
Pyridynes and indolynes as building blocks for functionalized heterocycles and natural products
Adam E.
Goetz
,
Tejas K.
Shah
and
Neil K.
Garg
*
Department of Chemistry and Biochemistry, University of California, 607 Charles Young Drive East, Box 951569, Los Angeles, CA 90095, USA. E-mail: neilgarg@chem.ucla.edu
First published on 5th September 2014
Abstract
Heterocyclic arynes, or hetarynes, have been studied for over 100 years. However, challenges associated with observing these reactive species, as well as developing synthetically useful methods for their generation and trapping, have limited their use. This review provides a brief historical perspective on the field of hetarynes, in addition to a discussion of pyridyne and indolyne methodologies. Moreover, this review highlights the use of pyridynes, indolynes, and related strained intermediates in natural product synthesis.
 Adam E. Goetz | Dr Adam Goetz received his BA in Chemistry from Carleton College in Northfield, MN and conducted summer research with Professor Robert Kempton at Northern Kentucky University. After graduating, he spent a year as a research assistant for Segetis, Inc. in Minneapolis, MN. He has recently earned his doctorate with Professor Neil K. Garg at the University of California, Los Angeles. His dissertation studies were focused on synthetic methodology involving heterocyclic arynes, in addition to applications of hetaryne methodology for the synthesis of complex natural products. |
 Tejas K. Shah | Tejas Shah received his BA in Chemistry and Molecular Biology & Biochemistry from Rutgers University in New Brunswick, NJ, where he performed undergraduate research under Professor Daniel Seidel. He is currently a fourth year graduate student in Professor Neil K. Garg's laboratory at the University of California, Los Angeles. His graduate studies are focused on developing and utilizing heterocyclic arynes for the synthesis of complex natural products and new materials. |
 Neil K. Garg | Neil Garg is a Professor of Chemistry at the University of California, Los Angeles. Professor Garg received a BS in Chemistry from New York University where he did undergraduate research with Professor Marc Walters. During his undergraduate years, he spent several months in Strasbourg, France while conducting research with Professor Mir Wais Hosseini at Université Louis Pasteur as an NSF REU Fellow. Garg obtained his PhD in 2005 from the California Institute of Technology under the direction of Professor Brian Stoltz. He then joined Professor Larry Overman's research laboratory at the University of California, Irvine as an NIH Postdoctoral Scholar. Garg joined the faculty at UCLA in 2007. His laboratory develops novel synthetic strategies and methodologies to enable the total synthesis of complex bioactive molecules. |
Introduction and historical perspective
The importance of heterocyclic compounds in modern chemistry cannot be overstated. Methods for the synthesis of functionalized heterocyclic compounds are highly prized because of their applications in pharmaceutical chemistry, materials chemistry, natural products synthesis, organometallic chemistry, and many other fields. Among the various methods for functionalizing heterocycles, the use of highly reactive heterocyclic arynes, or hetarynes,1 offers a strategically different approach compared to many conventional methods. Arynes have a rich history, and despite being studied experimentally for over 60 years, many of these species are still treated as scientific curiosities rather than as useful synthetic intermediates. The majority of studies related to arynes focus on carbocyclic arynes, such as ortho-benzyne (1),2 rather than heterocyclic arynes (e.g., 2–6, Fig. 1).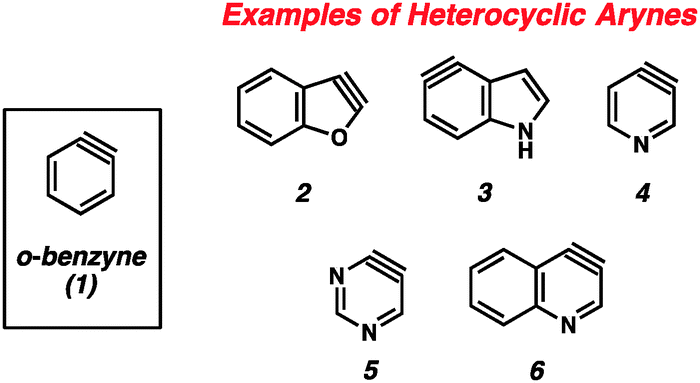 | ||
| Fig. 1 Examples of arynes. | ||
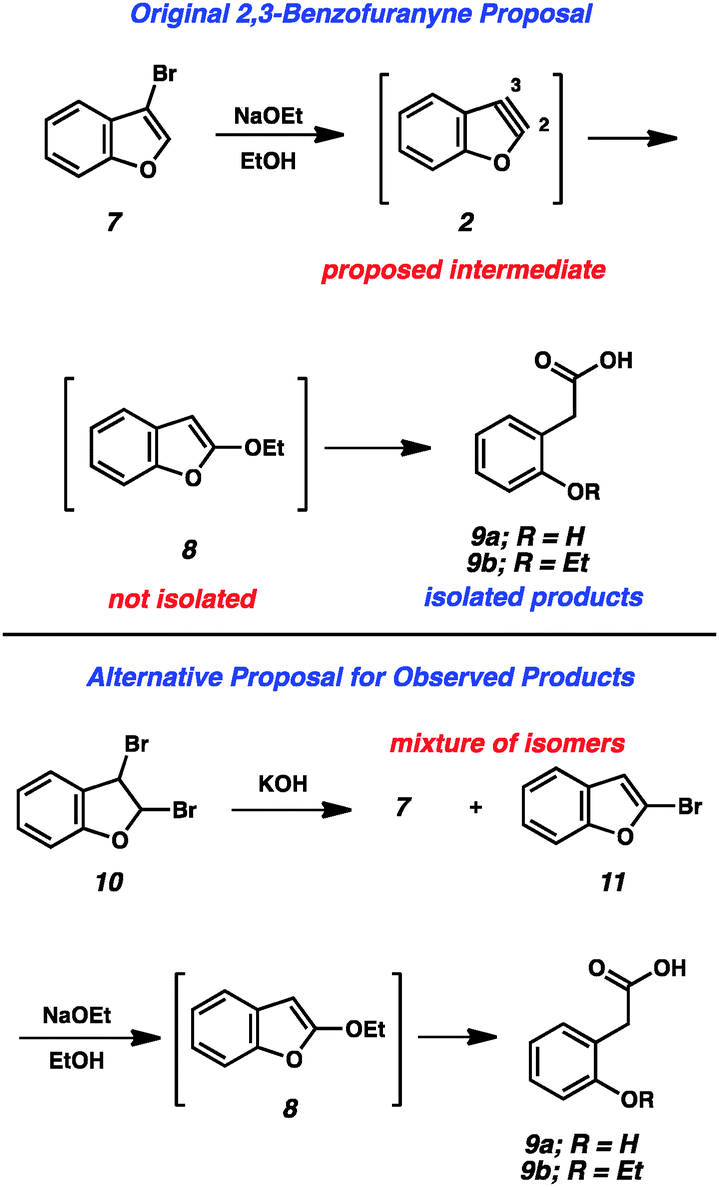
The first appearance of any type of aryne in the literature comes from Stoermer and Kahlert's 1902 report invoking 2,3-benzofuranyne (2) as an intermediate (Fig. 2).3 The authors proposed that treatment of 3-bromobenzofuran (7) with sodium ethoxide led to hetaryne 2, which was trapped by excess ethoxide to give 8. This proposal, despite its influence on the field of aryne chemistry, has been called into question.1b In the original report, compound 8 was not isolated, but rather was inferred based on the formation of 9a and 9b, which were thought to arise from ring opening of 8. Alternative proposals suggest that contamination of precursor 7 with the isomeric 2-bromobenzofuran (11) would readily give the proposed intermediate 8 through an SNAr pathway.1b Given that bromide 7 was prepared through elimination of dibromide 10, the alternate mechanism is certainly plausible.
 | ||
| Fig. 2 Initial proposal of 2,3-benzofuranyne formation. | ||
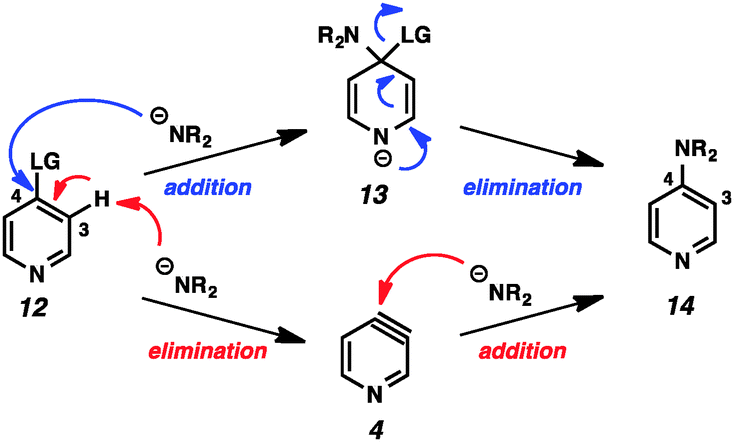
This example highlights one of the inherent challenges with the study of arynes and hetarynes; specifically, the fact that they cannot be readily observed. Typically, the strongest evidence for the presence of an aryne intermediate is the identity of the products obtained. However, even this can lead to incorrect conclusions. As shown in Fig. 3, it is possible to arrive at so-called “aryne products” through pathways that do not involve aryne formation. This possibility is especially relevant when strong bases, which are also nucleophilic, are employed to generate arynes through dehydrohalogenation, as is demonstrated using pyridine 12. In what is termed the addition/elimination pathway, nucleophilic attack by the amide on the electrophilic C4 position generates stabilized adduct 13, which then undergoes ejection of the leaving group to give product 14. This is in contrast to the elimination/addition pathway. In this route, pyridine 12 can undergo deprotonation at C3 followed by elimination of the leaving group to generate 3,4-pyridyne (4). Attack on this species by a nucleophile at C4 followed by protonation then provides product 14. Additionally, it is also possible that both aryne and non-aryne mechanisms are operating competitively in a given reaction. Since many of the early aryne studies relied on this method of aryne generation, not all claims of aryne generation are likely accurate, and care must be taken in interpretation of results. Previous reviews have sought to identify certain aryne precursors that are prone to favor one mechanism over the other.1
 | ||
| Fig. 3 Competing aryne and non-aryne mechanisms. | ||
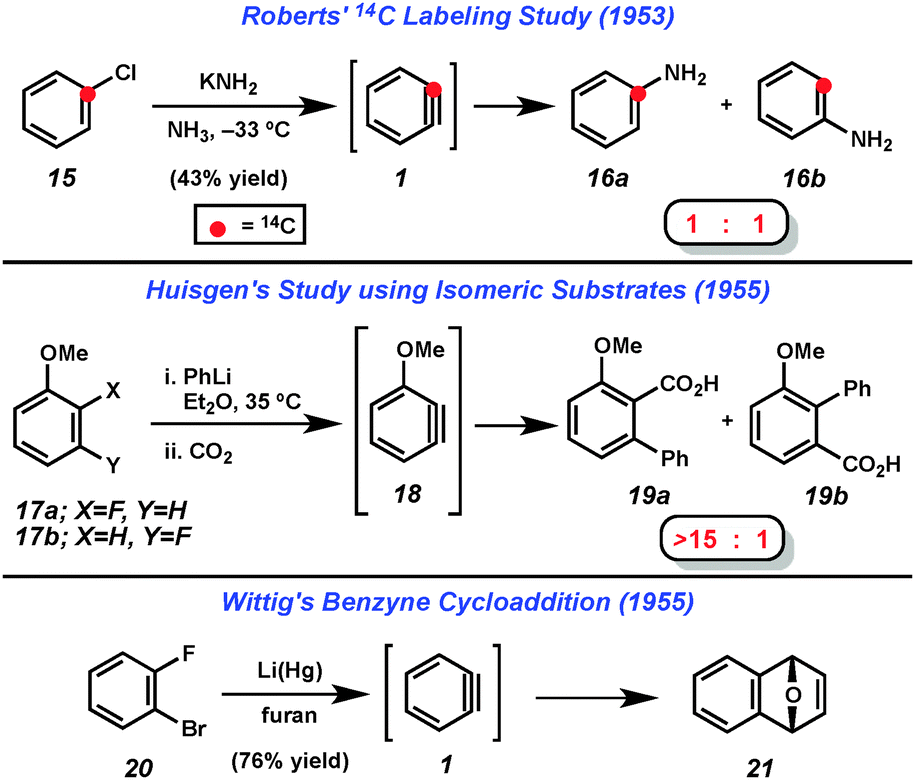
Despite uncertainty over the generation of arynes such as 2, solid evidence for the intermediacy of these highly reactive species was obtained in the 1950s. Following several reports of unexplained “rearrangements” in reactions of halobenzenes with amide bases,4 Roberts demonstrated that treatment of 14C-labeled chlorobenzene (15) with potassium amide gave an equimolar mixture of products 16a and 16b (Fig. 4).5 This was rationalized by the presence of the symmetrical “benzyne” (1) intermediate. Shortly thereafter, Huisgen demonstrated that treating either 2- or 3-fluoroanisole (17a or 17b) with phenyllithium followed by CO2 led to the isolation of compounds 19a and 19b with a strong preference for 19a (Fig. 4).6 Similar to Roberts' reasoning, Huisgen noted that since the two isomeric starting materials gave the same major product with similar ratios, a common intermediate, 18, was likely involved. He also noted that the π-system of the proposed intermediate 18 would be orthogonal to the aromatic system. Finally, Wittig demonstrated that treatment of dihalobenzene 20 with lithium amalgam in furan led to the isolation of cycloadduct 21, which further suggested the intermediacy of benzyne (1).7 These three studies represent the first solid evidence for the intermediacy of any aryne and inspired the development of much of the hetaryne chemistry reported since.
 | ||
| Fig. 4 Early ortho-benzyne studies. | ||
Hetaryne experimental results
Throughout the years, many heterocyclic arynes have been proposed as intermediates. As discussed above, it is likely that some of the reported reactions do not actually involve the reported aryne as an intermediate.1 Additionally, aryne reactions frequently give low yields of products, likely due to a combination of the high instability of these intermediates, as well as the harsh conditions that have been traditionally used to generate these species. As a result, the following sections focus on the aryne derivatives of pyridines and indoles, two of the most well-studied classes of hetarynes. Both of these families of hetarynes have undergone an evolution from their initial discovery to their current uses as versatile intermediates for organic synthesis. Emphasis is placed on developments that have sought to address key challenges related to the utility of these species. This article also highlights the application of hetarynes in the total synthesis of natural products, including our own laboratory's recent syntheses of tubingensin A, the [4.3.1]-bicyclic welwitindolinones, and several indolactam alkaloids.Pyridyne methodology
Considering all classes of heterocyclic arynes, pyridynes have been the most intensely studied over the past 60 years. Of the three pyridyne isomers shown in Fig. 5, 3,4-pyridyne (4) is suggested computationally8 to be the most stable, followed by 2,3-pyridyne (22). Attempts to study 1,2-pyridyne (23, alternatively represented as the 2-pyridyl cation) in solution have been unsuccessful,9 however, studies in the gas phase have shown evidence of this species.10 All of these isomers have long been recognized as promising intermediates for the preparation of substituted pyridines and, collectively, have been the target of much synthetic effort.11,12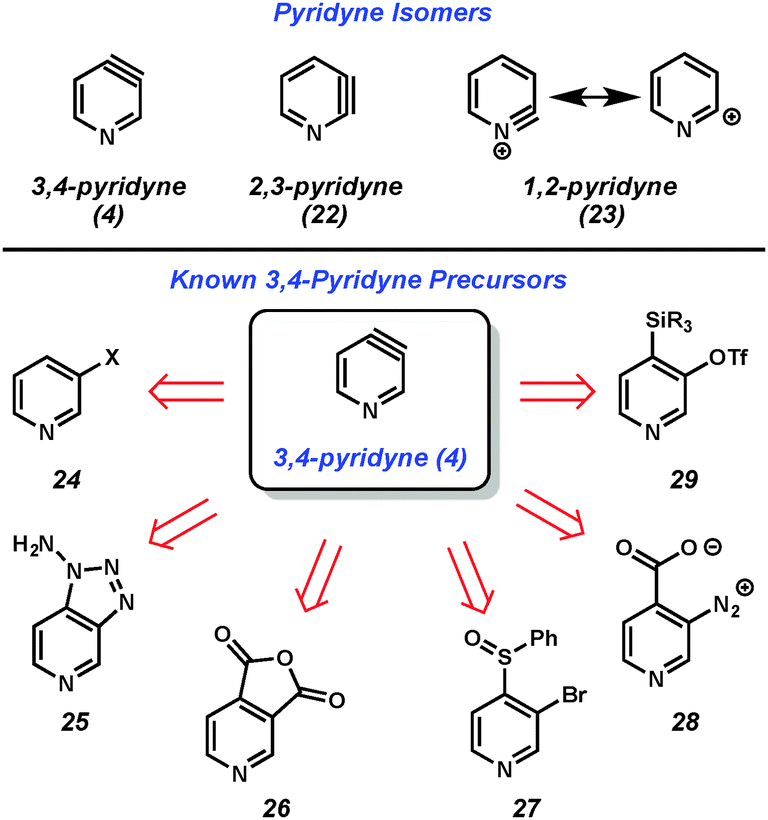 | ||
| Fig. 5 Pyridyne isomers and methods of generation. | ||
The 3,4-pyridyne can be used to illustrate the wide range of methods typically used to generate arynes. Most of the methods are derived from analogous precursors to o-benzyne, illustrating that similar strategies could be applicable for the generation of other hetarynes. For example, 3,4-pyridyne (4) is accessible from 3-halopyridines upon treatment with strong base (24 → 4),13 oxidation of aminotriazoles with Pb(OAc)4 (25 → 4),14 photolysis of anhydrides (26 → 4),15 magnesium-sulfur exchange followed by elimination (27 → 4),16 thermolysis of diazonium carboxylates (28 → 4),17 or fluoride-induced elimination of ortho-trialkylsilyl pyridyl triflates (29 → 4).18 Of note, this latter method, which was first pioneered by Kobayashi for o-benzyne generation,19 has demonstrated the most functional group tolerance in terms of both the trapping agents that can be utilized and the substituents present on the precursors. Although there is always the possibility that some precursors may react through non-aryne mechanisms, the isolation of pyridyne 4 in an N2 matrix and subsequent characterization by IR spectroscopy20 demonstrated that pyridyne 4 is a valid intermediate.
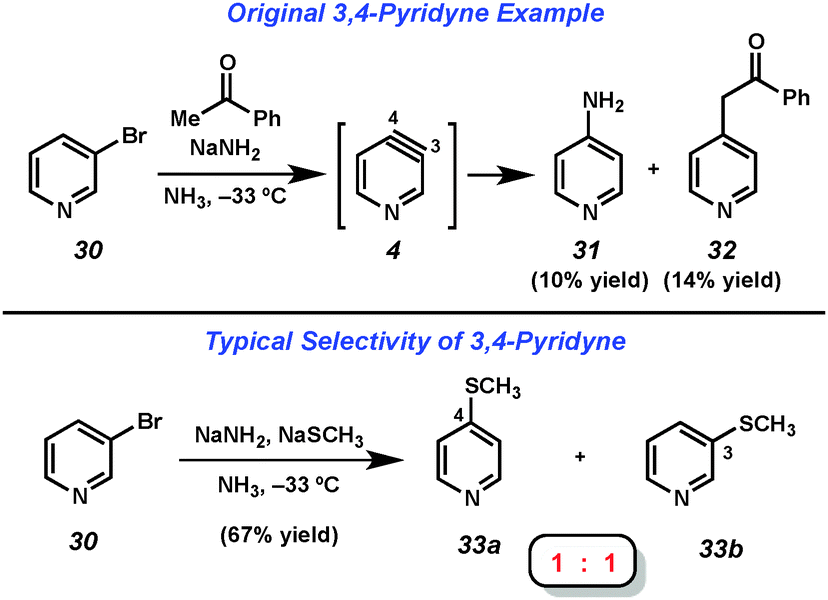
The 3,4-pyridyne has been demonstrated to be useful for the preparation of 3- and 4-substituted pyridines as well as 3,4-disubstituted derivatives.21 Leake and Levine reported the first generation of 3,4-pyridyne (4) utilizing 3-bromopyridine (30),22 which upon treatment with acetophenone and sodium amide in liquid ammonia gave low yields of 4-aminopyridine (31) and adduct 32, the latter of which arises from attack by the enolate of acetophenone (Fig. 6). Many early studies that relied on dehydrohalogenation for pyridyne generation were limited in the scope of the trapping agent employed. In order to minimize the byproducts resulting from attack by the base used for pyridyne generation, strongly nucleophilic groups such as alkoxides, amide salts, and thiolates were often employed. In an example of the latter reported by Zoltewicz, treatment of bromopyridine 30 with sodium methanethiolate and sodium amide led to an equimolar mixture of adducts 33a and 33b.23 This lack of regioselectivity is characteristic of pyridyne 4, and has limited the synthetic utility of this intermediate.
 | ||
| Fig. 6 Examples of 3,4-pyridyne studies. | ||
Several studies have focused on the development of methods to control regioselectivity in reactions of 3,4-pyridynes,18,24 and include a report by our laboratory in 2013, which systematically explored the use of substituents to control selectivity via aryne distortion.25 We prepared three pyridyne precursors, 34, 36, and 38, and then performed a series of pyridyne generation and trapping experiments (Fig. 7). For example, using 3,4-pyridyne precursor 34 in a nitrone cycloaddition yielded products 35a and 35b in a 1.9 to 1 ratio. However, by employing the sulfamoylated precursor 36 in the corresponding reaction, higher regioselectivity could be achieved favoring attack at C4. In a complementary sense, selectivity could be reversed by use of the brominated precursor 38 in the aryne trapping experiment to now favor attack at C3. Of note, after performing aryne trapping experiments of silyltriflates 36 and 38, the sulfamate and bromides remaining may be used in Ni- or Pd-catalyzed cross-coupling experiments.26,27 For example, after performing pyridyne trapping experiments with 1,3-dimethyl-2-imidazolidinone,28 similar to trapping experiments recently report by Sato and coworkers,2940 and 41 were obtained using catalytic C–C and C–N bond forming processes. Pyridyne precursors 34 and 38 are now available commercially.30
 | ||
| Fig. 7 Our laboratory's 3,4-pyridyne studies. | ||
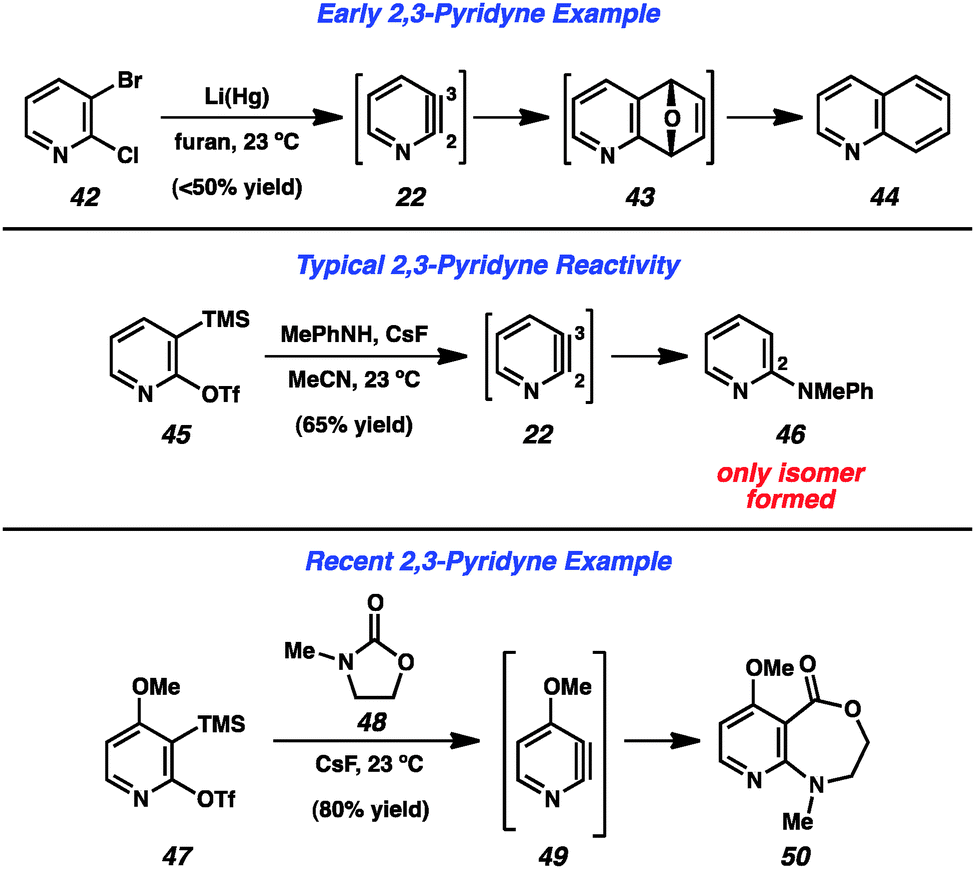
Early attempts to generate the 2,3-pyridyne (22) were more ambiguous than those targeting the 3,4-pyridyne (Fig. 8). Dehydrohalogenation cannot be easily utilized, as 3-halopyridine precursors (e.g., 30, Fig. 6) have been shown to lead to formation of the 3,4-pyridyne (4) selectively,13 and the use of 2-halopyridines makes it difficult to determine whether products arise through an aryne pathway or through an SNAr pathway. However, in 1962, Martens and den Hertog demonstrated that treatment of dihalide 42 with lithium amalgam in the presence of furan led to the isolation of quinoline (44) in moderate yield.31 The formation of this product was rationalized by a pathway involving 2,3-pyridyne (22) formation, cycloaddition to give 43, followed by reduction. The development of silyltriflate precursor 45, which is now commercially available,30 has allowed for a reliable means of generating the 2,3-pyridyne under mild conditions32 and can be used to demonstrate the high regioselectivity that is typically observed in these reactions. Treatment of precursor 45 with CsF leads to aryne 22, which undergoes attack by N-Me-aniline to exclusively give the 2-amino product 46.33 The high selectivity typically obviates the need to separate isomeric products, and can be used to form 2,3-disubstituted pyridines. Additionally, 50 was obtained through a formal C–N bond insertion reaction between pyridyne 49 into carbamate 48.29
 | ||
| Fig. 8 Examples of 2,3-pyridyne studies. | ||
Indolyne methodology
Given the importance of the indole heterocycle, attempts to utilize the corresponding indolyne to prepare substituted indole derivatives has received significant attention. Of the four possible indolyne isomers, three contain the reactive triple bond within the benzenoid ring (3, 51, and 52, Fig. 9), whereas the 2,3-indolyne (53) contains the triple bond in the pyrrole ring. The first report concerning indolynes came from Julia and coworkers, who in 1967 showed that treatment of 5-bromoindole (54) with potassium amide led to a mixture of 4- and 5-aminoindole products (56a and 56b), likely arising through indolyne intermediate 55.34 Despite the promise demonstrated by this early work,35 indolynes saw little attention over the subsequent 40 years.36 In a series of reports beginning in 2007, Buszek and co-workers reported elegant studies of indolyne Diels–Alder reactions.37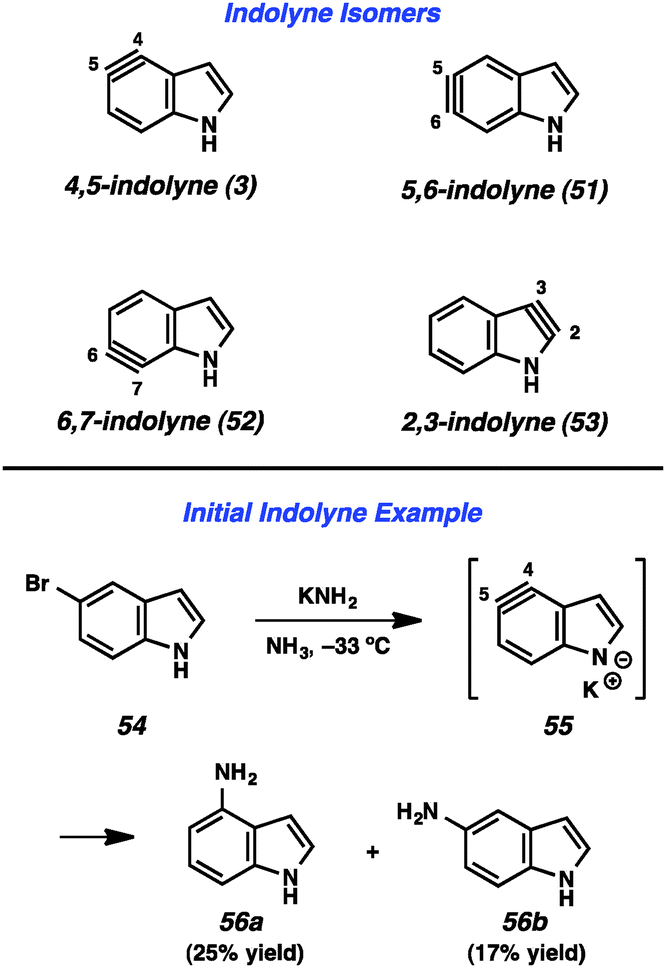 | ||
| Fig. 9 Indolyne isomers and seminal indolyne example. | ||
Our laboratory has focused on mild methods for indolyne generation and trapping, with the hope that indolynes could be better exploited by synthetic chemists. We have shown that 4,5-, 5,6-, and 6,7-indolynes 57–59 can be generated from silyltriflate precursors 60–62, respectively (Fig. 10).38 These precursors were accessible as N-Me, N-Boc, or N-H variants for each indolyne isomer and also left C3 available for later functionalization.38c Indolyne formation could be achieved upon treatment of the indolyne precursors with fluoride sources such as TBAF or CsF. Buszek has also reported an alternative strategy for the preparation of similar silyltriflate precursors that contain a phenyl substituent at C3.37b
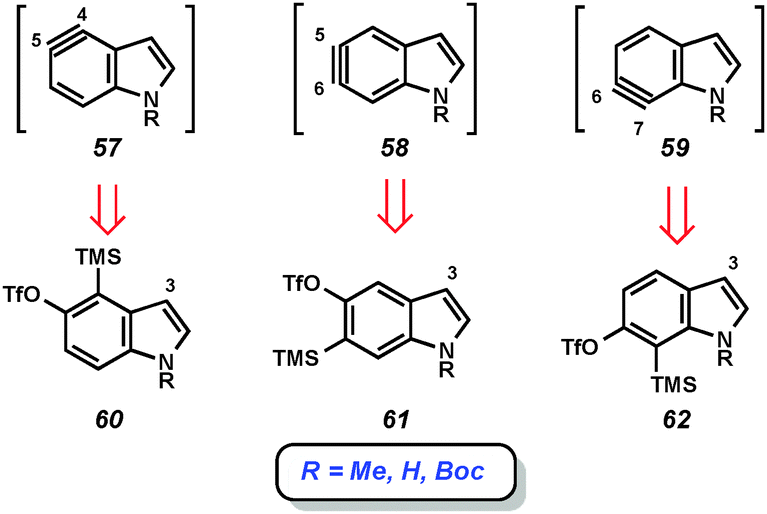 | ||
| Fig. 10 Silyltriflates as indolyne precursors. | ||
The mild fluoride-based conditions for indolyne generation allowed for the scope of trapping agents to be significantly expanded compared to prior indolyne studies. As seen in Fig. 11, treatment of 4,5-indolyne precursor 64 with CsF in the presence of aniline led to the formation of C5 adduct 63 preferentially over the formation of the corresponding C4-substituted product in a ratio of 12.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Trapping of the same indolyne with benzyl azide demonstrated the compatibility of cycloaddition reactions and provided triazole product 65 and its regioisomer in a 2.4:1 ratio. Further demonstrations of the scope of indolyne reactions can be seen in products 66–69. The regioselectivity observed in the formation of products 63, 65, 68, and 69 was explained using the Aryne Distortion Model, pioneered by Houk and coworkers.38b,c,39 Generally speaking, both the 4,5- and 5,6-indolynes preferentially give products resulting from initial attack at C5, while the 6,7-indolyne typically gives products resulting from attack at C6.
:1. Trapping of the same indolyne with benzyl azide demonstrated the compatibility of cycloaddition reactions and provided triazole product 65 and its regioisomer in a 2.4:1 ratio. Further demonstrations of the scope of indolyne reactions can be seen in products 66–69. The regioselectivity observed in the formation of products 63, 65, 68, and 69 was explained using the Aryne Distortion Model, pioneered by Houk and coworkers.38b,c,39 Generally speaking, both the 4,5- and 5,6-indolynes preferentially give products resulting from initial attack at C5, while the 6,7-indolyne typically gives products resulting from attack at C6.
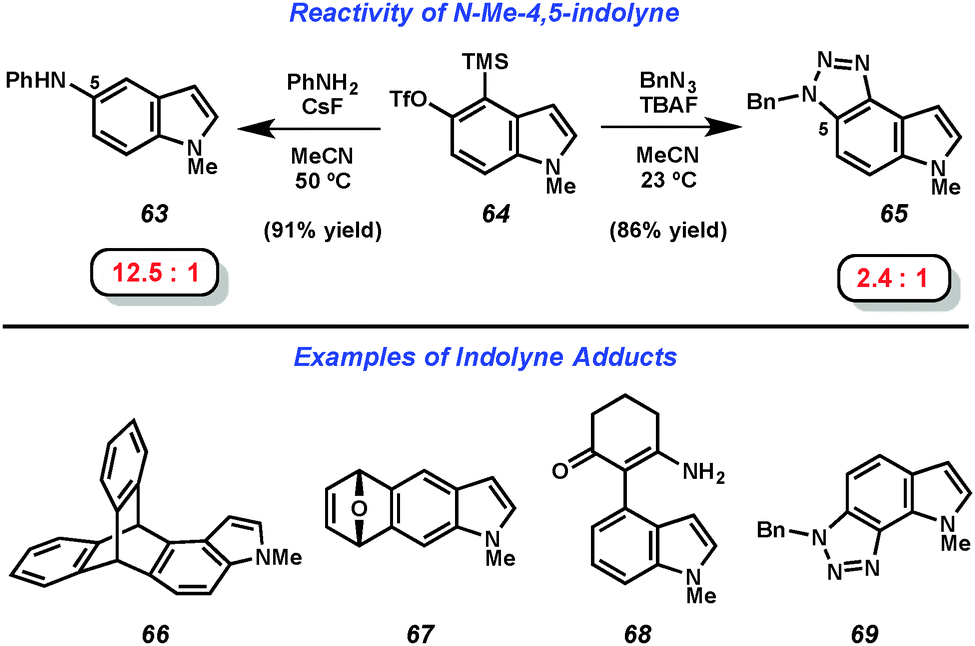 | ||
| Fig. 11 Sample reactivity of indolyne intermediates. | ||
Recent reports of indolynes and related species have further demonstrated the utility of these hetarynes for the assembly of complex indole derivatives. Lautens and coworkers reported that treatment of bromoindole 70 with LDA led to the generation of 6,7-indolyne 71 (Fig. 12). This species was found to undergo an intramolecular ene reaction involving the homoprenyl group on the indole nitrogen to afford tricyclic product 72.40 Interestingly, the observed regioselectivity is due to the fact that the homoprenyl group is tethered to the indole nitrogen.
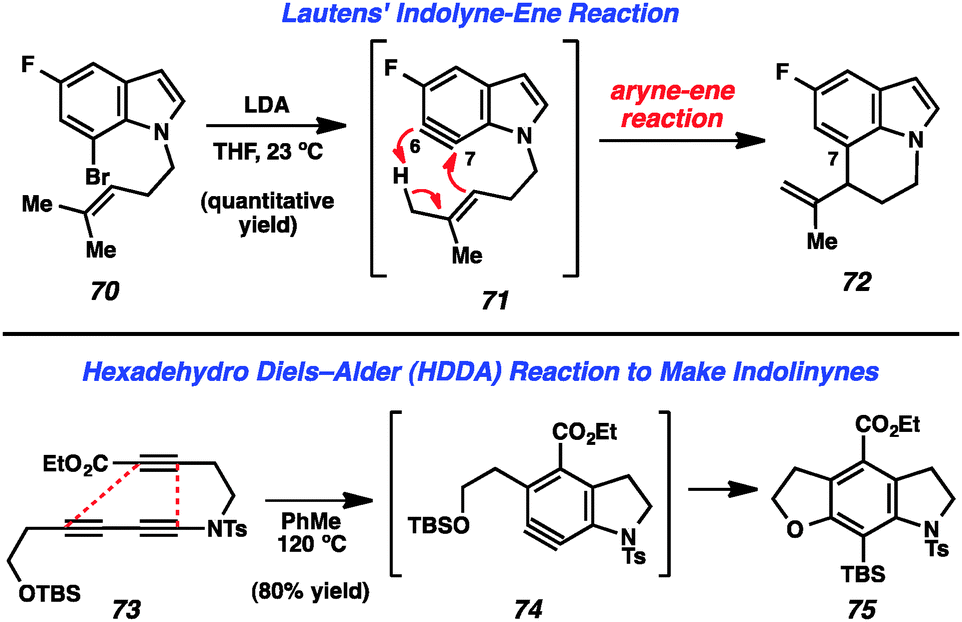 | ||
| Fig. 12 Recent examples of indolyne reactivity and indolinone generation. | ||
The Hoye group has recently reported a novel method for the generation of arynes in which a molecule containing three alkynes undergoes a “Hexadehydro Diels–Alder (HDDA) Reaction” to provide the reactive intermediate.41 The authors demonstrated that molecules such as tri-yne 73 could be used to form indolinyne 74, which further reacted to furnish product 75 (Fig. 12).41a Of note, this reaction can be used to prepare indoline systems in which the benzenoid ring is fully substituted. Almost simultaneously, Lee and coworkers have reported a nearly identical strategy for the generation of indolinynes and explored a variety of reactions of these species.42
In contrast to the benzenoid indolyne isomers discussed above, the 2,3-indolyne (53, Fig. 13) likely experiences extreme strain due to the presence of the triple bond within the five-membered ring. Gribble has reported an extensive study of unsuccessful attempts to generate 5343 and, while there are claims of this intermediate being involved in reactions,44 unambiguous evidence has yet to be offered. In fact, there have been many attempts over the years to generate various 5-membered hetarynes, however, most claims are lacking in conclusive proof.1b In addition to the 2,3-indolyne (53), attempts to generate the 3,4-pyrrolyne (76) have been made, resulting only in <5% yield of any products suggestive of an aryne mechanism.45 The strongest case for formation of any of the 5-membered hetarynes can be made for thiophynes 77–79, especially the 3,4-thiophyne (78).46 It was suggested that the longer C–S bond would help relieve some of the strain present in these molecules compared to other 5-membered hetarynes. Unfortunately, isolated yields of cycloadducts suggestive of 78 are ≤30%. Thus, although these species may be accessible, their synthetic utility is currently limited.
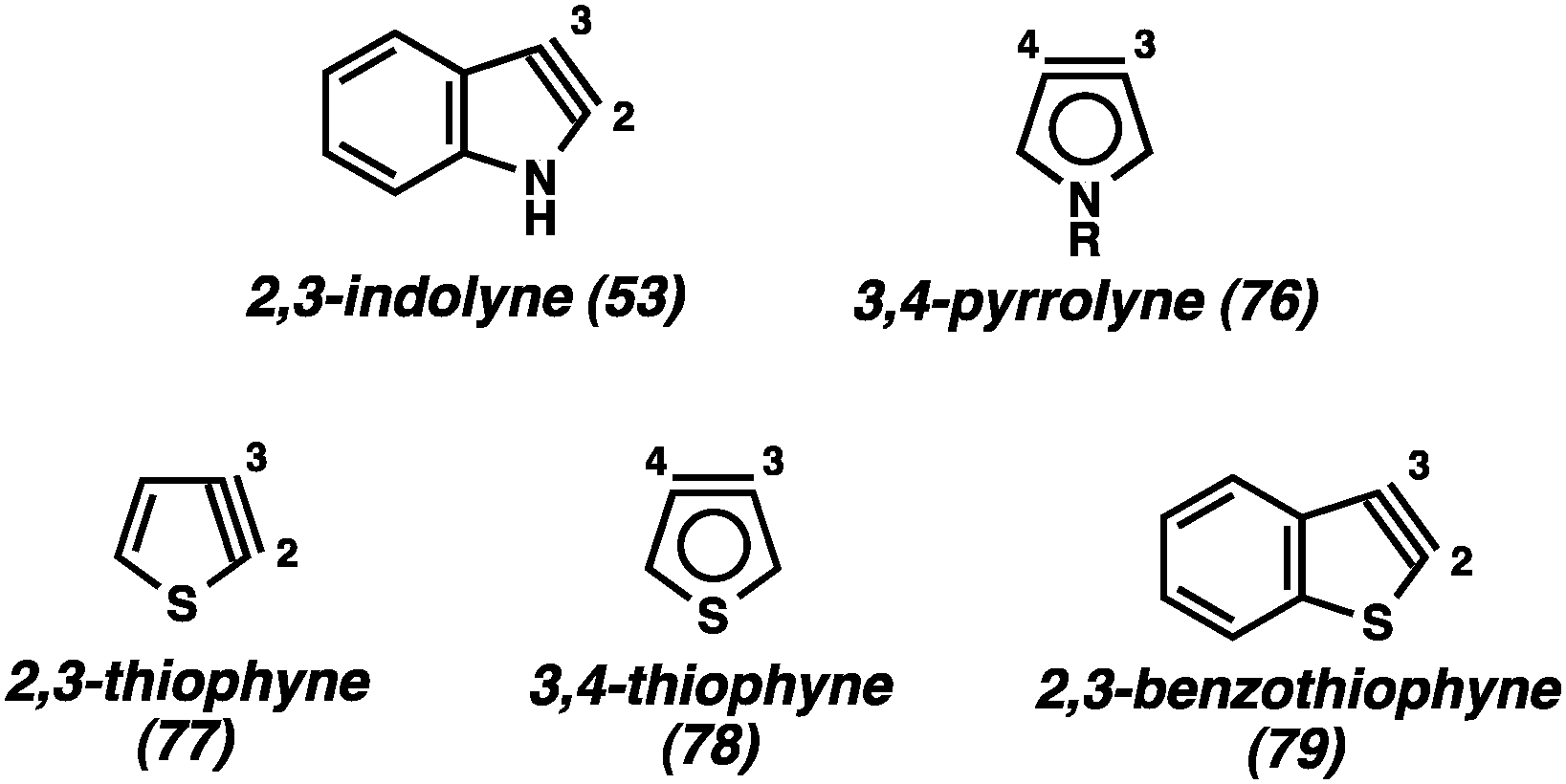 | ||
| Fig. 13 2,3-Indolyne and other 5-membered hetarynes. | ||
Use of hetarynes in total synthesis
Despite early limitations, hetarynes are now useful tools for the total synthesis of complex molecules. Many of the targets that have been accessed using hetaryne chemistry are shown in Fig. 14. Indolynes (or related species) have found use in the synthesis of C4-substituted indole derivatives,47 including lysergic acid (80),35c various makaluvamines (81),36 and members of the welwitindolinone (82)48 and indolactam (83)49 alkaloid families. All of these syntheses utilize the electrophilic nature of the indolyne to functionalize C4 of the indole ring, a position that is often difficult to access. Additionally, the indole diterpenoid, tubingensin A (84) was prepared by our laboratory using a related carbazolyne intermediate.50 Finally, indolynes have been utilized as cycloaddition partners for the synthesis of cis-trikentrin A (85) and herbindole B (86).37c,e Pyridynes have found use mainly as cycloaddition partners for the synthesis of molecules such as ellipticine (87)24b,d,51 and macrostomine (88),52 but also for intramolecular arylation reactions to build perlolidine (89)53 and eupolauramine (90).54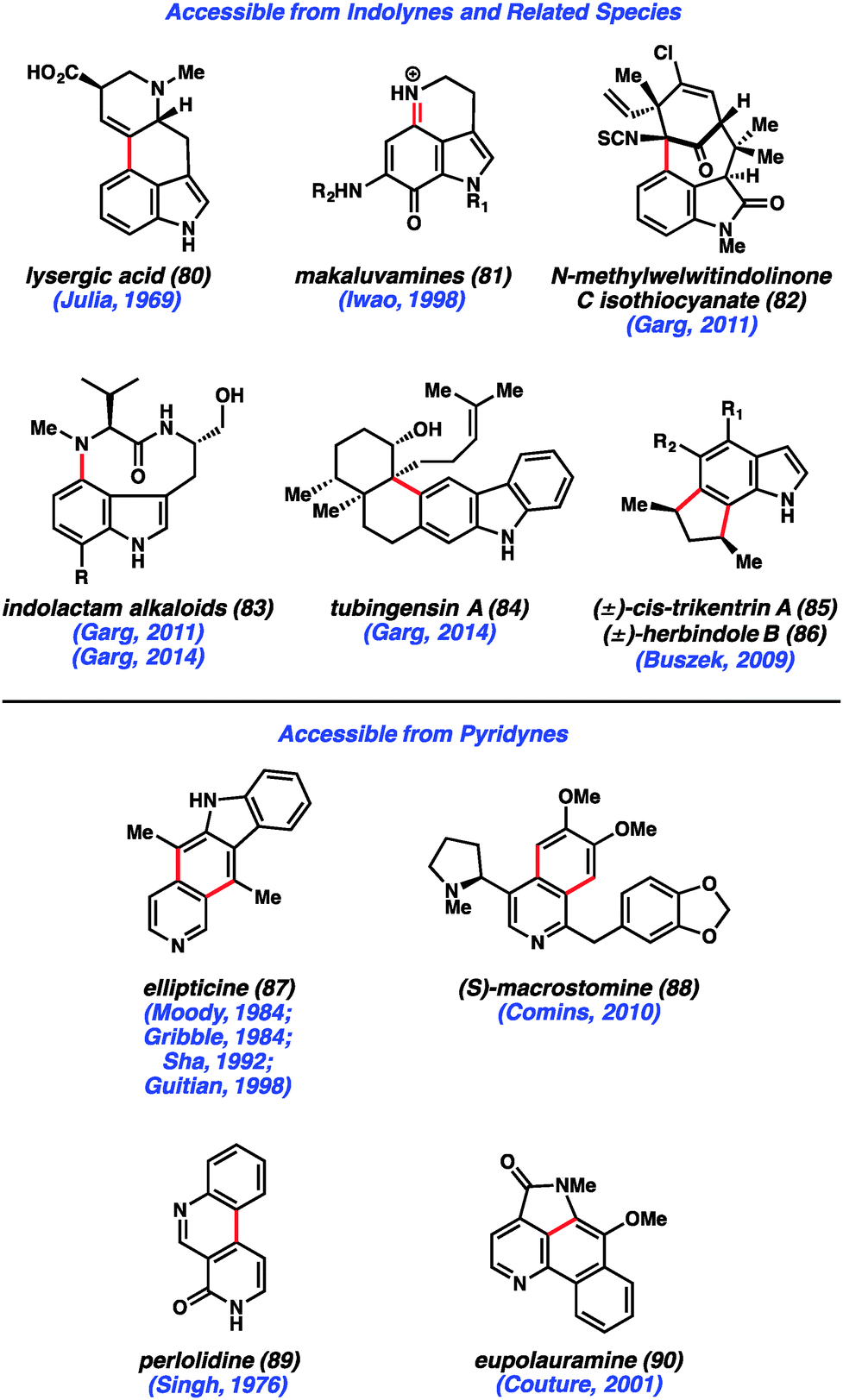 | ||
| Fig. 14 Natural products synthesized using heterocyclic arynes. | ||
Pyridynes have a rich history in total synthesis dating back to the seminal report by Singh and coworkers in 1976 on the synthesis of the grass alkaloid perlolidine (89, Scheme 1).53 Starting from readily available 2-amino-3-methylpyridine (91), the authors were able to quickly access compound 92. Exposure of this compound to standard aryne-forming conditions resulted in formation of the biaryl linkage to provide 94 in good yield. It is proposed that the aniline ring in compound 92 undergoes a Friedel–Crafts type intramolecular arylation reaction with the highly reactive tethered pyridyne (see transition structure 93). Compound 94 was successfully elaborated to perlolidine (89) by oxidation, followed by benzyl ether cleavage.
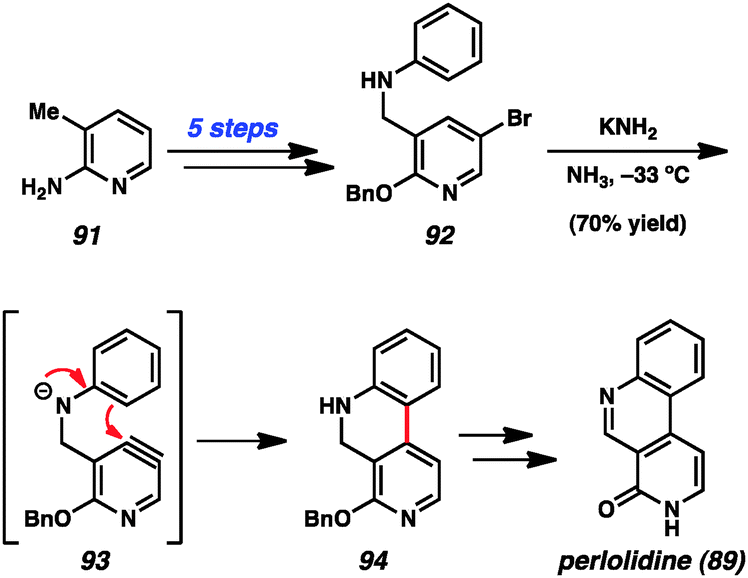 | ||
| Scheme 1 Singh's synthesis of perlolidine. | ||
Another prominent example utilizing pyridynes in total synthesis comes from studies of ellipticine (87), which has been targeted by multiple groups.24b,d,51 In an exciting approach, Guitián examined the effect of employing substituted 3,4-pyridynes as the dienophile with a variety of dienes (Scheme 2).24b,d Although other substituted pyridynes showed variable levels of selectivity depending on the diene present, when 2-chloro-3,4-pyridyne precursor 95 was reacted with CsF and decorated furan 96, the desired regioisomer 97 was formed as the major product.55 Compound 97 was then elaborated to ellipticine (87).
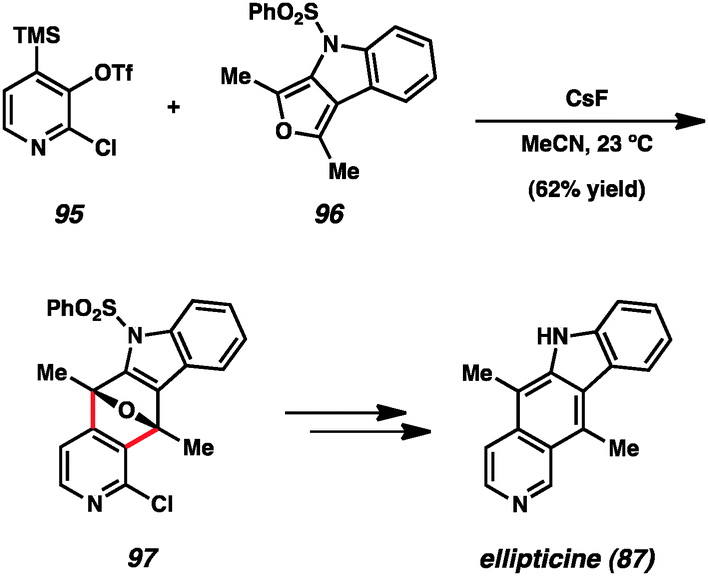 | ||
| Scheme 2 Guitián's synthesis of ellipticine. | ||
Likewise, indolynes and related intermediates have been judiciously exploited in total synthesis. A seminal example was reported by Julia and coworkers in their formal synthesis of lysergic acid (80) in 1969 (Scheme 3).35c The authors found that treatment of 5-bromoindoline 98 with NaNH2 led to cyclized product 100 in a modest 15% yield. Tetracycle 100 presumably arises from attack of the vinylogous enolate onto the C4 position of the indolinyne (see transition structure 99), where the regiochemistry is controlled by geometrical constraints. Of note, the authors reported that the use of C3-epi-108 did not provide any of the corresponding product when reacted under the same conditions. Tetracycle 110 could be elaborated to the natural product following the procedure reported by Woodward and coworkers.56 Despite the low yield in the key aryne step, this synthesis validated the notion that indolyne-type intermediates could be used to build complex heterocyclic compounds.
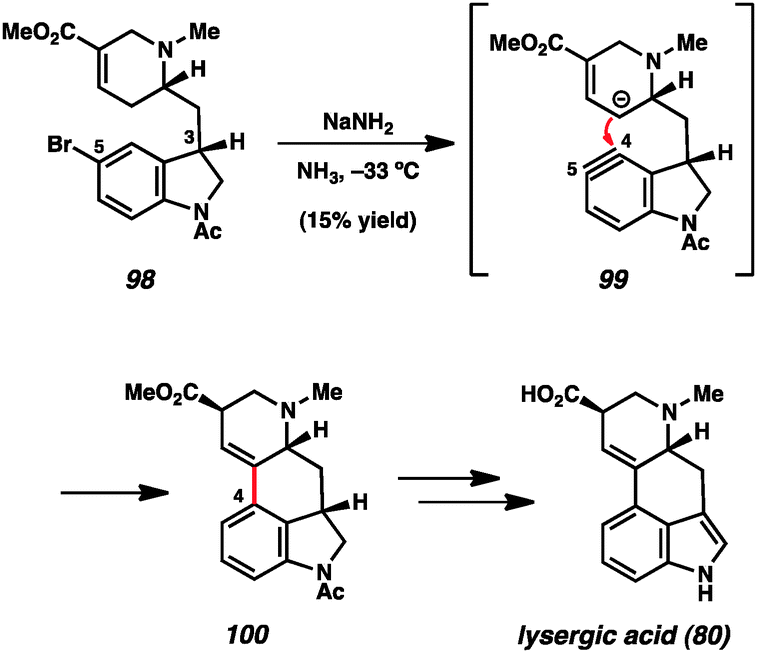 | ||
| Scheme 3 Julia's formal synthesis of lysergic acid. | ||
As mentioned earlier, our laboratory has been interested in the development of indolyne methodology, and we have recently employed these intermediates in several total syntheses. One class of compounds that appeared ideally suited for a test of indolyne methodology was the indolactam alkaloids (e.g., Fig. 15, 101–104).57 These compounds have been widely studied for decades because of their interesting structures and pharmacological properties. Although these compounds are most well known for their use in studies aimed at better understanding tumor growth, indolactam V (101) is also known to function as a stem cell differentiator.58 Structurally, these alkaloids are characterized by several unique features including a conformationally-flexible 9-membered lactam and a 3,4-disubstituted indole core. Additionally, compounds 102–104 possess sp2–sp3 linkages at C7, which present a significant challenge for synthesis.
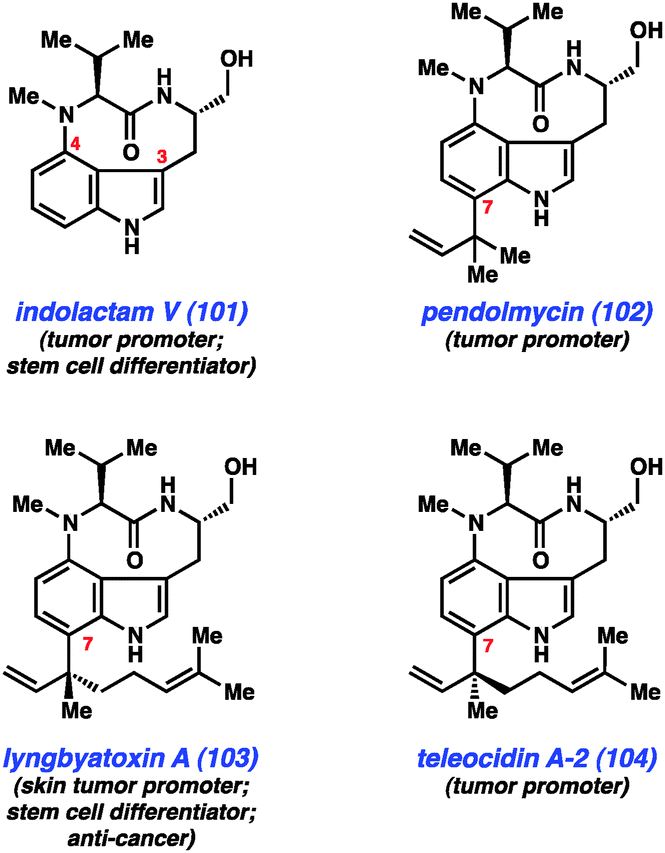 | ||
| Fig. 15 Indolactam alkaloids. | ||
With the goal of accessing the four natural products 101–104 through common late-stage intermediates, we first pursued an efficient means to assemble the common core of these targets (Scheme 4).49 Beginning from commercially available 5-benzyloxyindole (105), we were able to access multi-gram quantities of bromosilyltriflate 106. Of note, this efficient conversion proceeded in 7 steps with an overall yield of 62%. With silyltriflate 106 in hand, we were poised to build the C4–N linkage using indolyne chemistry. We found that exposure of silyltriflate 106 to CsF and peptide 107 furnished adduct 109 in 75% yield, presumably by way of 4,5-indolyne intermediate 108. Although 4,5-indolynes typically react to give products of C5 substitution, the presence of the C6 bromide in 108 overturns the usual preference by perturbing the distortion of the aryne.49a Indolyne adduct 109 could be elaborated to the natural product 101 in several steps including reductive removal of the aryl bromide and ring closure at C3.
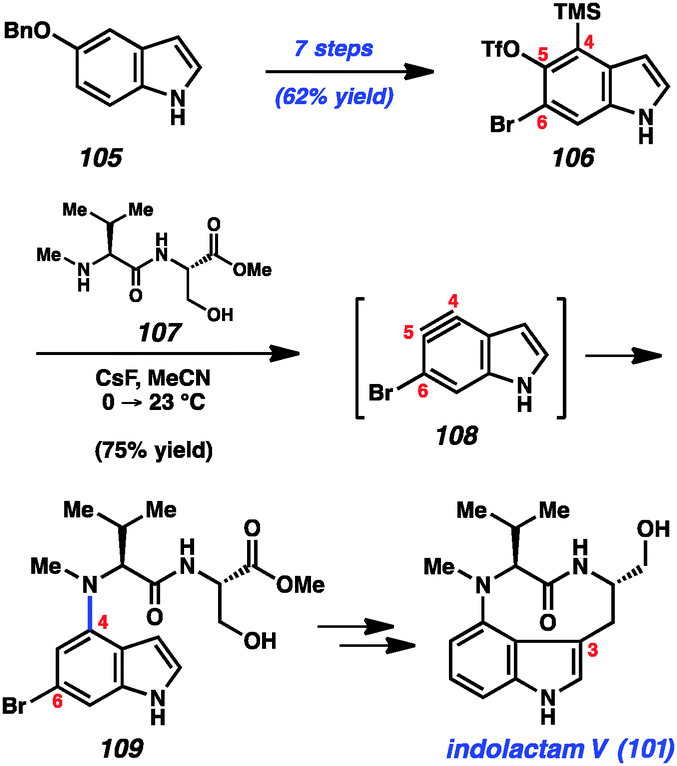 | ||
| Scheme 4 Synthesis of indolactam V. | ||
Having synthesized indolactam V (101) using indolyne methodology, we used 101 as a stepping-stone toward indolactam alkaloids 102–104 (Fig. 16).49b To access pendolymycin (102), 101 was elaborated to bromide 110 using a simple two-step sequence. We then surveyed a variety of reaction conditions to assemble the critical sp2–sp3 linkage at C7. We were delighted to find that bromide 110 underwent coupling with zinc enolate 111 to furnish 112 in 61% yield using Hartwig's enolate coupling methodology.59 It is notable that this complexity-generating reaction proceeds smoothly, despite the presence of a tertiary amine and the free NHs present in 110. From 112, a three-step sequence involving amide reduction with Schwartz's reagent, olefination of the resulting aldehyde, and deprotection of the primary alcohol furnished pendolmycin (112). An analogous approach was used to prepare lyngbyatoxin A (113), in addition to its congener teleocidin A-2 (114) as suggested in Fig. 16.
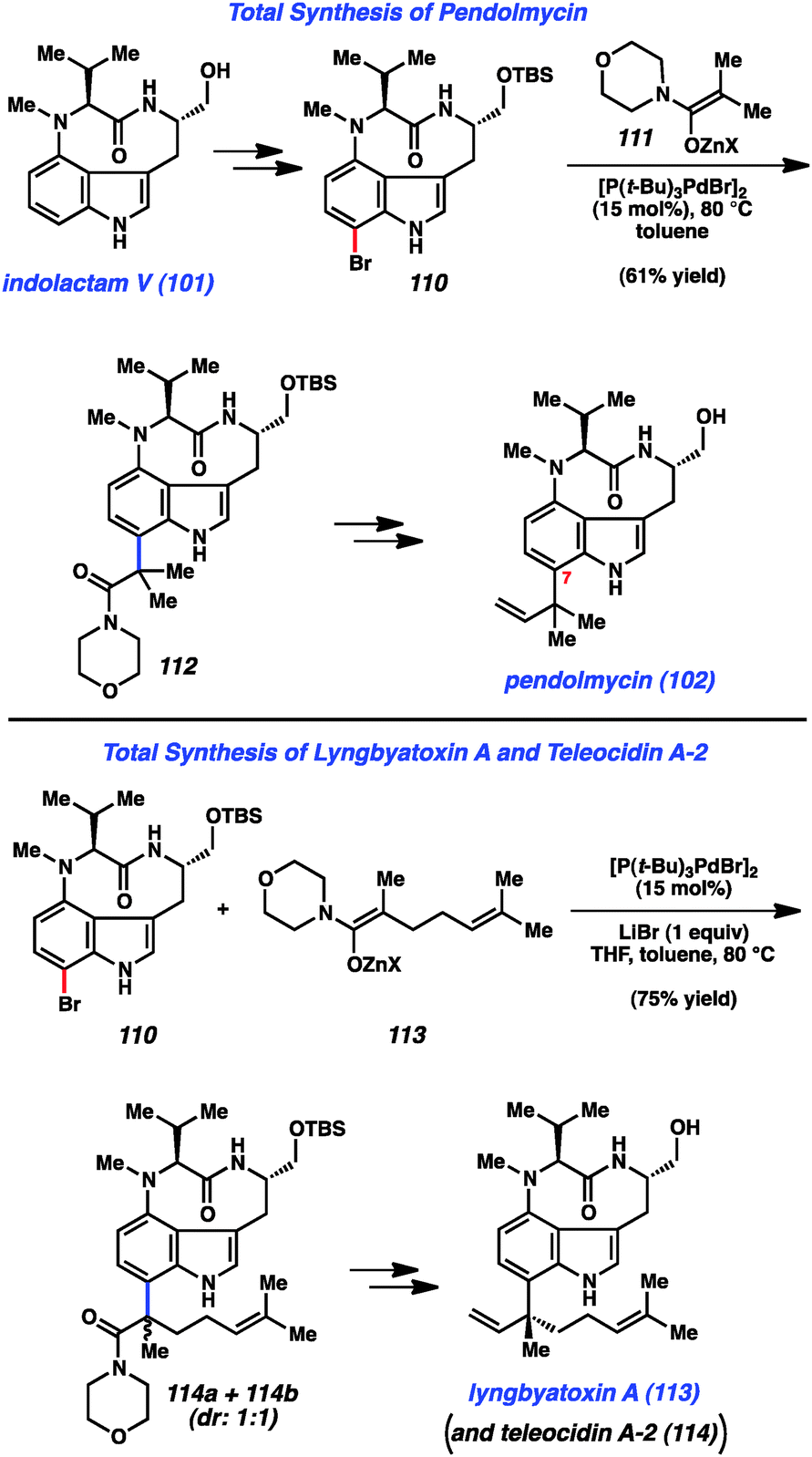 | ||
| Fig. 16 Synthesis of C7 substituted indolactam alkaloids. | ||
In a more recent example, our laboratory has reported what is arguably one of the most complex aryne cyclizations to date while pursuing the welwitindolinone alkaloids that bear a bicyclic [4.3.1] core (e.g., 82 and 115–119, Fig. 17). The isolation of these compounds was reported by Moore and co-workers through two publications in 1994 and 1999.60 Although the structures of these compounds are relatively compact, these natural products bear highly functionalized cores that pose tremendous challenges to synthetic chemists. Over the past two decades, at least fifteen laboratories have reported approaches toward these captivating natural products61 and several recent total syntheses have been reported.48,62,63
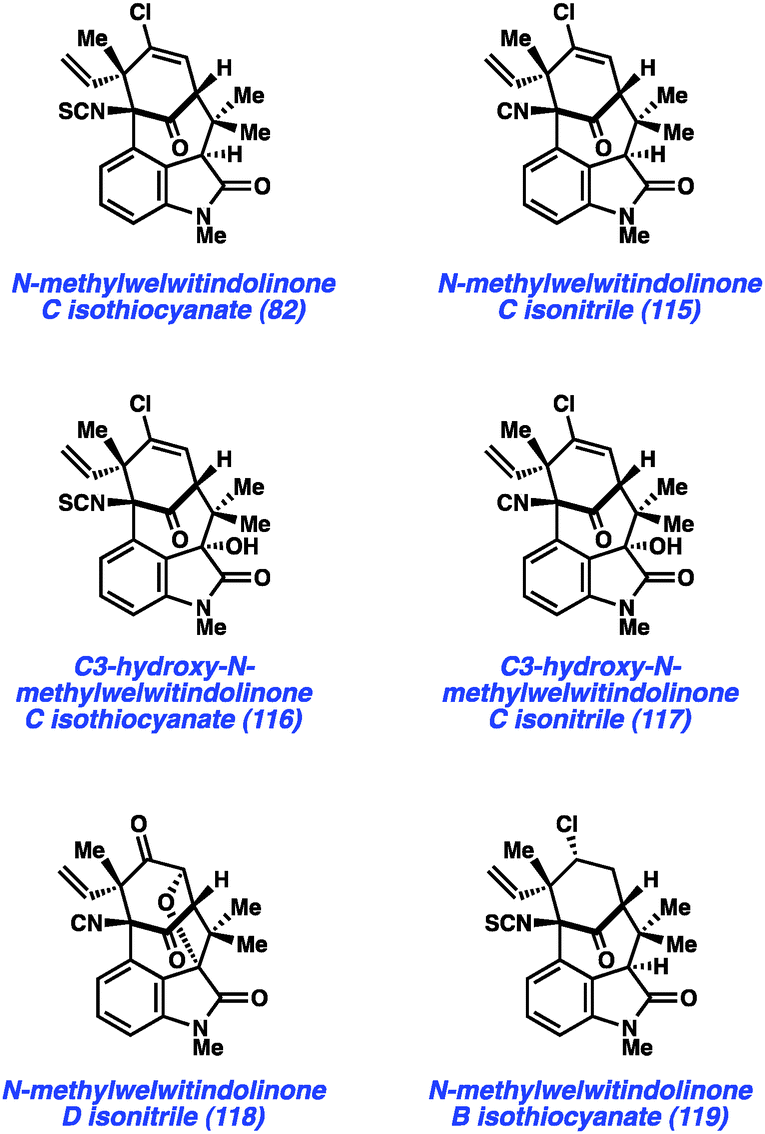 | ||
| Fig. 17 [4.3.1]-Bicyclic welwitindolinone natural products. | ||
As shown in Scheme 5, our synthesis began from known enone 120, which is prepared using a modification of Natsume's five-step procedure reported in the enantiomeric series.64 Cleavage of the pivalate ester, followed by iodine-catalyzed conjugate addition of 5-bromo-N-methylindole provided indole 121 in 54% yield over two steps. Silyl protection of the alcohol then furnished TBS ether 122, the key substrate for constructing the complex [4.3.1]-bicyclic core. Exposure of bromoindole 122 to the complex base conditions first developed by Caubere65 led to generation of intermediate 123, which contains both an enolate and 4,5-indolyne. Cyclization led to the desired C-arylated product 124 in 33% yield, as well as the corresponding O-arylated product in 13% yield. Product 124 possesses the necessary tetracyclic core, including the congested linkage at C4 of the indole ring. This intermediate was successfully converted to the elusive natural product N-methylwelwitindolinone C isothiocyanate (82).48 The indolyne cyclization was subsequently utilized for the synthesis of a number of related family members including isonitrile derivative 115 and the C3-hydroxy derivatives 116 and 117.48b Additionally, indolyne cyclization product 114 has been converted to another family member, N-methylwelwitindolinone D isonitrile (118),48d which contains an additional ethereal linkage.
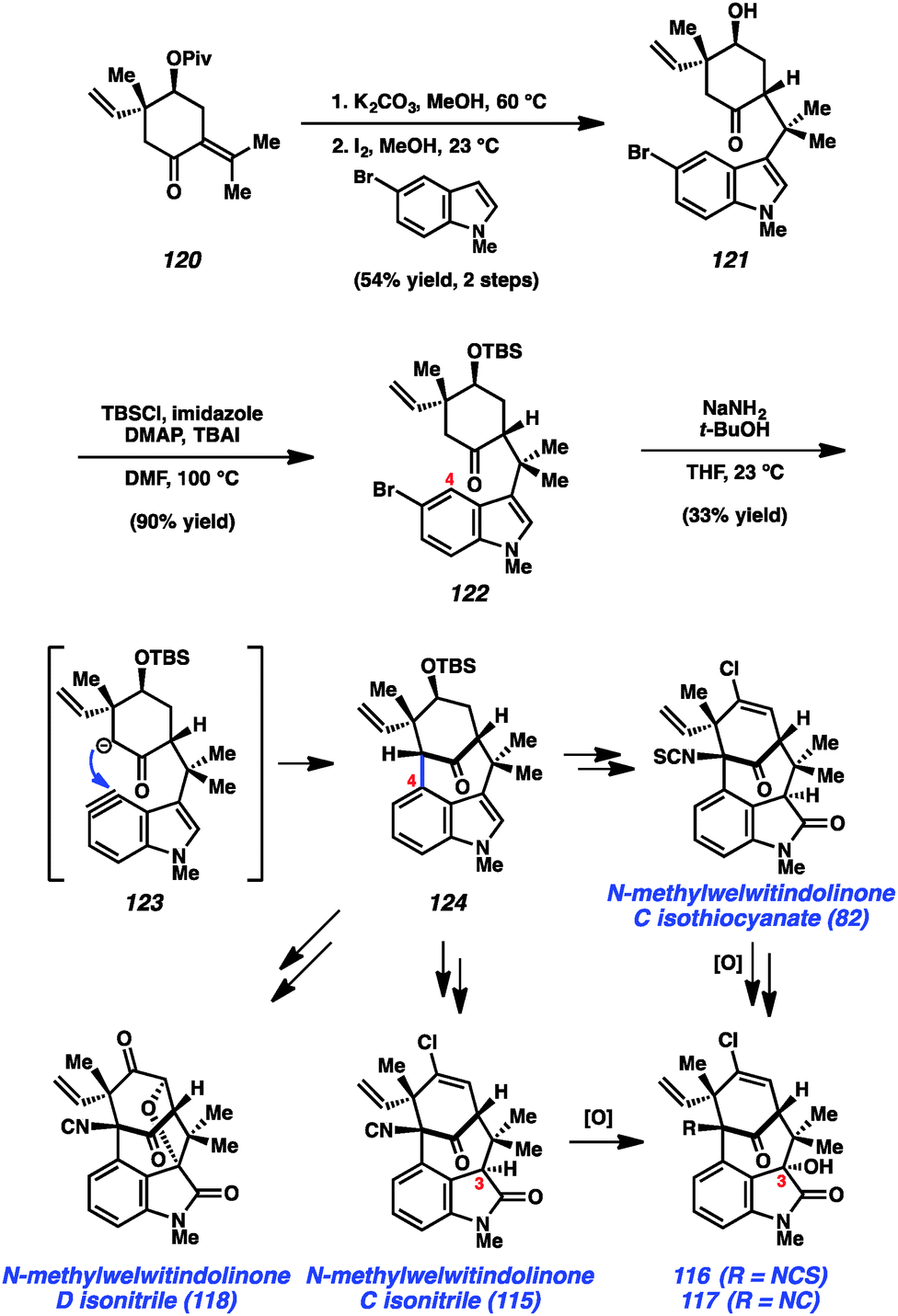 | ||
| Scheme 5 Total synthesis of welwitindolinones. | ||
Most recently, we have further tested the viability of using heterocyclic arynes to construct complex scaffolds in our pursuit of the tubingensin natural products (84 and 125, Fig. 18). Both tubingensins A and B were isolated in 1989 by Gloer and co-workers from the fungus Aspergillus tubingensis.66,67 In addition to possessing interesting biological activities (e.g., antiviral, anticancer properties), these compounds have unique and challenging structures. For example, tubingensin A (84) contains a densely functionalized cis-decalin scaffold, appended to a disubstituted carbazole unit. The decalin contains four stereogenic centers, all of which are contiguous and two of which are vicinal and quaternary. Although arynes have not previously been used to assemble vicinal quaternary stereocenters, we envisioned the use of a ‘carbazolyne’ intermediate to achieve this goal and ultimately establish the tubingensin A skeleton.50,68
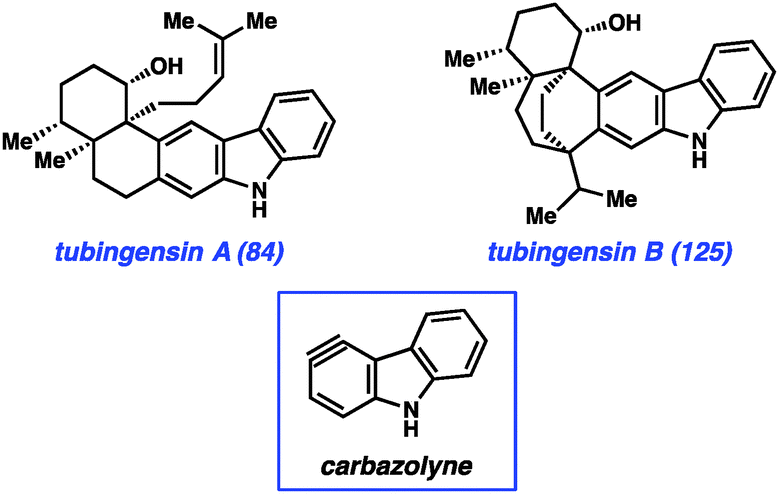 | ||
| Fig. 18 Tubingensins A and B. | ||
Our concise total synthesis of tubingensin A (84) is summarized in Scheme 6. Beginning from silyl enol ether 126, which was readily obtained from (+)-dihydrocarvone, we implemented a hydroboration/B-alkyl Suzuki–Miyaura coupling sequence. Thus, 126 was first treated with 9-BBN to generate the requisite alkyl boron intermediate. Subsequent coupling with bromotriflate 127 in the presence of catalytic Pd(PPh3)4 furnished product 128 in 64% yield. Of note, neither the bromide or silyl enol ether were disturbed in this transformation. With the C12–C13 linkage intact, 128 was elaborated to silyl enol ether 129 in three steps. In the critical carbazolyne cyclization (see transition structure 130), exposure of 129 to NaNH2/t-BuOH delivered the desired product 131 in 84% yield. The transformation proceeds with complete diastereoselectivity and, importantly, introduces the challenging vicinal quaternary stereocenters of the natural product. Finally, a deprotection/reduction sequence furnished the natural product 84. The total synthesis of tubingensin A (84) requires only 9 steps (longest linear sequence), beginning from known compounds, and highlights the efficiency by which heterocyclic arynes may be used to build stereocenter-rich architectures.
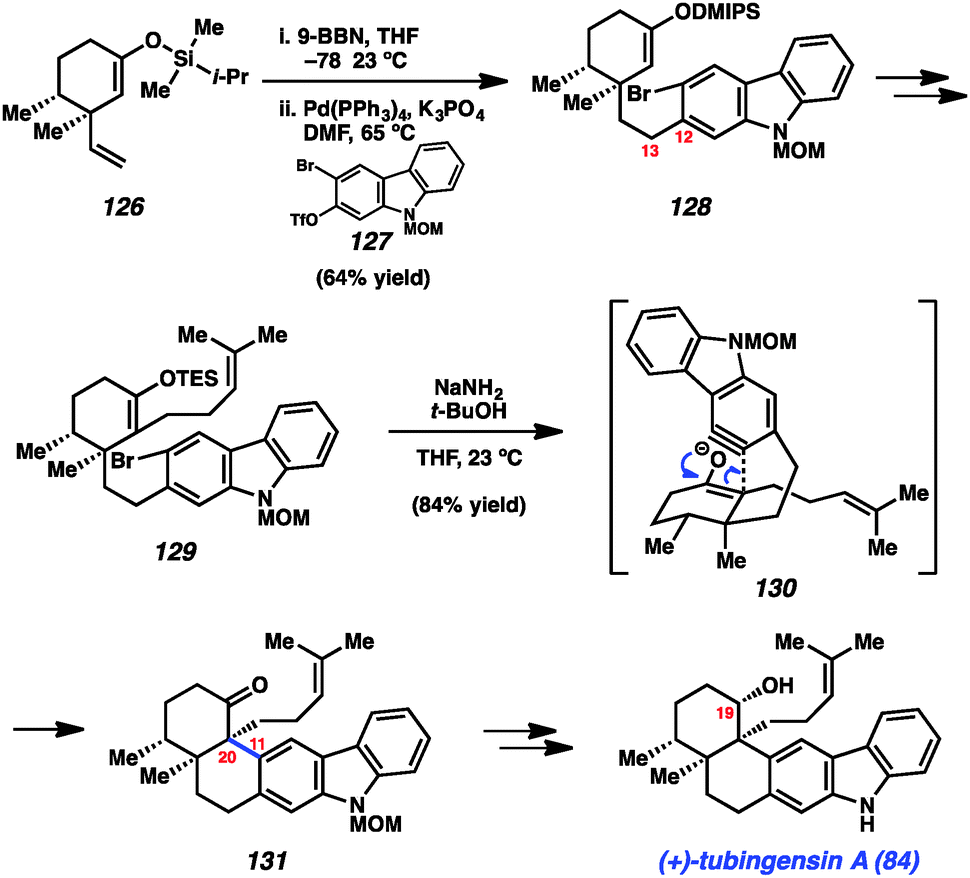 | ||
| Scheme 6 Synthesis of tubingensin A. | ||
Conclusions
Over the last 60 years, the chemistry of heterocyclic arynes has lagged behind that of their all-carbon counterparts such as ortho-benzyne. Although many of the early claims were questionable regarding the generation of these reactive intermediates, more recent studies have demonstrated that these species are not only accessible, but offer a unique alternative for the synthesis of substituted heterocycles. The increasing popularity of easy-to-handle precursors, such as o-trimethylsilyl triflates, allow for the generation of hetarynes under mild conditions that allow for a significantly expanded scope of trapping agents to be employed. In fact, some heterocyclic aryne silyltriflate precursors are now commercially available.30 Additionally, the high reactivity of hetarynes can be exploited for the construction of complex motifs that would otherwise be difficult to access. It is envisioned that the development of even more exotic heterocyclic arynes and trapping agents will be seen in the future, along with more challenging applications in the synthesis of drugs and natural products.Acknowledgements
The authors are grateful to Boehringer Ingelheim, Bristol–Myers Squibb, DuPont, Eli Lilly, Amgen, AstraZeneca, Roche, the A.P. Sloan Foundation, the S. T. Li Foundation, the Dreyfus Foundation, and the University of California, Los Angeles, for financial support. A.E.G. thanks the Foote Family, the ACS Division of Organic Chemistry, and the UCLA Graduate Division's DYF Program for fellowship support.Notes and references
- For reviews of hetarynes, see: (a) T. Kauffmann, Angew. Chem., Int. Ed. Engl., 1965, 4, 543–618 CrossRef; (b) M. G. Reinecke, Tetrahedron, 1982, 38, 427–498 CrossRef CAS; (c) S. M. Bronner, A. E. Goetz and N. K. Garg, Synlett, 2011, 2599–2604 CAS; (d) A. E. Goetz and N. K. Garg, J. Org. Chem., 2014, 79, 846–851 CrossRef CAS PubMed.
- For reviews of ortho-benzyne, see: (a) A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Org. Biomol. Chem., 2013, 11, 191–218 RSC; (b) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550–3577 CrossRef CAS PubMed; (c) C. M. Gampe and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3766–3778 CrossRef CAS PubMed; (d) A. Bhunia, S. R. Yetra and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140–3152 RSC; (e) R. Sanz, Org. Prep. Proced. Int., 2008, 40, 215–291 CrossRef CAS; (f) H. Pellisier and M. Santelli, Tetrahedron, 2003, 59, 701–730 CrossRef.
- R. Stoermer and B. Kahlert, Ber., 1902, 35, 1633–1640 CrossRef CAS.
- (a) H. Gilman and S. Avakian, J. Am. Chem. Soc., 1945, 67, 349–351 CrossRef CAS; (b) H. Gilman and R. H. Kyle, J. Am. Chem. Soc., 1948, 70, 3945–3946 CrossRef CAS; (c) R. A. Benkeser and R. G. Severson, J. Am. Chem. Soc., 1949, 71, 3838–3839 CrossRef CAS; (d) R. A. Benkeser and W. E. Buting, J. Am. Chem. Soc., 1952, 74, 3011–3014 CrossRef CAS.
- J. D. Roberts, H. E. Simmons, Jr., L. A. Carlsmith and C. W. Vaughan, J. Am. Chem. Soc., 1953, 75, 3290–3291 CrossRef CAS.
- (a) R. Huisgen and H. Rist, Naturwissenschaften, 1954, 41, 358–359 CrossRef CAS; (b) R. Huisgen and H. Rist, Justus Liebigs Ann. Chem., 1955, 594, 137–158 CrossRef CAS.
- G. Wittig and L. Pohmer, Angew. Chem., 1955, 67, 348 CrossRef CAS.
- (a) H. H. Nam, G. E. Leroi and J. F. Harrison, J. Phys. Chem., 1991, 95, 6514–6519 CrossRef CAS; (b) S. L. Debbert and C. J. Cramer, Int. J. Mass Spectrom., 2000, 201, 1–15 CrossRef CAS; (c) N. Mariet, M. Ibrahim-Ouali, J.-L. Parrain and M. Santelli, THEOCHEM, 2004, 679, 53–58 CrossRef CAS PubMed; (d) N. J. Rau and P. G. Wenthold, J. Phys. Chem. A, 2011, 115, 10353–10362 CrossRef CAS PubMed; (e) G. Sánchez-Sanz, I. Alkorta, C. Trujillo and J. Elguero, Tetrahedron, 2012, 68, 6548–6556 CrossRef PubMed.
- J. F. Bunnett and P. Singh, J. Org. Chem., 1981, 46, 4567–4569 CrossRef CAS.
- (a) F. C. Gozzo and M. N. Eberlin, J. Org. Chem., 1999, 64, 2188–2193 CrossRef CAS; (b) R. Sparrapan, M. A. Mendes, M. Carvalho and M. N. Eberlin, Chem. – Eur. J., 2000, 6, 321–326 CrossRef CAS.
- For the corresponding 2,3- and 3,4-pyridyne-N-oxides, see: (a) T. Kato and T. Niitsuma, Chem. Pharm. Bull., 1965, 13, 963–968 CrossRef CAS; (b) R. J. Martens and H. J. den Hertog, Recl. Trav. Chim. Pays-Bas, 1967, 86, 655–669 CrossRef CAS; (c) T. Kato and T. Niitsuma, Heterocycles, 1973, 1, 233–236 CrossRef CAS.
- For studies pertaining to the related quinolynes, see ref. 29, 33, and the following: (a) T. Kauffmann, F.-P. Boettcher and J. Hansen, Justus Liebigs Ann. Chem., 1962, 659, 102–109 CrossRef; (b) I.-C. Grig-Alexa, E. Garnier, A.-L. Finaru, L. Ivan, C. Jarry, J.-M. Léger, P. Caubère and G. Guillaumet, Synlett, 2004, 2000–2004 CAS; (c) G. E. Collis and A. K. Burrell, Tetrahedron Lett., 2005, 46, 3653–3656 CrossRef CAS PubMed; (d) H. N. Pangamte and R. H. D. Lyngdoh, J. Phys. Chem. A, 2010, 114, 2710–2726 CrossRef CAS PubMed.
- Treatment of 3-halopyridines with strong bases has been demonstrated to selectively generate 3,4-pyridyne (4) over 2,3-pyridyne (22), see: J. A. Zoltewicz and C. L. Smith, Tetrahedron, 1969, 25, 4331–4337 CrossRef CAS.
- G. W. J. Fleet and I. Fleming, J. Chem. Soc. C, 1969, 1758–1763 RSC.
- J. Kramer and R. S. Berry, J. Am. Chem. Soc., 1972, 94, 8336–8347 CrossRef CAS.
- N. Furukawa, T. Shibutani and H. Fujihara, Tetrahedron Lett., 1987, 28, 2727–2730 CrossRef CAS.
- T. Kauffmann and F.-P. Boettcher, Chem. Ber., 1962, 95, 949–955 CrossRef CAS.
- M. Tsukazaki and V. Snieckus, Heterocycles, 1992, 33, 533–536 CrossRef CAS PubMed.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 12, 1211–1214 CrossRef.
- Pyridyne 4 showed a strong absorbance at 2085 cm−1, similar to the signal observed at 2082 cm −1 for o-benzyne (1), which is attributed to the C–C triple bond; see: H.-H. Nam and G. E. Leroi, J. Am. Chem. Soc., 1988, 110, 4096–4097 CrossRef CAS.
- For metal-catalyzed [2+2+2] cycloadditions involving 3,4-pyridynes, see: (a) T. Iwayama and Y. Sato, Chem. Commun., 2009, 5245–5247 RSC; (b) Y. Sato and T. Iwayama, Heterocycles, 2010, 80, 917–924 CrossRef PubMed.
- R. Levine and W. W. Leake, Science, 1955, 121, 780 CAS.
- J. A. Zoltewicz and C. Nisi, J. Org. Chem., 1969, 34, 765–766 CrossRef CAS.
- (a) K. Vinter-Pasquier, B. Jamart-Grégoire and P. Caubère, Heterocycles, 1997, 45, 2113–2129 CrossRef CAS; (b) M. T. Díaz, A. Cobas, E. Guitián and L. Castedo, Synlett, 1998, 157–158 CrossRef PubMed; (c) A. Wang, S. Tandel, H. Zhang, Y. Huang, T. C. Holdeman and E. R. Biehl, Tetrahedron, 1998, 54, 3391–3400 CrossRef CAS; (d) M. T. Díaz, A. Cobas, E. Guitián and L. Castedo, Eur. J. Org. Chem., 2001, 4543–4549 CrossRef.
- A. E. Goetz and N. K. Garg, Nat. Chem., 2013, 5, 54–60 CrossRef CAS PubMed.
- For recent reviews of nickel-catalyzed couplings involving C–O bonds, see: (a) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346–1416 CrossRef CAS PubMed; (b) B.-J. Li, D.-G. Yu, C.-L. Sun and Z.-J. Shi, Chem. – Eur. J., 2011, 17, 1728–1759 CrossRef CAS PubMed; (c) T. Mesganaw and N. K. Garg, Org. Process Res. Dev., 2013, 17, 29–39 CrossRef CAS; (d) J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org. Chem., 2013, 19–30 CrossRef CAS.
- For the Ni-catalyzed amination of aryl sulfamates, see: (a) S. D. Ramgren, A. L. Silberstein, Y. Yang and N. K. Garg, Angew. Chem., Int. Ed., 2011, 50, 2171–2173 CrossRef CAS PubMed; (b) T. Mesganaw, A. L. Silberstein, S. D. Ramgren, N. F. Fine Nathel, X. Hong, P. Liu and N. K. Garg, Chem. Sci., 2011, 2, 1766–1771 RSC; (c) L. Hie, S. D. Ramgren, T. Mesganaw and N. K. Garg, Org. Lett., 2012, 14, 4182–4185 CrossRef CAS PubMed.
- H. Yoshida, E. Shirakawa, Y. Honda and T. Hiyama, Angew. Chem., Int. Ed., 2002, 41, 3247–3249 CrossRef CAS.
- N. Saito, K. Nakamura, S. Shibano, S. Ide, M. Minami and Y. Sato, Org. Lett., 2013, 15, 386–389 CrossRef CAS PubMed.
- Several silyltriflate hetaryne precursors are available from Aldrich Chemical Co., Inc. The precursors and their product numbers are as follows: 3,4-pyridyne precursor 34: L511633; bromopyridine precursor 38: L511641; 2,3-pyridyne precursor 45: L511668; 60 (R = H) 4,5-indolyne precursor: L511625.
- R. J. Martens and H. J. den Hertog, Tetrahedron Lett., 1962, 3, 643–645 CrossRef.
- M. A. Walters and J. J. Shay, Synth. Commun., 1997, 27, 3573–3579 CrossRef CAS.
- Y. Fang and R. C. Larock, Tetrahedron, 2012, 68, 2819–2826 CrossRef CAS PubMed.
- M. Julia, Y. Huang and J. Igolen, C. R. Acad. Sci., Ser. C, 1967, 265, 110–112 CAS.
- (a) M. Julia, F. Le Goffic, J. Igolen and M. Baillarge, C. R. Acad. Sci., Ser. C, 1967, 264, 118–120 CAS; (b) J. Igolen and A. Kolb, C. R. Acad. Sci., Ser. C, 1969, 269, 54–56 CAS; (c) M. Julia, F. Le Goffic, J. Igolen and M. Baillarge, Tetrahedron Lett., 1969, 10, 1569–1572 CrossRef; (d) M. Julia, J. Igolen and M. Kolb, C. R. Acad. Sci., Ser. C, 1971, 273, 1776–1777 CAS.
- One exception is the work of Iwao and coworkers in the synthesis of a variety of Makaluvamine natural products: M. Iwao, O. Motoi, T. Fukuda and F. Ishibashi, Tetrahedron, 1998, 54, 8999–9010 CrossRef CAS.
- (a) K. R. Buszek, D. Luo, M. Kondrashov, N. Brown and D. VanderVelde, Org. Lett., 2007, 9, 4135–4137 CrossRef CAS PubMed; (b) N. Brown, D. Luo, D. VanderVelde, S. Yang, A. Brassfield and K. R. Buszek, Tetrahedron Lett., 2009, 50, 63–65 CrossRef CAS PubMed; (c) K. R. Buszek, N. Brown and D. Luo, Org. Lett., 2009, 11, 201–204 CrossRef CAS PubMed; (d) A. N. Garr, D. Luo, N. Brown, C. J. Cramer, K. R. Buszek and D. VanderVelde, Org. Lett., 2010, 12, 96–99 CrossRef CAS PubMed; (e) N. Brown, D. Luo, J. A. Decapo and K. R. Buszek, Tetrahedron Lett., 2009, 50, 7113–7115 CrossRef CAS PubMed; (f) P. D. Thornton, N. Brown, D. Hill, B. Neuenswander, G. H. Lushington, C. Santini and K. R. Buszek, ACS Comb. Sci., 2011, 13, 443–448 CrossRef CAS PubMed; (g) A. Nerurkar, N. Chandrasoma, L. Maina, A. Brassfield, D. Luo, N. Brown and K. R. Buszek, Synthesis, 2013, 1843–1852 CAS.
- (a) S. M. Bronner, K. B. Bahnck and N. K. Garg, Org. Lett., 2009, 11, 1007–1010 CrossRef CAS PubMed; (b) P. H.-Y. Cheong, R. S. Paton, S. M. Bronner, G.-Y. J. Im, N. K. Garg and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 1267–1269 CrossRef CAS PubMed; (c) G.-Y. J. Im, S. M. Bronner, A. E. Goetz, R. S. Paton, P. H.-Y. Cheong, K. N. Houk and N. K. Garg, J. Am. Chem. Soc., 2010, 132, 17933–17944 CrossRef CAS PubMed.
- For additional references regarding the aryne distortion model, see ref. 1c, 1d, 25, and the following: (a) S. M. Bronner, J. L. Mackey, K. N. Houk and N. K. Garg, J. Am. Chem. Soc., 2012, 134, 13966–13969 CrossRef CAS PubMed; (b) A. E. Goetz, S. M. Bronner, J. D. Cisneros, J. M. Melamed, R. S. Paton, K. N. Houk and N. K. Garg, Angew. Chem., Int. Ed., 2012, 51, 2758–2762 CrossRef CAS PubMed.
- D. A. Candito, D. Dobrovolsky and M. Lautens, J. Am. Chem. Soc., 2012, 134, 15572–15580 CrossRef CAS PubMed.
- (a) T. R. Hoye, B. Baire, D. Niu, P. H. Willoughby and B. P. Woods, Nature, 2012, 490, 208–212 CrossRef CAS PubMed; (b) D. Niu, P. H. Willoughby, B. P. Woods, B. Baire and T. R. Hoye, Nature, 2013, 501, 531–534 CrossRef CAS PubMed; (c) J. Chen, B. Baire and T. R. Hoye, Heterocycles, 2014, 88, 1191–1200 CrossRef CAS PubMed; (d) D. Niu and T. R. Hoye, Nat. Chem., 2014, 6, 34–40 CrossRef CAS PubMed; (e) T. R. Hoye, B. Baire and T. Wang, Chem. Sci., 2014, 5, 545–550 RSC; (f) D. Niu, T. Wang, B. P. Woods and T. R. Hoye, Org. Lett., 2014, 16, 254–257 CrossRef CAS PubMed.
- (a) S. Y. Yun, K.-P. Wang, N.-K. Lee, P. Mamidipalli and D. Lee, J. Am. Chem. Soc., 2013, 135, 4668–4671 CrossRef CAS PubMed; (b) K.-P. Wang, S. Y. Yun, P. Mamidipalli and D. Lee, Chem. Sci., 2013, 4, 3205–3211 RSC; (c) R. Karmakar, P. Mamidipalli, S. Y. Yun and D. Lee, Org. Lett., 2013, 15, 1938–1941 CrossRef CAS PubMed; (d) R. Karmakar, S. Y. Yun, K.-P. Wang and D. Lee, Org. Lett., 2014, 16, 6–9 CrossRef CAS PubMed.
- S. C. Conway and G. W. Gribble, Heterocycles, 1992, 34, 2095–2108 CrossRef CAS.
- (a) H. Muller, Dissertation, University of Heidelberg, 1964 Search PubMed; (b) J. Bergman and N. Eklund, Tetrahedron Lett., 1980, 36, 1439–1443 CrossRef CAS.
- J.-H. Liu, H.-W. Chan, F. Xue, Q.-G. Wang, T. C. W. Mak and H. N. C. Wong, J. Org. Chem., 1999, 64, 1630–1634 CrossRef CAS PubMed.
- (a) X.-S. Ye, W.-K. Li and H. N. C. Wong, J. Am. Chem. Soc., 1996, 118, 2511–2512 CrossRef CAS; (b) M. G. Reinecke, J. G. Newsom and L.-J. Chen, J. Am. Chem. Soc., 1981, 103, 2760–2769 CrossRef CAS.
- A recent Reaxys search showed >700 natural products contain a C4-substituted indole motif.
- (a) A. D. Huters, K. W. Quasdorf, E. D. Styduhar and N. K. Garg, J. Am. Chem. Soc., 2011, 133, 15797–15799 CrossRef CAS PubMed; (b) K. W. Quasdorf, A. D. Huters, M. W. Lodewyk, D. J. Tantillo and N. K. Garg, J. Am. Chem. Soc., 2012, 134, 1396–1399 CrossRef CAS PubMed; (c) E. D. Styduhar, A. D. Huters, N. A. Weires and N. K. Garg, Angew. Chem., Int. Ed., 2013, 52, 12422–12425 CrossRef CAS PubMed.
- (a) S. M. Bronner, A. E. Goetz and N. K. Garg, J. Am. Chem. Soc., 2011, 133, 3832–3835 CrossRef CAS PubMed; (b) N. F. Fine Nathel, T. K. Shah, S. M. Bronner and N. K. Garg, Chem. Sci., 2014, 5, 2184–2190 RSC.
- A. E. Goetz, A. L. Silberstein, M. A. Corsello and N. K. Garg, J. Am. Chem. Soc., 2014, 136, 3036–3039 CrossRef CAS PubMed.
- (a) C. May and C. J. Moody, J. Chem. Soc., Chem. Commun., 1984, 926–927 RSC; (b) G. W. Gribble, M. G. Saulnier, M. P. Sibi and J. A. Obaza-Nutaitis, J. Org. Chem., 1984, 49, 4518–4523 CrossRef CAS; (c) C. May and C. J. Moody, J. Chem. Soc., Perkin Trans. 1, 1988, 247–250 RSC; (d) C.-K. Sha and J.-F. Yang, Tetrahedron, 1992, 48, 10645–10654 CrossRef CAS.
- M. F. Enamorado, P. W. Ondachi and D. L. Comins, Org. Lett., 2010, 12, 4513–4515 CrossRef CAS PubMed.
- (a) S. V. Kessar, Y. P. Gupta, P. S. Pahwa and P. Singh, Tetrahedron Lett., 1976, 17, 3207–3208 CrossRef; (b) S. V. Kessar and P. Singh, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2001, 40, 1129–1131 Search PubMed.
- C. Hoarau, A. Couture, H. Cornet, E. Deniau and P. Grandclaudon, J. Org. Chem., 2001, 66, 8064–8069 CrossRef CAS PubMed.
- A mixture of cycloaddition adducts was obtained in a 2.4:1 ratio.
- E. C. Kornfeld, E. J. Fornefeld, G. B. Kline, M. J. Mann, D. E. Morrison, R. G. Jones and R. B. Woodward, J. Am. Chem. Soc., 1956, 78, 3087–3114 CrossRef CAS.
- For pertinent reviews on (−)-indolactam V isolation, tumor-promoting activity, and derivatives, see: (a) K. Irie and K. Koshimizu, Mem. Coll. Agric., Kyoto Univ., 1988, 132, 1–59 CAS; (b) K. Irie and K. Koshimizu, Comments Agric. Food Chem., 1993, 3, 1–25 CAS; (c) K. Irie, Nippon Nogei Kagaku Kaishi, 1994, 68, 1289–1296 CrossRef CAS; (d) K. Irie, Y. Nakagawa and H. Ohigashi, Curr. Pharm. Des., 2004, 10, 1371–1385 CrossRef CAS; (e) K. Irie, Y. Nakagawa and H. Ohigashi, Chem. Rec., 2005, 5, 185–195 CrossRef CAS PubMed.
- S. Chen, M. Borowiak, J. L. Fox, R. Maehr, K. Osafune, L. Davidow, K. Lam, L. F. Peng, S. L. Schreiber, L. L. Rubin and D. Melton, Nat. Chem. Biol., 2009, 5, 258–265 CrossRef CAS PubMed.
- T. Hama, D. A. Culkin and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 4976–4985 CrossRef CAS PubMed.
- (a) K. Stratmann, R. E. Moore, R. Bonjouklian, J. B. Deeter, G. M. L. Patterson, S. Shaffer, C. D. Smith and T. A. Smitka, J. Am. Chem. Soc., 1994, 116, 9935–9942 CrossRef CAS; (b) J. L. Jimenez, U. Huber, R. E. Moore and G. M. L. Patterson, J. Nat. Prod., 1999, 62, 569–572 CrossRef CAS PubMed.
- A. D. Huters, E. D. Styduhar and N. K. Garg, Angew. Chem., Int. Ed., 2012, 51, 3758–3765 CrossRef CAS PubMed ; see also references therein.
- For formal total syntheses, see: (a) T.-h. Fu, W. T. McElroy, M. Shamszad and S. F. Martin, Org. Lett., 2012, 14, 3834–3837 CrossRef CAS PubMed; (b) T.-h. Fu, W. T. McElroy, M. Shamszad, R. W. Heidebrecht, Jr., B. Gulledge and S. F. Martin, Tetrahedron, 2013, 69, 5588–5603 CrossRef CAS PubMed.
- (a) V. Bhat, K. M. Allan and V. H. Rawal, J. Am. Chem. Soc., 2011, 133, 5798–5801 CrossRef CAS PubMed; (b) K. M. Allan, K. Kobayashi and V. H. Rawal, J. Am. Chem. Soc., 2012, 134, 1392–1395 CrossRef CAS PubMed.
- M. Sakagami, H. Muratake and M. Natsume, Chem. Pharm. Bull., 1994, 42, 1393–1398 CrossRef CAS.
- P. Caubere, Acc. Chem. Res., 1974, 7, 301–308 CrossRef CAS.
- M. R. TePaske, J. B. Gloer, D. T. Wicklow and P. F. Dowd, J. Org. Chem., 1989, 54, 4743–4746 CrossRef CAS.
- M. R. TePaske, J. B. Gloer, D. T. Wicklow and P. F. Dowd, Tetrahedron Lett., 1989, 30, 5965–5968 CrossRef CAS.
- For an elegant total synthesis of 84, see: M. Bian, Z. Wang, X. Xiong, Y. Sun, C. Matera, K. C. Nicolaou and A. Li, J. Am. Chem. Soc., 2012, 134, 8078–8081 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |