I. Borrell and J. Llop, Eur. J. Med. Chem., 2010, 45, 5318 CrossRef CAS PubMed; (b) V. Gaja, V. Gomez-Vallejo, M. Puigivila, C. Perez-Campana, A. Martin, A. Garcia-Osta, T. Calvo-Fernandez, M. Cuadrado-Tejedor, R. Franco and J. Llop, Mol. Imaging Biol., 2014, 16, 5380 Search PubMed.
- A. A. Siddiki, B. S. Takale and V. N. Telvekar, Tetrahedron Lett., 2013, 54, 1294–1297 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision B.1., Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed; (b) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- (a) S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef; (b) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- F. Weigend and R. Ahirichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- K. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- (a) K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS; (b) P. W. Ayers, W. Yang and L. J. Bartolotti, The Fukui Function, in Chemical Reactivity Theory: A Density Functional View, ed. P. Chattaraj, Taylor & Francis, Boca Ratón, 2009, pp. 255–267 Search PubMed.
- (a) A. de Cózar and F. P. Cossío, Phys. Chem. Chem. Phys., 2011, 13, 10858–10868 RSC; (b) I. Fernández, F. P. Cossío and F. M. Bickelhaupt, J. Org. Chem., 2011, 76, 2310–2314 CrossRef PubMed.
- C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471–2474 CrossRef CAS.
- CPMD code (http://www.cpmd.org): Copyright MPI für Festkçrperforschung, Stuttgart, and IBM Zürich Research Laboratory, 1990–2006.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; (b) C. Lee, W. Yang and R. C. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- See movie SM1 in the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for the synthesis of 13N-labeled phenyl azide, and identification of the labelled species using radio-HPLC and GC-MS. Energies, zero-point vibrational energies, Gibbs energy corrections and Cartesian coordinates of all the stationary points discussed in this work. Movie including the CPMD simulations. Full ref. 18. See DOI: 10.1039/c5cc01913c |
| This journal is © The Royal Society of Chemistry 2015 |