
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NMR crystallography of monovalent cations in inorganic matrixes: Li+ siting and the local structure of Li+ sites in ferrierites†
P.
Klein
ab,
J.
Dedecek
a,
H. M.
Thomas
a,
S. R.
Whittleton
a,
V.
Pashkova
a,
J.
Brus
c,
L.
Kobera
c and
S.
Sklenak
*a
aJ. Heyrovský Institute of Physical Chemistry, The Czech Academy of Sciences, Dolejškova 3, CZ 182 23 Prague 8, Czech Republic. E-mail: stepan.sklenak@jh-inst.cas.cz
bDepartment of Inorganic Technology, Faculty of Chemical Technology, University of Pardubice, Doubravice 41, Pardubice, CZ 532 10, Czech Republic
cInstitute of Macromolecular Chemistry, The Czech Academy of Sciences, Heyrovsky sq. 2, Prague 6, CZ 162 06, Czech Republic
First published on 23rd April 2015
Abstract
7Li–7Li correlation MAS NMR spectroscopy, interpreted using periodic DFT including molecular dynamics conformational sampling of Li+ sites, is employed to obtain the siting of Li+ at exchangeable positions of ferrierites and the local structure of these Li+ sites. The former is controlled by the Al siting in the zeolite framework.
Zeolites represent the most important group of heterogeneous catalysts widely applied in industry. Besides the Y and USY zeolites of the faujasite structure, silicon-rich zeolites (Si/Al > 8) such as ZSM-5, the beta zeolite, ferrierite, MCM-22, and mordenite exhibit the highest industrial impact.1 These crystalline microporous aluminosilicates with 3D channel structures are made of corner-sharing TO4 tetrahedra (T = Si or Al−). Isomorphous framework Al/Si substitutions result in a negative charge of AlO4− tetrahedra, which is balanced by extra-framework cationic species representing active sites for numerous redox- or base-catalyzed reactions.
The cations can occupy extra-framework sites in silicon-rich zeolites which differ in the coordination, arrangement of the near environment, and location in zeolite channels.1 These properties are controlled by the siting of the Al atoms of the negatively charged AlO4− tetrahedra in the framework crystallographic T sites. There are properties of silicon-rich zeolites, which strongly limit the applicability of diffraction methods to determine the siting of cations: (i) a medium (e.g. ferrierite (4) and mordenite (4)) or high (e.g. MCM-22 (8), the beta zeolite (9), and ZSM-5 (12 or 24)) number of crystallographically distinguishable framework T sites of which only some are partly occupied by Al atoms, (ii) low number of Al atoms in the framework (Si/Al > 8), and (iii) large unit cells.2 Therefore the knowledge regarding the siting of cations and the local structure of cationic sites in silicon-rich zeolites is very limited.1,3 However, this knowledge is essential to evaluate the catalytic and sorption properties of cation exchanged zeolites. Scarce crystallographic studies deal with monovalent cations,4–6 but the studies are rather limited to heavy ions (Cs+ (ref. 7–9) and Tl+ (ref. 10)) not very attractive for catalysis. Li is a very light element which is hardly detected by X-ray diffraction in silicon-rich zeolites employing powder diffraction, and therefore, neutron diffraction was used to detect the siting of Li+ ions in silicon-rich matrixes.5
The siting of divalent metal cations was determined by X-ray crystallography for ferrierites11,12 with Si/Al 8.5 and mordenites,3 while for zeolites with a lower Al content in the framework a methodology based on the Vis spectra of Co2+ ions was developed.1,13–15 Moreover, X-ray experiments provide only the positions of cations in the framework but not the local structure of the cationic sites as the coordinates of the cations are combined with the averaged coordinates of the framework reflecting mainly empty cationic sites and also the corresponding siliceous structures (i.e., without the framework Al/Si substitutions). Conversely, 6,7Li MAS NMR spectroscopy represents a powerful tool to identify Li+ siting.16–19 The issue of incorporation of Li+ ions into various matrixes has attracted particular attention due to its crucial importance in various fields such as, for example, energy storage20 and CO2 capture.21
In this communication, we demonstrate a new approach to the determination of the siting of Li+ and the local structure of Li+ sites in the crystalline aluminosilicate matrixes based on the 7Li–7Li correlation MAS NMR spectroscopy in tandem with periodic DFT calculations of the structure of Li+ sites and subsequent DFT cluster computations of the 7Li NMR shielding. The calculations of the structure include extensive molecular dynamics conformational sampling of the siting of Li+ ions. The Li+ siting and its variability in silicon-rich ferrierites (Si/Al ≥ 20) is determined and subsequently independently verified using the previous results regarding the Al siting in the same ferrierite samples.22
The significant differences between the 7Li MAS NMR spectra (Fig. S1, ESI†) clearly reflect the variations in the Li+ siting caused by the different siting of Al atoms in the framework T sites.22 However, a detailed analysis of these spectra is limited due to the narrow range of the 7Li chemical shifts23 and low resolution of 7Li MAS NMR spectra. Zeolites are typical materials for which the broadening of NMR resonances (e.g. for 29Si and 27Al) can originate from the variability of middle- and long-range orderings of the zeolite framework due to isomorphous substitutions of Al into the silicate framework. The observed NMR signal therefore represents a Gaussian envelope of a number of close NMR resonances.24–27 The observed 7Li broadening is due to neither the anisotropy of the chemical shift nor weak quadrupolar interaction.23 In this case, the application of 6Li MAS NMR spectroscopy (not shown in the figures) does not result in the increase of the spectral resolution. Thus, also the application of an ultra-high field does not represent a viable route to increase the spectral resolution. Multiple quantum (MQ) MAS NMR experiments, which provide a significant increase of the spectral resolution for quadrupolar nuclei, cannot be applied on 7Li either due to the small quadrupolar interaction of 7Li nucleus.23 Our experiments show that also applications of various one-dimensional MAS NMR pulse sequences (solid echo, 1H decoupled single-pulse, and triple-quantum filtered MAS NMR experiments) do not result in the increase of the resolution of the 7Li MAS NMR spectra (not shown in the figures). In contrast, we show that analysis of non-diagonal cross-peaks in the 2D 7Li–7Li correlation spectra28–31 provides an increase of the spectral resolution and thus the accuracy of the estimation of the number of 7Li NMR resonances (Fig. 1).
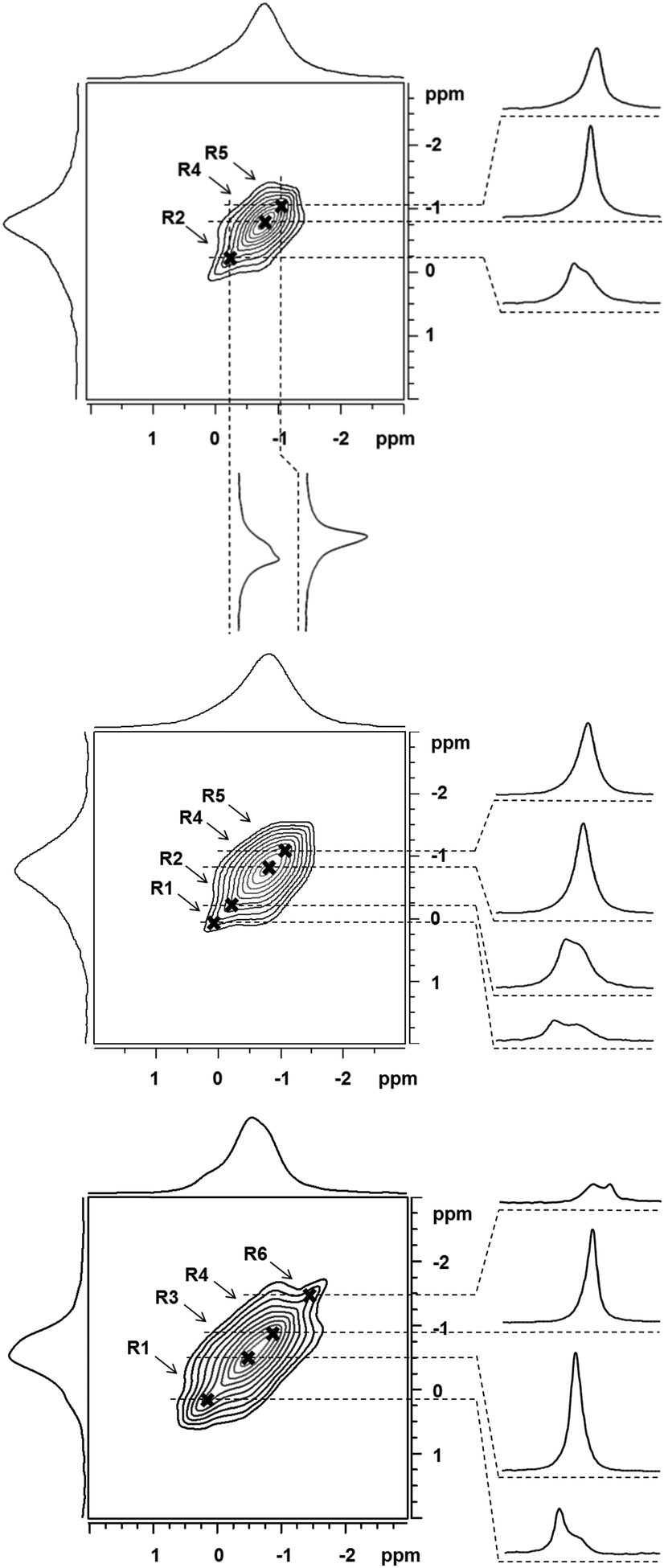 | ||
| Fig. 1 2D projection of 7Li–7Li correlation MAS NMR spectrum of the dehydrated FER/20 (top), FER/27 (middle), and FER/30 (bottom) samples, spectrum projection, selected slices, and marked positions of non-diagonal cross-peaks. | ||
As the long-range 7Li–7Li correlations are evolved during the mixing period (50 ms), the cross-sections through the shoulders apparent in the skyline projections allow refinement of 7Li isotropic chemical shifts of the dipolarly coupled partners. Our preliminary calibration tests indicated that the upper limit of 7Li⋯7Li distances that can be effectively probed by the applied correlation experiment is ca. 4.0–4.3 Å.
The 7Li NMR resonances identified for FER/20–FER/30 using the 7Li–7Li correlation experiments are shown in Table 1 and Fig. 2. The latter also depicts the relative concentrations of the Li+ ions corresponding to the individual 7Li NMR resonances obtained by the simulation of 7Li MAS NMR spectra employing the 7Li chemical shifts from the 7Li–7Li correlation experiments.
| Sample | 7Li NMR resonances (ppm) | |||||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | R6 | |
| FER/20 | −0.23 | −0.80 | −1.07 | |||
| FER/27 | 0.17 | −0.20 | −0.80 | −1.05 | ||
| FER/30 | 0.12 | −0.55 | −0.85 | −1.50 | ||
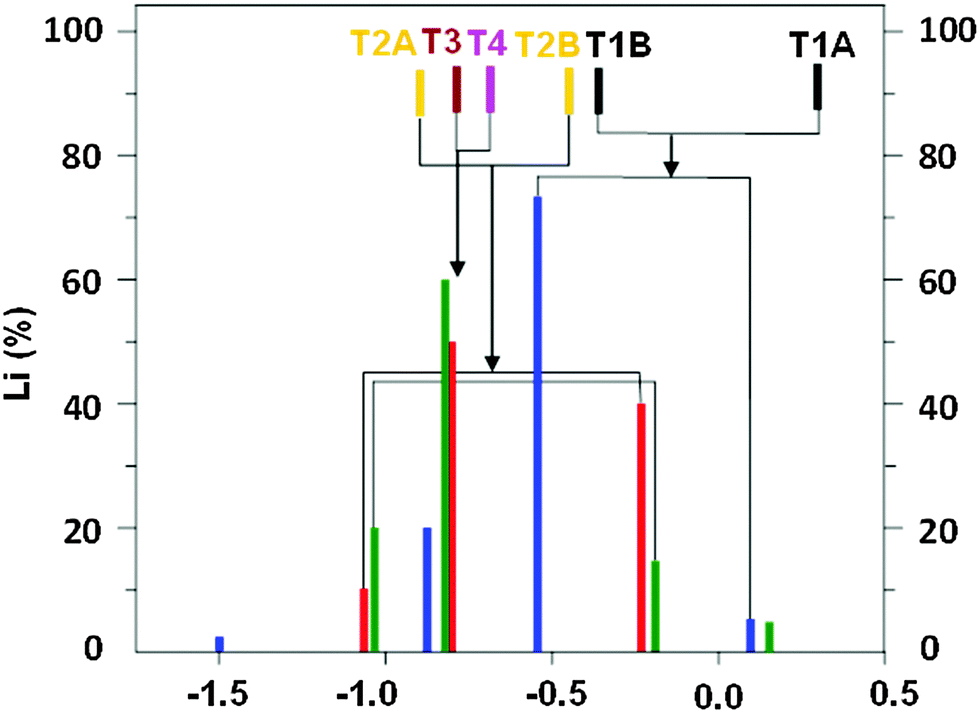 | ||
Fig. 2 Experimental 7Li chemical shifts and their intensities in the spectra of the dehydrated FER/20–FER/30 samples, 7Li chemical shifts calculated for Li+ ions balancing Al atoms in the T1–T4 sites, and their assignments to the experimental data. FER/20 ( ), FER/27 ( ), FER/27 ( ), and FER/30 ( ), and FER/30 ( ); Li+ balancing Al in the T1 ( ); Li+ balancing Al in the T1 ( ), T2 ( ), T2 ( ), T3 ( ), T3 ( ), and T4 ( ), and T4 ( ) sites. ) sites. | ||
Periodic DFT calculations including molecular dynamics conformational sampling of all possible Li+ sites for Al(T1a),22 Al(T1b),22 Al(T2), Al(T3), and Al(T4) yield two low energy (i.e., their relative energy ≤2.0 kcal mol−1 with respect to the most stable Li+ site for Al in a particular T site) Li+ sites for T1 and T2 while only one for T3 and T4. The Li+ sites for Al(T1a)22 and Al(T1b)22 are the same. Subsequently the 7Li NMR shieldings are calculated using seven shell clusters cut out from the optimized structures and converted into 7Li chemical shifts (Fig. 2). This combination of employing periodic DFT for the structure determination and cluster DFT for the evaluation of the NMR shielding, which is a local property, has been successfully used in our prior study.22 The calculated relative energies of the other Li+ sites are significantly higher and therefore they are not populated.32 The optimized structures of the low energy Li+ sites together with the relative energies and the corresponding 7Li chemical shifts are shown in Fig. 3.
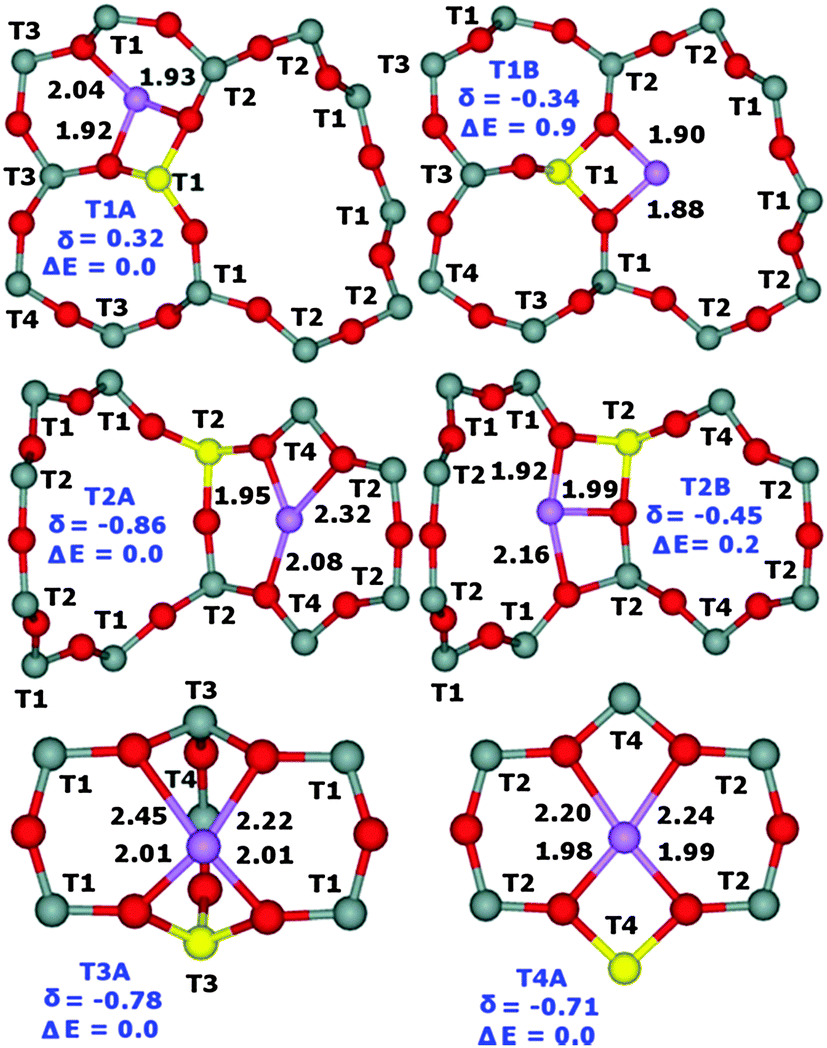 | ||
| Fig. 3 Optimized structures (Li–O distances in Å) with the designations of the T sites of the low energy Li+ sites (T1A and T1B for Al(T1), T2A and T2B for Al(T2), T3A for Al(T3), and T4A for Al(T4)), the relative energies in kcal mol−1, and the corresponding 7Li chemical shifts in ppm converted using the 7Li NMR shielding of 90.16 ppm for Li+(H2O)4 with the 7Li chemical shift at 0.00 ppm.33 Silicon atoms are in gray, oxygen atoms in red, aluminum atoms in yellow, and lithium in violet. | ||
Our computational results show that Li+ ions are coordinated to two O atoms (only one O atom for the T2A site) of the AlO4− framework (Li–OAl distances from 1.88 to 2.01 Å) and up to two O atoms of the SiO4 framework (Li–OSi distances from 2.04 to 2.45 Å) in dehydrated zeolites and that the Li+ siting is controlled by the location of Al atoms in the framework T sites. Two Li+ sites are occupied concurrently for Al(T1) and Al(T2) while only one for Al(T3) and Al(T4).
Fig. 2 compares the experimental 7Li chemical shifts with the calculated ones. The patterns of the experimental and predicted 7Li chemical shifts in Fig. 2 show significant similarities. The R5 resonance observed for the FER/20 and FER/27 samples (Table 1) can be safely assigned to the T2A site. Since there are two low energy Li+ sites for Al(T2), the R2 resonance measured for the same samples corresponds to the T2B site. Then, it follows that the R4 resonance observed also for FER/20 and FER/27 belongs to either one of the T3A and T4A sites or to both of them. The calculated 7Li chemical shifts of T3A and T4A are too close to each other to be distinguishable by 7Li MAS NMR spectroscopy. The R1 resonance observed for FER/27 and FER/30 can be safely assigned to the T1A site. There are three more 7Li NMR resonances measured for FER/30 (Table 1). R4 corresponds to either one of the T3A and T4A sites or to both of them while R3 belongs to the T1B site which is the other low energy Li+ site for Al(T1). Fig. 2 shows the assignment of the observed 7Li NMR resonances to the Li+ sites. The shift deviations are ca. ±0.2 ppm. The remaining very low intensity R6 resonance at −1.50 ppm, which is observed only for FER/30, cannot be assigned to any of the calculated low energy Li+ sites and its origin is unknown. The ranges of the observed (without the unassigned R6) and calculated 7Li chemical shifts (1.24 and 1.18 ppm, respectively) are in very good agreement.
There are no experimental data based on diffraction methods regarding the siting of Li+ in ferrierites. However, the knowledge of the Al siting in the three ferrierite samples used22 permits a verification of the siting of Li+ ions obtained in this study. The Al atoms occupy the T sites in the samples (for details see Fig. 6 of ref. 22) as follows: T2, T3, and T4 in FER/20; T1, T2, T3, and T4 in FER/27; and T1, T3, and T4 in FER/30. Fig. 4 compares the relative concentration of Al atoms (in %) corresponding to the T sites obtained from (i) the corresponding Li+ siting analyzed using 7Li MAS NMR and (ii) 27Al MAS NMR experiments.22
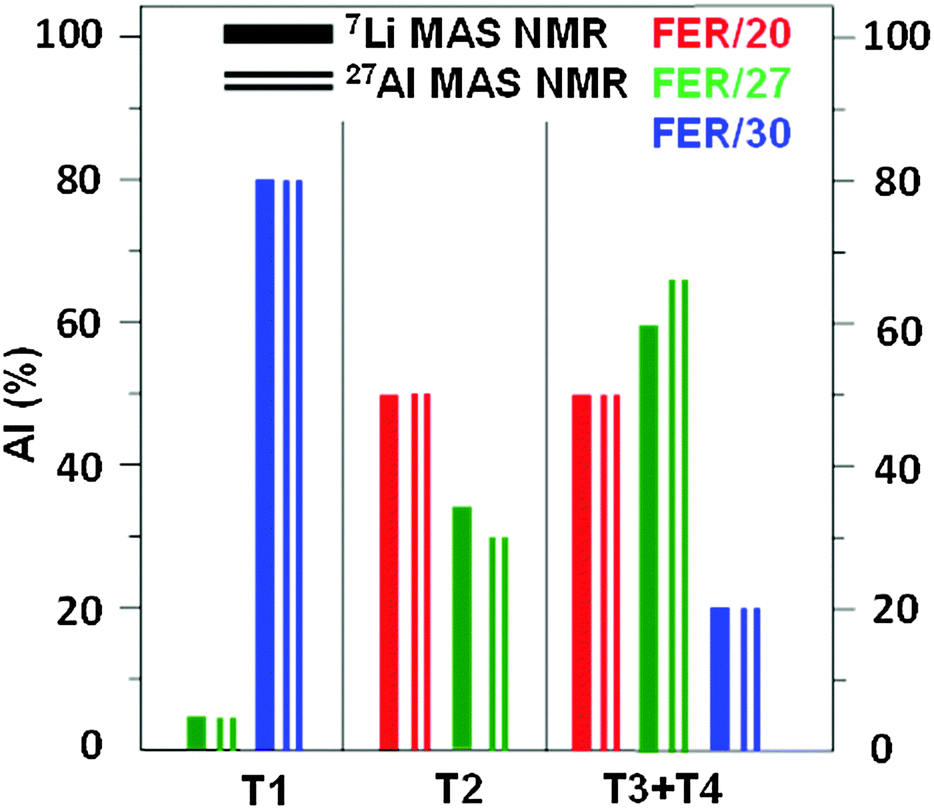 | ||
| Fig. 4 Relative concentration of Al atoms (in %) corresponding to the distinguishable framework T sites obtained from (i) the corresponding Li+ siting analyzed using 7Li MAS NMR and (ii) 27Al MAS NMR.22 | ||
The agreement between the results obtained by 7Li and 27Al MAS NMR is very good and confirms the assignment of the experimental 7Li NMR resonances to the Li+ sites related to Al in the individual T sites. It substantiates that the siting of Li+ is controlled by the siting of Al atoms in the zeolite framework. Moreover, the agreement supports the reliability of (i) the periodic DFT calculations of the local structure of Li+ sites using extensive conformational sampling and (ii) DFT cluster computations of the 7Li NMR shielding. Moreover, only the 27Al 3Q MAS NMR experiments allow the distinction between the T3 and T4 sites and reveal that the 7Li R4 resonance observed for all the three samples belongs to Li+ in both the T3A and T4A sites. Therefore, the combination of 7Li MAS NMR and 27Al 3Q MAS NMR experiments in tandem with DFT calculations is suggested to represent the optimal approach to the analysis of the Al and Li+ sitings in the framework T and extra-framework sites, respectively, in silicon-rich zeolites.
Li+ ions in many crystalline oxides are assigned to tetrahedral and octahedral sites based on their 6Li chemical shifts.34 According to 6Li chemical shift correlation data,35 Li+ ions in the former and latter positions exhibit positive and negative 6Li chemical shifts, respectively. However, our study on a silicon-rich zeolite reveals that the coordination of Li+ ions (also showing positive and negative 7Li chemical shifts) exhibits low symmetry and it is different from tetrahedral or octahedral. The local structure of Li+ sites is controlled by the framework structure and by the location of Al atoms in the framework T sites.
In summary, we present a newly developed method to determine the siting of Li+ and the local structure of Li+ sites in crystalline aluminosilicate matrixes based on a combination of 7Li–7Li correlation MAS NMR spectroscopy and periodic DFT calculations of the structure of Li+ sites and subsequent DFT cluster computations of the 7Li NMR shielding. The developed approach can be in general applied to Li+ ions in other zeolites and various crystalline matrixes with large unit cells and a low concentration of Li+ and also to other NMR-active cations without a significant limitation of their concentration. It should be stressed that calculations with an extensive conformational sampling of the cation are required (due to the absence of experimental data regarding the siting of the cation) to obtain the accurate siting of the cation, i.e. employing only optimizations of the structure of the cationic sites in the zeolite framework is not sufficient.
This work was supported by the Grant Agency of the Czech Republic (# GA 15-14007S), COST Action MP1202 HINT (Ministry of Education, Youth and Sports LD14010) and the RVO:61388955 and RVO:61389013. This work was supported by the IT4Innovations Centre of Excellence project (CZ.1.05/1.1.00/02.0070), funded by the European Regional Development Fund and the national budget of the Czech Republic via the Research and Development for Innovations Operational Programme, as well as Czech Ministry of Education, Youth and Sports via the project Large Research, Development and Innovations Infrastructures (LM2011033).
Notes and references
- J. Dedecek, Z. Sobalik and B. Wichterlova, Catal. Rev., 2012, 54, 135–223 CAS.
- http://www.iza-structure.org/databases .
- W. J. Mortier, Extraframework cationic positions in zeolites, Elsevier, Amsterdam, 1982 Search PubMed.
- I. J. Pickering, P. J. Maddox, J. M. Thomas and A. K. Cheetham, J. Catal., 1989, 119, 261–265 CrossRef CAS.
- B. F. Mentzen, J. Phys. Chem. C, 2007, 111, 18932–18941 CAS.
- B. F. Mentzen and G. Bergeret, J. Phys. Chem. C, 2007, 111, 12512–12516 CAS.
- D. H. Olson, N. Khosrovani, A. W. Peters and B. H. Toby, J. Phys. Chem. B, 2000, 104, 4844–4848 CrossRef CAS.
- B. F. Mentzen, G. Bergeret, H. Emerich and H. P. Weber, J. Phys. Chem. B, 2006, 110, 97–106 CrossRef CAS PubMed.
- C. W. Kim, N. H. Heo and K. Seff, J. Phys. Chem. C, 2011, 115, 24823–24838 CAS.
- N. H. Heo, C. W. Kim, H. J. Kwon, G. H. Kim, S. H. Kim, S. B. Hong and K. Seff, J. Phys. Chem. C, 2009, 113, 19937–19956 CAS.
- M. C. Dalconi, G. Cruciani, A. Alberti, P. Ciambelli and M. T. Rapacciuolo, Microporous Mesoporous Mater., 2000, 39, 423–430 CrossRef CAS.
- M. C. Dalconi, A. Alberti, G. Cruciani, P. Ciambelli and E. Fonda, Microporous Mesoporous Mater., 2003, 62, 191–200 CrossRef CAS.
- J. Dedecek and B. Wichterlova, J. Phys. Chem. B, 1999, 103, 1462–1476 CrossRef CAS.
- D. Kaucky, J. Dedecek and B. Wichterlova, Microporous Mesoporous Mater., 1999, 31, 75–87 CrossRef CAS.
- S. Sklenak, P. C. Andrikopoulos, S. R. Whittleton, H. Jirglova, P. Sazama, L. Benco, T. Bucko, J. Hafner and Z. Sobalik, J. Phys. Chem. C, 2013, 117, 3958–3968 CAS.
- M. Feuerstein and R. F. Lobo, Chem. Mater., 1998, 10, 2197–2204 CrossRef CAS.
- L. J. Smith, H. Eckert and A. K. Cheetham, J. Am. Chem. Soc., 2000, 122, 1700–1708 CrossRef CAS.
- L. J. Smith, H. Eckert and A. K. Cheetham, Chem. Mater., 2001, 13, 385–391 CrossRef CAS.
- H. Oka, S. Kasahara, T. Okada, S. Yoshida, A. Harada, H. Ohki and T. Okuda, Microporous Mesoporous Mater., 2002, 51, 1–5 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- T. D. Pham, Q. Liu and R. F. Lobo, Langmuir, 2013, 29, 832–839 CrossRef CAS PubMed.
- J. Dedecek, M. J. Lucero, C. Li, F. Gao, P. Klein, M. Urbanova, Z. Tvaruzkova, P. Sazama and S. Sklenak, J. Phys. Chem. C, 2011, 115, 11056–11064 CAS.
- K. J. D. Mackenzie and A. M. E. Smith, Multinuclear Solid State NMR of Inorganic Materials, Elsevier, 2002 Search PubMed.
- C. A. Fyfe, G. C. Gobbi, J. Klinowski, J. M. Thomas and S. Ramdas, Nature, 1982, 296, 530–533 CrossRef CAS.
- S. Sklenak, J. Dedecek, C. B. Li, B. Wichterlova, V. Gabova, M. Sierka and J. Sauer, Angew. Chem., Int. Ed., 2007, 46, 7286–7289 CrossRef CAS PubMed.
- S. Sklenak, J. Dedecek, C. Li, B. Wichterlova, V. Gabova, M. Sierka and J. Sauer, Phys. Chem. Chem. Phys., 2009, 11, 1237–1247 RSC.
- J. Dedecek, S. Sklenak, C. Li, F. Gao, J. Brus, Q. Zhu and T. Tatsumi, J. Phys. Chem. C, 2009, 113, 14454–14466 CAS.
- L. S. Cahill, Y. Iriyama, L. F. Nazar and G. R. Goward, J. Mater. Chem., 2010, 20, 4340–4346 RSC.
- H. Hain, M. Scheuermann, R. Heinzmann, L. Wuensche, H. Hahn and S. Indris, Solid State Nucl. Magn. Reson., 2012, 42, 9–16 CrossRef CAS PubMed.
- Y.-Y. Hu, Z. Liu, K.-W. Nam, O. J. Borkiewicz, J. Cheng, X. Hua, M. T. Dunstan, X. Yu, K. M. Wiaderek, L.-S. Du, K. W. Chapman, P. J. Chupas, X.-Q. Yang and C. P. Grey, Nat. Mater., 2013, 12, 1130–1136 CrossRef CAS PubMed.
- A. Kuhn, S. Dupke, M. Kunze, S. Puravankara, T. Langer, R. Poettgen, M. Winter, H.-D. Wiemhoefer, H. Eckert and P. Heitjans, J. Phys. Chem. C, 2014, 118, 28350–28360 CAS.
- By 3–7 kcal mol−1 for Al(T1), 5–6 kcal mol−1 for Al(T2), 5–6 kcal mol−1 for Al(T3), and 12–19 kcal mol−1 for Al(T4).
- The calculated NMR shielding values are as follows: 89.84 ppm (T1A), 90.50 ppm (T1B), 91.02 ppm (T2A), 90.61 ppm (T2B), 90.94 ppm (T3A), and 90.87 ppm (T4A).
- M. Vijayakumar, S. Kerisit, K. M. Rosso, S. D. Burton, J. A. Sears, Z. G. Yang, G. L. Graff, J. Liu and J. Z. Hu, J. Power Sources, 2011, 196, 2211–2220 CrossRef CAS PubMed.
- Z. Xu and J. F. Stebbins, Solid State Nucl. Magn. Reson., 1995, 5, 103–112 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details regarding (i) sample preparation and characterization, (ii) 7Li NMR experiments, (iii) computational models and methods and (iv) 7Li MAS NMR spectra sections. See DOI: 10.1039/c5cc01830g |
| This journal is © The Royal Society of Chemistry 2015 |