
Dual doping effects (site blockage and electronic promotion) imposed by adatoms on Pd nanocrystals for catalytic hydrogen production†
S.
Jones
a,
S. M.
Fairclough
ab,
M.
Gordon-Brown
a,
W.
Zheng
a,
A.
Kolpin
a,
B.
Pang
c,
W. C. H.
Kuo
d,
J. M.
Smith
b and
S. C. E.
Tsang
*a
aDepartment of Chemistry, University of Oxford, OX1 3QR, UK. E-mail: Edman.Tsang@chem.ox.ac.uk
bDepartment of Materials, University of Oxford, OX1 3PH, UK
cMetallurgy and Materials, University of Birmingham, B15 2TT, UK
dJohnson Matthey, Blount’s Court, Sonning Common, Reading, RG4 9NH, UK
First published on 11th September 2014
Abstract
Three distinctive doping effects to modify the electronic and geometric properties of Pd nanocrystals for HCOOH decomposition to H2/CO2 are presented: Bi atoms take preferable residence on higher index sites, which leads to a reduction in HCOOH dehydration; Te atoms dwell favourably on terrace sites, which reduces the rate of dehydrogenation; Ag atoms, without site specificity, induce strong electronic effects to promote the activity on the dwindling number of surface Pd sites at high coverage.
Metallic nanoparticles are the key components of many catalysts, the activity and selectivity of which is highly dependent on surface properties.1 Control of size, shape and bimetallic structure (core–shell, alloy, etc.) is widely used to produce nanoparticles with optimized electronic and geometric properties.2 We have recently reported a facile approach, using polymer based additives, to modify the electronic structure of metal nanoparticles. Adsorption of pendant groups, such as amines, results in electron donation to the metal surface influencing catalytic activity.3 It has long been reported that addition of foreign metal adatoms of up to a monolayer can also dramatically modify the catalytic and sensing properties of metallic nanoparticles.4,5 However, there is a lack of fundamental understanding about the doping effects at the atomic level. For example, the activity and selectivity of a selected reaction can be enhanced or reduced depending on the type, amount, and method of addition of metal dopants, reaction conditions and the nature of the support used but the interplay between these factors is rather complex to resolve.6 As a result, we are particularly interested in elucidating the interaction(s) between metal and the dopant without a support using more well defined Pd nanocrystals and adatoms with progressive coverage under ambient conditions, the knowledge of which may facilitate rational nanocatalyst design in the future. Catalytic decomposition of formic acid to hydrogen over metallic Pd nanoparticles is hereby selected as a probe reaction. This reaction has exciting potential in hydrogen storage to supply cleaner power for small electrical devices at room temperature via Proton Exchange Membrane (PEM) micro-fuel cells.7 The decomposition and related electrochemical reaction have been studied intensively in recent years and are reported to proceed via two clear cut mechanisms dependent on the nature of metal sites; namely the desirable dehydrogenation to produce hydrogen and carbon dioxide on flat surface metal sites (eqn (1)), and undesirable dehydration to form carbon monoxide and water on low coordinate sites of a Pd nanocrystal even in the presence of a stabilizing polymer (eqn (2)).8 The strongly adsorbed HCOOH and CO (higher adsorption energy than PVP) can quickly migrate to other metal sites leading to deactivation, hence an urgent need for surface tailoring of the catalyst.9
| HCOOH → H2 + CO2 ΔG = −48.4 kJ mol−1 | (1) |
| HCOOH → H2O + CO ΔG = −28.5 kJ mol−1 | (2) |
With tailoring the Pd colloid size, careful rate measurement, surface diagnosis coupled with understanding of formic acid decomposition pathways and HRTEM/STEM and simulation analysis on unsupported Pd nanocrystals as models, in this communication, we report the effects of three classes of adatoms on Pd. This can lead to the specific modification of the electronic and geometric doping on the atomic scale and a significant alteration to the catalytic activity. Firstly, Bi adatoms are found to dwell specifically on low coordinate sites (corners/edges) in the Pd crystal model. This site blockage, in competition with the dehydration reaction (eqn (2)), reduces deactivation of Pd, resulting in a higher rate; further surface coverage beyond the corner/edge sites rapidly attenuates activity, giving a typical volcano response. However, in the case of Te, deposition is favourable on terrace sites, competing with the active sites for the dehydrogenation reaction (eqn (1)), resulting in a much reduced activity. Finally, Ag adatoms present no particular site preference on the Pd surface. Despite a high degree of site blockage, the strong electronic promotion effect by Ag adatoms (ligand effect) overrides the dwindling number of Pd sites, resulting in a dramatic enhancement in the catalytic rate.
Doped Pd nanocrystals were synthesized by a two-step process. First, PVP stabilized Pd nanocrystals were synthesized using a modified method of Bonet et al. by ethylene glycol reduction and treated with hydrogen. Subsequently the metal dopant was added and fully reduced onto the Pd core using NaBH4 (details in ESI†).10 X-ray diffraction (XRD) showed the average PVP stabilized Pd size to be 3.2 nm, consistent with results of transmission electron microscopy (TEM) which showed the nanocrystals to be spherical or cubo-octahedral by the dark field imaging (ESI†). Using calculations by van Hardeveld and Hartog for modelling cubo-octahedrons, the reported most stable shape, approximately 30% of surface atoms are at edge or corner sites, which is roughly 11% of the total atoms in the Pd nanocrystal embracing within the weak coordinating PVP matrix.11 Decorating with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.15 mole ratio of Pd to adatom, assuming a one to one coverage and ignoring small size variation, theoretically results in coverage of ∼35% of the particle surface sufficient to cover all low coordinated sites. The effects of a range of metal additives were examined on the activity of Pd-PVP colloids, with a 1:0.15 ratio of Pd to metal additive for decomposition of formic acid, as shown in Table 1 and in the ESI.† Typically, the initial gas production rate (evaluated in the first 10 min) is higher than the final rate (measured in the final 20 min) for a 2 hour reaction. The lower rate at the end of the experiment can be explained by substrate consumption but more importantly, by a continuous formation of CO poison due to the dehydration route (eqn (2)), leading to deactivation of the catalyst.12 Of all the metal additives investigated only Ag and Bi produced more gas than the unmodified Pd colloid over the 2 hours (the activity 3.20 and 1.35 times that of Pd-PVP, respectively), but they appear to operate with different promotion mechanisms. As observed in Table 1 (and ESI†) the initial gas production rates of all metal additives investigated, except Ag, are reduced compared to that of unmodified Pd colloid, indicative of coverage of the active Pd crystal by the additives. However, Ag clearly exerts a strong promotional effect on Pd enhancing both initial and final activities for the gas production, despite coverage of the Pd surface (Ag nanocrystals are inactive for the decomposition reaction). On the other hand, Bi does not promote the initial rate but gives a higher final rate, suggesting a decrease in the deactivation rate. To further investigate this, we have studied the structures of these additives in more detail to correlate with their activities.
:0.15 mole ratio of Pd to adatom, assuming a one to one coverage and ignoring small size variation, theoretically results in coverage of ∼35% of the particle surface sufficient to cover all low coordinated sites. The effects of a range of metal additives were examined on the activity of Pd-PVP colloids, with a 1:0.15 ratio of Pd to metal additive for decomposition of formic acid, as shown in Table 1 and in the ESI.† Typically, the initial gas production rate (evaluated in the first 10 min) is higher than the final rate (measured in the final 20 min) for a 2 hour reaction. The lower rate at the end of the experiment can be explained by substrate consumption but more importantly, by a continuous formation of CO poison due to the dehydration route (eqn (2)), leading to deactivation of the catalyst.12 Of all the metal additives investigated only Ag and Bi produced more gas than the unmodified Pd colloid over the 2 hours (the activity 3.20 and 1.35 times that of Pd-PVP, respectively), but they appear to operate with different promotion mechanisms. As observed in Table 1 (and ESI†) the initial gas production rates of all metal additives investigated, except Ag, are reduced compared to that of unmodified Pd colloid, indicative of coverage of the active Pd crystal by the additives. However, Ag clearly exerts a strong promotional effect on Pd enhancing both initial and final activities for the gas production, despite coverage of the Pd surface (Ag nanocrystals are inactive for the decomposition reaction). On the other hand, Bi does not promote the initial rate but gives a higher final rate, suggesting a decrease in the deactivation rate. To further investigate this, we have studied the structures of these additives in more detail to correlate with their activities.
:0.15 mole ratio of adatoms for formic acid decomposition
| Pd-PVP + adatoms | Initial ratea (L g(Pd)−1 min−1) | Final rateb (L g(Pd)−1 min−1) | Total gasc (L g(Pd)−1) |
|---|---|---|---|
| a Rate of gas production in the first 10 min. b Rate of gas production in the final 20 min. c Total gas produced in 2 hours (0.15 × 10−3 moles of Pd in 15.86 mL of 1.4 M formic acid stirred at 25 °C). | |||
| Unmodified | 0.106 | 0.0066 | 2.81 |
| Ag, 1:0.15 |
0.264 | 0.0250 | 8.98 |
| Bi, 1:0.15 |
0.070 | 0.0200 | 3.80 |
| Te, 1:0.15 |
0.010 | 0.0038 | 0.59 |
XRD of the Bi decorated Pd (0–15% Bi) showed a fcc structure matching that expected for Pd crystals, with no Bi peaks observed (ESI†). HRTEM also shows lattice spacing matching that of Pd(111) and energy dispersive X-ray (EDX) analysis shows that the Pd to Bi ratio is close to expected from synthesis with no separate Bi rich particles/areas found (ESI†). Thus, the Bi atoms are quantitatively deposited on the Pd nanocrystals with no phases separated from the Pd-PVP. It is envisaged that the adsorption of metal dopants such as Bi is more favourable than the weak PVP. A small shift in the Pd(111) peak value is observed with increasing Bi ratio, which appears to be smaller than those of typical Bi–Pd alloys and also considerably smaller than that calculated, using a method similar to Fairclough et al., for alloys with the specified ratios of Pd to Bi (ESI†).13–15 This is in agreement with Wenkin et al. who found that Bi had a preference to reside on the Pd surface due to its lower surface energy and stronger adsorption energy.16 It is thought that the small shift maybe caused by the lattice mismatch upon surface decoration or by the presence of interstitial hydrogen atoms.17,18 Probing CO adsorbed on nanoparticle surfaces, using Fourier transform infrared (FTIR) spectroscopy, has been widely reported for characterizing both electronic and geometric properties of metal nanoparticles.19,20 Thus, CO adsorption was used as a chemical marker to examine the Pd surface features modified by the Bi adatoms. It is known that, at low surface coverage as the conditions of low partial pressure CO we used, CO binds on (111) terrace Pd sites in a bridging mode (1910–1960 cm−1) but in a linear fashion on low coordinate Pd sites (2040–2070 cm−1).21Fig. 1(a), normalized for the bridging peak, shows that Bi decoration modified the Pd surface in two ways: firstly a red shift is seen, demonstrating an electronic donation from Bi to Pd increasing its ability to back donate to CO (the red shift value is greater than those of site-dependent coverage, see ESI†). Secondly, a clear decrease in the linear peak shows that the low coordinate Pd sites, on which linear CO species reside, are selectively blocked with an increasing ratio of Bi. For the sample with a 1:0.05 ratio of Pd to Bi, low coordinate sites are not yet completely blocked but a decrease in linear bound CO intensity is observed whilst with a 1:0.15 ratio (calculated as sufficient to cover all low coordinate sites), no linear CO peak can be seen. At a 1:0.6 ratio of Pd to Bi (sufficient to give over a monolayer of coverage), no CO peaks are observed indicating full coverage of the Pd surface. The presence of weaker coordinating PVP and the amount (varied) did not seem to affect the adsorption values.
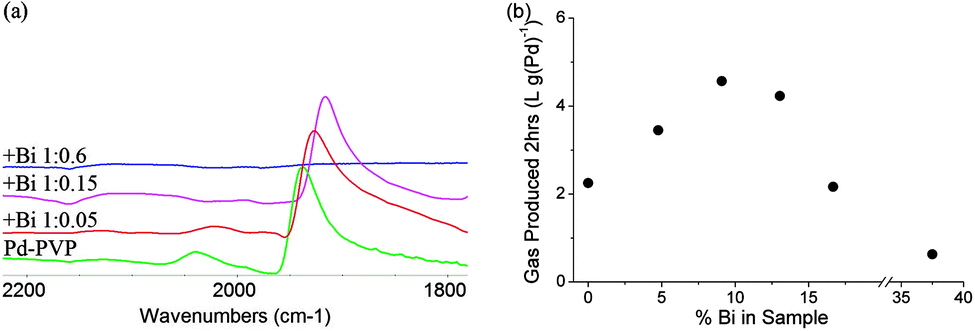 | ||
| Fig. 1 (a) FTIR of CO on Bi decorated Pd-PVP with Pd:Bi ratios of 1:0, 1:0.05, 1:0.15, 1:0.6; (b) total gas production (2 h) of Bi decorated Pd-PVP with increasing Bi content. | ||
Testing the activity for total gas production of the Bi decorated Pd-PVP nanocrystals with increasing Bi content may reflect the promotion mechanism. As seen from Fig. 1(b), the optimal activity is around 10% Bi (a 1:0.1 molar ratio) and then decreases, giving a typical volcano response. This correlates well with the 11% of Pd atoms in corner or edge positions predicted for 3.2 nm particles by the Hartogg and Hardeveld model, indicating that they are preferentially blocked by Bi atoms with a 1:1 ratio even in the presence of PVP matrix.11 However, the lower initial activity compared to unmodified Pd suggests that the electronic promotion effect from Bi is not sufficient to override the loss in Pd sites. At excess Bi, the rapid deactivation indicates that Bi atoms begin to take residence on terrace sites, blocking the active sites for dehydrogenation (eqn (1)).
In contrast, addition of Ag to Pd-PVP increases both the initial and final activity for formic acid decomposition, implying a totally different doping mechanism when compared to Bi. Modified Pd-PVP nanoparticles with a 1:0.15 mole ratio of Ag were initially synthesized with Ag reduced by ethylene glycol at 120 °C, a significant shift of the ‘Pd(111)’ peak is observed implying alloying, but both a room temperature synthesis (to reduce atom mobility) and the calculated XRD for Pd core–Ag shell structures indicate a similar degree of shift (ESI†). The strain induced by the formation of a core–shell structure has been shown to produce distinctive changes in the lattice parameter, which can account for our observed XRD shift.17,22
For Ag decoration of carbon supported Pd (no PVP), the Pd(111) peak position does not change as much upon the Ag deposition even at high coverage, suggesting that they are not subject to the same degree of strain for larger (5.5 nm) particles, but they still display the same trend in activity promotion as Ag doped PVP-Pd, suggesting that strain is not the cause (ESI†). Ag decorated Pd-PVP particles were also probed using CO FTIR, Fig. 2(a). Bridging CO on Pd is still observed even at high coverage of Ag adatoms whilst identification of linear CO on Pd is obscured by the presence of CO adsorbed on Ag.8 However, the change in the rate profile for Ag decorated Pd, reflected by the ratio of initial to final rates, is similar to unmodified Pd-PVP indicating that the low coordinate sites are not selectively blocked as demonstrated in the case of Bi. The trend with increasing the Pd:Ag molar ratio is also different to Bi. The maximum activity is sustained with increasing Ag content, reaching an optimum with a ratio of 1:0.15 corresponding to ∼40% coverage of the Pd surface without any decrease in activity with ever increasing coverage. Although, the deposition causes aggregation of Ag to clusters with a lower degree of spreading than with Bi on the Pd surface, these adatoms are clearly capable of modifying the remaining exposed Pd atoms for enhanced catalysis. It should be noted that a similar observation was made by Tsang et al. who reported the promotion effect to Pt nanoparticle by Co which exerted a short range electronic influence to nearby Pt atoms.5 In Table 1 and Fig. 3(a) a third doping effect is described: the addition of a 1:0.15 of Pd to Te ratio severely inhibits the overall activity of the Pd-PVP. Again using CO FTIR as a surface probe, it is clearly shown that there is a dramatic loss in bridging CO this time; for ideal crystals and a monolayer coverage, the 1:0.15 ratio would cover ∼50% of terrace sites on 3.2 nm crystals, blocking the active sites for the dehydrogenation reaction preferentially. In addition, the severe loss of activity, over a factor of two, may also suggest an additional electronic demoting effect: the shift in the linear CO peak to higher wave-number, indicative of electron withdrawal from Pd to Te. The electronic effect on these low coordinate sites may be localized and is complex, and as such will not be discussed further.9
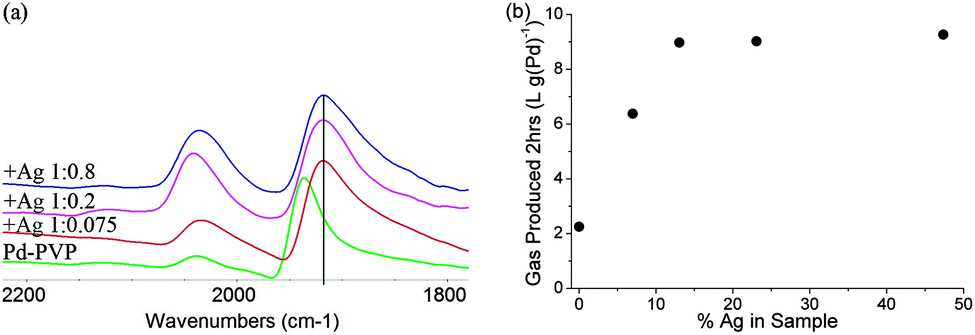 | ||
| Fig. 2 (a) FTIR of CO on Ag decorated Pd-PVP with Pd:Ag ratios of 1:0, 1:0.075, 1:0.2, 1:0.8; (b) total gas production (2 h) of Ag decorated Pd-PVP with increasing Ag content. | ||
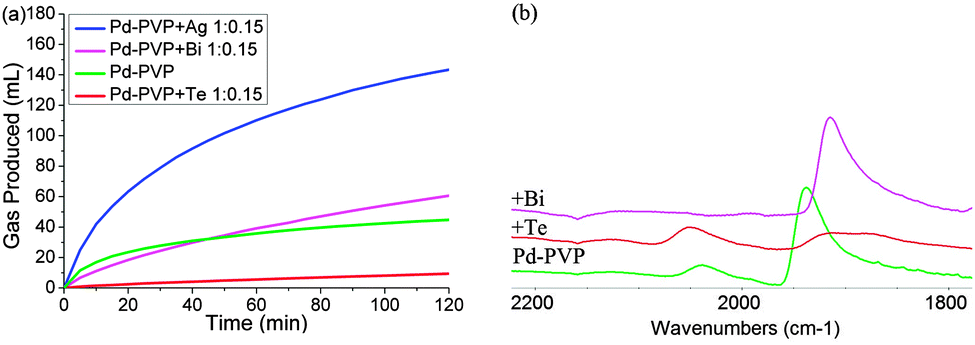 | ||
| Fig. 3 (a) Activity plots of Pd-PVP nanoparticles with and without a 1:0.15 Pd to adatom (Ag, Bi, Te) ratio; (b) FTIR of CO on Pd-PVP with and without a 1:0.15 Pd to adatom (Bi, Te) ratio. | ||
The high-Angle Annular Dark Field (HADDF) scanning image shown in Fig. 4(a) suggests that there are two distinctive types of lattices observed for the first time in a single crystal of Te doped Pd. The inner crystal with smaller lattice is covered with a larger lattice (EDX confirms both Pd and Te signals). The inner crystal lattice is coherent with the outer surface lattice which has a larger parameter. It should be noted that the d-spacing of the outer surface lattice fits with the Te hcp structure whereas the inner crystal fits with the Pd fcc structure. This crystal simulation also supports the postulation of face deposition of Te atoms on a Pd crystal (ESI†). In contrast to PdTe we did not find extensive deposition with a surface lattice of Bi atoms but instead individual Bi atoms selectively deposited on the upper part of the Pd lattice were found. A typical low energy high-angle annular dark field image of a Pd crystal doped with Bi (EDX confirms both Pd and Bi signals) with Fourier transformation and a corresponding line profiling on the surface deposition are shown in Fig. 4(b) and ESI.† The Bi atoms appear brighter than Pd since the atomic number of Bi is higher than that of Pd. The corresponding crystal simulation confirms that the Bi atoms are deposited on the Pd around the edge/corner regions of the [110] orientation of the model PdBi (ESI†). Due to the extensive characterization of this nanocrystal and the extremely weak contrast difference between Ag and Pd (ESI†), we did not perform the STEM analysis of PdAg. But, according to the studies of PdAg in the literature, the surface from the deposition of Ag atoms on Pd is characterised with both Pd and Ag surface atoms almost randomly distributed in the topmost bimetallic layer, as also suggested by our present work (ESI†). Thus, from detailed HRTEM/STEM and crystal simulations, the results are consistent with our proposed models of selective depositions.
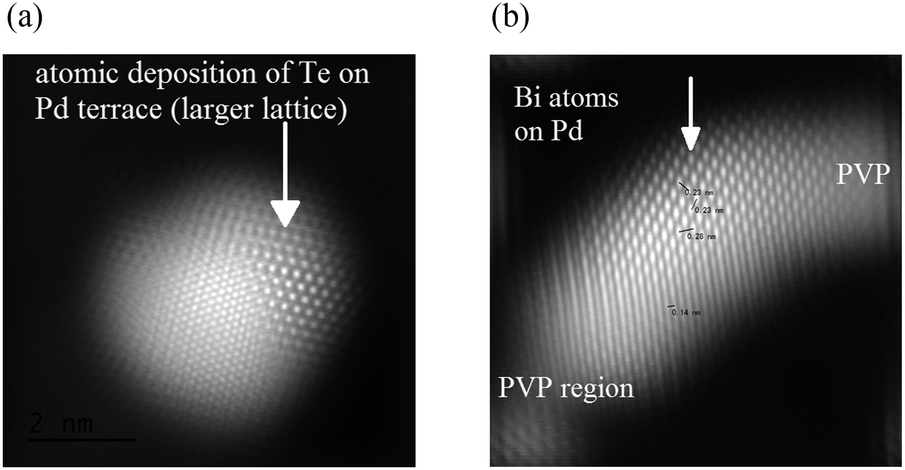 | ||
| Fig. 4 (a) Fourier transformed filtered-FFT STEM image of PVP + Te (1:0.15) showing a single Pd(Te) crystal; (b) filtered-FFT STEM image of PVP + Bi (1:0.15) showing a single Pd(Bi) crystal partially covered with PVP. | ||
At this point, there is an important question that has been raised: why do Bi atoms bind selectively at low coordinate sites, Te atoms at terrace sites and Ag atoms exhibit no preference?
We believe that the nature of the chemical precursor and the reduction mechanism play an important role in the binding sites of adatoms. Bi3+ nitrate was used as a chemical precursor for deposition, although we do not yet know the nature of ligands of Bi3+ during the deposition in ethylene glycol, its characteristic localized lone pair electrons, often entitled ‘stereochemical lone pair’ is well-known and expected.23 Following our previous findings for amines and the reports of Lee et al., the single lone pair of electrons bind favourably atop metal sites (d–p mixing) with the preference dependent on coordination, corners > edges > terrace.3,24 We expect that the same principle would apply in the case of Bi3+ doping for selective blockage of higher index Pd sites before it is further reduced to elemental adatoms, which have been observed by our STEM imaging of the well defined Pd crystals. On the other hand, we have also reported that organic binders (carboxylate, sulphonate) with two or more lone pairs would prefer to bind on terrace sites to maximise surface interactions. Thus, in the case of the Te4+ salt used, it is envisaged that the higher reduction potential of Te4+ than Bi3+ (0.57 versus 0.317 eV) would render an initial partial reduction of this to a Te2+ species, followed by further reduction to metallic adatoms. The two lone pairs of the Te2+ will thus show a preference for binding on the terrace sites as evidenced in the STEM image as terrace site deposition, hence blocking the dehydrogenation step. On the other hand, Ag+, from the nitrate salt, as a late transition metal (Group IB) with no stereochemical lone electrons will be readily reduced by the Pd–H surface with no site specificity, agreeing with previous reports.20
The present assignments of selective edge site blockage by Bi, terrace site blockage by Te and unselective site blockage by Ag on Pd nanoparticles (three different decorating mechanisms) under our doping conditions, without much atom mobility or scrambling on the surface at room temperature, with confidence are mainly attributed to the tailored control of high quality Pd colloids, careful rate analysis, high CO FTIR diagnosis value coupled with an understanding of formic acid decomposition pathways on Pd sites. In addition, HRTEM/STEM and crystal simulation analyses (ESI†) of the decorated models can allow the rationalization of the doping effects at an atomic level. On the other hand, in catalysis and electrocatalysis communities, doping effects of post-transition metal, PTM such as Bi and Pb for low coordination site blockage are generally known but the broad Pd size distributions, atomic restructuring at elevated temperatures and/or under influence of charge/electric fields preclude the effects for further elucidation at an atomic level.25,26
Finally, it is believed that surface tailoring for optimized catalysis can be done using the same underlying principle: all chemical species maximize their surface interactions (bonding) with nanoparticle surfaces in order to reduce the overall energy of the system. In this case, adsorbed formate in a bridging mode (from dissociative adsorption of formic acid with two pairs of electrons) on Pd is the key surface intermediate for the desired dehydrogenation route.8 An electron richer Pd surface favours the rate determining C–H activation of the ‘bridging’ formate on terrace metal sites to give hydrogen and carbon dioxide whereas the linear formate adsorption on high index sites gives rise to CO.8 Thus, we have identified Ag and Bi as effective additives for this surface reaction. On the other hand, should catalysis take place on corner sites rather than terrace sites as the desirable route, Te should be invoked. The competitive adsorption of substrate, CO, dopant, and stabilizer depends on the relative adsorption strength and binding mode. Further differentiating the crude site classifications and elucidating their fundamental catalytic pathways will lead to rational designs of catalysts with superior performance in future.
Notes and references
- D. Astruc, L. Feng and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed
.
- K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS
- S. Jones,
et al.
, Angew. Chem., Int. Ed., 2012, 51, 11275–11278 CrossRef CAS PubMed
- Liu,
et al.
, Phys. Chem. Chem. Phys., 2012, 14, 16415–16423 RSC
- S. C. Tsang,
et al.
, ACS Nano, 2008, 2, 2547–2553 CrossRef CAS PubMed
- M. D. Macia, E. Herrero and J. M. Feliu, Electrochim. Acta, 2002, 47, 3653–3661 CrossRef CAS
- S. Enthaler, ChemSusChem, 2008, 1, 801–804 CrossRef CAS PubMed
- K. Tedsree,
et al.
, Nat. Nanotechnol., 2001, 6, 302–307 CrossRef PubMed
- K. W. Zilm, L. Bonneviot, G. L. Haller, O. H. Han and M. Kermarec, J. Phys. Chem., 1990, 94, 8495–8498 CrossRef CAS
- F. Bonet,
et al.
, Nanostruct. Mater., 1999, 11, 1277–1284 CrossRef CAS
- R. Van Hardeveld and F. Hartog, Surf. Sci., 1969, 15, 189–230 CrossRef CAS
- X. Zhou,
et al.
, Chem. Commun., 2008, 10, 3540–3542 RSC
- F. A. Al-Odail, A. Anastasopoulos and B. E. Hayden, Top. Catal., 2011, 77–82 CrossRef CAS
- M. Wenkin, R. Touillaux, P. Ruiz, B. Delmon and M. Devillers, Appl. Catal., A, 1996, 148, 181–199 CrossRef CAS
- S. Fairclough,
et al.
, J. Phys. Chem. C, 2012, 116, 26898–26907 CAS
- M. Wenkin, P. Ruiz, B. Delmon and M. Devillers, J. Mol. Catal. A: Chem., 2002, 180, 141–159 CrossRef CAS
- J. Yang, J. Yang and J. Ying, ACS Nano, 2012, 6, 9373–9382 CrossRef CAS PubMed
- M. Simões, S. Branton and C. Coutanceau, Appl. Catal., B, 2011, 110, 40–49 CrossRef PubMed
- R. Liu, B. Tesche and H. Knozinger, J. Catal., 1991, 129, 402–413 CrossRef CAS
- F. Gauthard, F. Epron and J. Barbier, J. Catal., 2003, 220, 182–191 CrossRef CAS
- F. M. Hoffman, Surf. Sci. Rep., 1983, 3, 107–193 CrossRef
- P. Strasser,
et al.
, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed
- L. A. Olsen, J. Lopez-Solano, A. Garcia, T. Balic-Zunic and E. Makovicky, J. Solid State Chem., 2010, 183, 2133–2143 CrossRef CAS PubMed
- J. H. Ryu, S. S. Han, D. H. Kim, G. Henkelman and H. M. Lee, ACS Nano, 2011, 5, 8515–8522 CrossRef CAS PubMed
- J. A. Anderson, J. Mellor and R. P. K. Wells, J. Catal., 2009, 261, 208–216 CrossRef CAS PubMed
- L. V. Minevski and R. R. Adzic, J. Appl. Electrochem., 1988, 18, 240–244 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Details of sample preparation, testing and characterisation. See DOI: 10.1039/c4cc06195k |
| This journal is © The Royal Society of Chemistry 2015 |