
Fluorescence polarization immunoassays for monitoring nucleoside triphosphate diphosphohydrolase (NTPDase) activity
Amelie
Fiene
a,
Younis
Baqi
ab,
Joanna
Lecka
cd,
Jean
Sévigny
cd and
Christa E.
Müller
*a
aPharmaCenter Bonn, Pharmaceutical Institute, Pharmaceutical Chemistry I, University of Bonn, An der Immenburg 4, D-53121 Bonn, Germany. E-mail: christa.mueller@uni-bonn.de; Fax: +49 228 732567; Tel: +49 228 732301
bDepartment of Chemistry, Faculty of Science, Sultan Qaboos University, PO Box 36, Postal Code 123, Muscat, Oman
cDépartement de microbiologie-infectiologie et d'immunologie, Faculté de Médecine, Université Laval, Québec, Québec, Canada
dCentre de recherche du CHU de, Québec, Québec, Canada
First published on 21st October 2014
Abstract
The following members of the ecto-nucleoside triphosphate diphosphohydrolase family, NTPDase1 (CD39), NTPDase-2, -3, and -8, play an important role in purinergic signal transduction by regulating extracellular nucleotide levels. Potent and selective NTPDase inhibitors are required as pharmacological tools and have potential as novel drugs, e.g. for anti-cancer and anti-bacterial therapy. We have developed fast and sensitive NTPDase fluorescence polarization (FP) immunoassays using the natural substrates (ATP or ADP). During the NTPDase1-catalyzed reaction, the substrate is dephosphorylated to ADP which is further dephosphorylated yielding AMP as the final product (by NTPDase1). NTPDase3 and -8 yield AMP and ADP, while NTPDase2 results mainly in the formation of ADP. Direct quantification of the respective product, AMP or ADP, is achieved by displacement of an appropriate fluorescent tracer nucleotide from a specific antibody leading to a change in fluorescence polarization. The assays are highly sensitive and can be performed with low substrate concentrations (20 μM ATP or 10 μM ADP) below the KM values of NTPDases, which simplifies the identification of novel competitive inhibitors. Optimized antibody and enzyme concentrations allow the reproducible detection of 2 μM ADP and 1 μM AMP (at 10% substrate conversion). Validation of the assays yielded excellent Z′-factors greater than 0.70 for all investigated NTPDase subtypes indicating high robustness of the analytical method. Furthermore, we tested a standard inhibitor and performed a first exemplary screening campaign with a library consisting of >400 compounds (Z′-factor: 0.87, hit rate 0.5%). Thereby we demonstrated the suitability of the FP assay for IC50 value determination and high-throughput screening in a 384-well format. The new FP assays were shown to be superior to current standard assays.
Introduction
Extracellular nucleosides and nucleotides, such as adenosine, ATP, ADP, UTP and UDP act as signaling molecules in virtually all tissues and organs. They can modulate a variety of functions in the body including thromboregulation, neurotransmission, inflammation, immune response and cell proliferation by activating P1 (adenosine) or P2 (nucleotide) receptors.1,2 To prevent aberrant purinergic signaling events levels of extracellular nucleotides must be tightly controlled.3 Enzymes located at the cell surface hydrolyze nucleotides resulting in the formation of nucleosides and inorganic phosphates (Fig. 1). Ecto-nucleotidases are subdivided into four major groups or families: ecto-5′-nucleotidase (Ecto-5′-NT, CD73), alkaline phosphatases (APs), ecto-nucleotide pyrophosphatase/phosphodiesterases (E-NPPs) and ectonucleoside triphosphate diphosphohydrolases (E-NTPDases). Nucleoside 5′-tri- and di-phosphates (NTPs and NDPs) can be hydrolyzed by members of the E-NTPDase family with different abilities, the E-NPP family (NTPs), and by APs. Nucleoside 5′-monophosphates, such as AMP, are hydrolyzed by APs and Ecto-5′-NT.2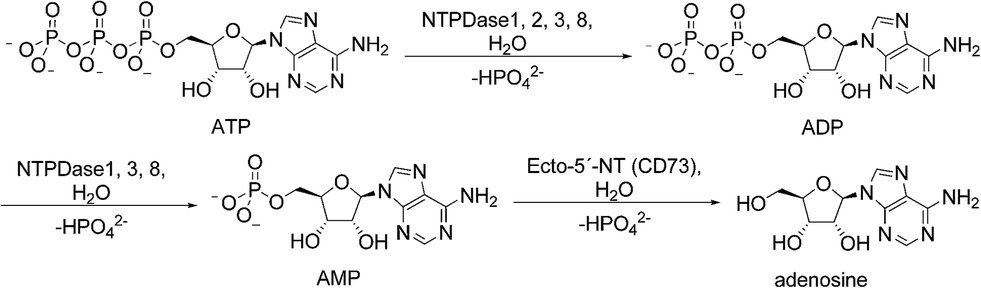 | ||
| Fig. 1 Hydrolysis of adenine nucleotides by ecto-nucleotidases. NTPDases convert ATP to ADP + Pi and ADP to AMP + Pi. Hydrolysis of AMP to adenosine is catalyzed by ecto-5′-NT. | ||
The E-NTPDase family comprises eight members, and of these NTPDase1 (CD39), NTPDase2 (CD39L1), NTPDase3 (CD39L3) and NTPDase8 are located on the cell surface catalyzing the hydrolysis of extracellular nucleotides. They are anchored in the cell membrane by two cell membrane-spanning helical domains. In contrast, NTPDase4-7 are located on intracellular membranes and organelles; secreted forms of NTPDase5 and NTPDase6 have also been reported.2,4 All NTPDases share five conserved sequence domains that are believed to be relevant for their catalytic activity. The E-NTPDase members hydrolyze nucleotides in the presence of millimolar concentrations of Ca2+ or Mg2+ at physiological pH values. They differ in their substrate preferences, e.g. for adenine versus uracil nucleotides, and for nucleoside triphosphates versus diphosphates. NTPDase1, -3 and -8 hydrolyze nucleoside tri- and diphosphates with ATP/ADP ratios of 1–2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 3–4:1 and 2:1, respectively, whereas NTPDase2 shows much higher (10- to 40-fold) preference for ATP over ADP, and therefore produces ADP as its main product.1,5,6 ATP is hydrolyzed by NTPDase1 via ADP to AMP, without significant release of ADP. In contrast, NTPDase2, -3 and -8 hydrolyze ATP to ADP, which is released from the enzyme, and in the case of NTPDase3 and NTPDase8, ADP is subsequently hydrolyzed to AMP.6–10 All NTPDases exhibit Michaelis–Menten kinetics. The KM values for ATP as the main substrate of human NTPDase1, -2 and -3 are 17, 70, and 75 μM, respectively.6 For human NTPDase8 KM values between 81–226 μM have been described.10,11 Since ADP is a poor substrate of NTPDase2, KM values for ADP have only been published for NTPDase1, -3 and -8. They are in the same low micromolar range as those for ATP.6,10,11
:1, 3–4:1 and 2:1, respectively, whereas NTPDase2 shows much higher (10- to 40-fold) preference for ATP over ADP, and therefore produces ADP as its main product.1,5,6 ATP is hydrolyzed by NTPDase1 via ADP to AMP, without significant release of ADP. In contrast, NTPDase2, -3 and -8 hydrolyze ATP to ADP, which is released from the enzyme, and in the case of NTPDase3 and NTPDase8, ADP is subsequently hydrolyzed to AMP.6–10 All NTPDases exhibit Michaelis–Menten kinetics. The KM values for ATP as the main substrate of human NTPDase1, -2 and -3 are 17, 70, and 75 μM, respectively.6 For human NTPDase8 KM values between 81–226 μM have been described.10,11 Since ADP is a poor substrate of NTPDase2, KM values for ADP have only been published for NTPDase1, -3 and -8. They are in the same low micromolar range as those for ATP.6,10,11
NTPDases are expressed in almost every tissue. They are upregulated under certain pathological conditions and therefore represent important potential drug targets for modulating purine receptor-mediated signaling pathways.12 For example, overexpression of Ecto-5′-NT and NTPDases has been described in tumor cells.13 Since ATP can act as a danger signaling molecule, and adenosine on the other hand promotes angiogenesis, tumor growth and immunosuppression, inhibition of NTPDases might be a new therapeutic strategy for the treatment of cancer and immunodeficiency disorders.13–16 Besides mammalian enzymes several important pathogenic microorganisms, such as Legionella pneumophila, have been described to express bacterial NTPDases, which are known to contribute to their virulence.17 Therefore NTPDase inhibitors have been proposed as novel anti-bacterial therapeutics.18 However, the (patho)physiological roles of the enzymes are not completely understood. Potent and selective inhibitors of NTPDases are required as pharmacological tools to investigate their roles in health and disease, and to validate NTPDases as novel drug targets. However, only few NTPDase inhibitors have been described to date. These include nucleotide derivatives and analogs derived from ATP, and non-nucleotidic inhibitors such as the non-selective drugs suramin or reactive blue 2. Therefore, potent and selective drug-like NTPDase inhibitors need to be developed.4,19
To identify novel subtype-selective inhibitors a sensitive and reproducible method, which allows the detection of NTPDase activity with its natural substrate, is required. So far various assays have been described including the malachite green assay, capillary electrophoresis (CE) based assays, luciferase assay, thin-layer-chromatography (TLC) assays and enzyme-coupled assays, all of which suffer from serious drawbacks. The malachite green assay is a colorimetric assay, which allows the detection of inorganic phosphate.20,21 NTPDases produce phosphate as a side-product of the enzymatic reaction. The released phosphate can be detected by the addition of molybdate and malachite green under acidic conditions (absorption at 623 nm).22,23 Increasing enzyme activity leads to an increased production of phosphate and therefore a higher absorption signal. An advantage of this frequently used assay is its easy handling allowing measurement in microtiter plates, which enables high-throughput screening (HTS). Furthermore, the required reagents are inexpensive. However, the assay only detects the side-product phosphate, but not the produced nucleotides. Phosphate contamination, which is common especially in biological samples, leads to a high background signal. The approach requires a high level of substrate consumption to generate an adequate signal-to-noise ratio and consequently does not permit the use of low substrate concentrations. NTPDases display low micromolar KM values. However, screening at substrate concentrations around the KM value would be advantageous for HTS assays. Indeed if high substrate concentrations well above the KM value are employed, the concentration of the competitive inhibitors needs also to be high in order to compete with the substrate. This can cause problems when screening libraries of compounds with moderate solubility or of compound fragments. Moreover, the detection of phosphate complicates measurement of NTPDase activity in biological systems due to the potential release of phosphate from multiple sources resulting in lacking signal specificity. Furthermore, because of the absorption measurement at 623 nm some colored compounds, or natural extracts containing such compounds, may interfere with the assay. Besides, Feng et al. recently discovered that hydrophobic amines, such as quinine and papaverine, cause negative interference with the classical malachite green assay where malachite green is added after molybdate to the acidified reaction solutions of phosphate. Thereby many false-positive results can be generated.24
In recent years CE has emerged as a versatile technique for enzyme assays. It is highly useful for investigating enzymatic reactions involving charged substrates or products, e.g. for the monitoring of dephosphorylation reactions. Its advantages over conventional methods of detecting NTPDase activity include a separation of substrate and product and a low sample volume requirement. It also allows high throughput by automation. Capillary electrophoresis methods for the characterization of recombinant NTPDase1, -2 and -3 and for the testing of NTPDase inhibitors have been developed and recently described by our research group.19,25,26 Nevertheless, this is a time-consuming method, which does not allow the detection of many data points at the same time. Furthermore the moderate sensitivity of CE-UV detection requires the use of high substrate concentration, well above the KM-values, which complicates the identification of competitive NTPDase inhibitors.
Radioactive TLC has also been used for the measurement of NTPDase activity. Its advantage is a very sensitive detection, and the possibility to work with low nanomolar substrate concentrations, but most pharmaceutical companies and many academic institutions try to avoid radioassays because of the associated disposal and regulatory issues.27,28 Furthermore, TLC is a time-consuming and expensive method. Therefore, it is not well suitable for the HTS of large compound libraries.
Luciferase-based assays are frequently used to monitor the consumption of ATP, which is the main substrate of NTPDases. The luminescent luciferase signal guarantees low interference with test compounds. Unfortunately, this approach requires a high level of substrate consumption in order to obtain adequate signal-to-noise ratios. This makes it difficult to work under initial velocity conditions.28–30
Various coupled enzyme assays may be used to monitor NTPDases, for example an enzyme-coupled fluorescent method for the determination of phosphate concentrations by using horseradish peroxidase and 10-acetyl-3,7-dihydroxyphenoxazine (Amplex® Red).27,31 However, in enzyme-coupled assays test compounds may interfere with the employed enzymes. Therefore, such assays do not represent the best choice for HTS campaigns.28,30
To avoid the problems associated with currently applied NTPDase assays we established and validated new assays for screening human NTPDase1, -2, -3 and -8 with a fluorescence polarization (FP) readout. The methodology is generally applicable for ADP-, AMP- or GMP-producing enzymes and has been used for monitoring kinases, phosphodiesterases and heat shock proteins.30,32,33 It enables the direct detection of the enzymatic reaction product ADP when using ATP as a substrate (for NTPDase2, NTPDase3 and NTPDase8) or of AMP upon using ADP as a substrate (for NTPDase1). The fluorescence polarization immunoassay (FPIA)-based assays rely on highly selective antibodies that can distinguish between adenosine mono-, di- and triphosphates. The signal is generated when ADP or AMP produced during the enzymatic reaction displaces a fluorescent tracer nucleotide from the antibody, which leads to a change in its fluorescent properties.34,35 The assays are highly sensitive, and therefore low substrate concentrations below the KM values of NTPDases are applicable. Although the required reagents are more expensive compared to the reagents used in standard assays, the new assays are the first reported NTPDase screening assays, which enable a fast, direct detection of the enzymatic reaction products in a homogenous format and makes them therefore ideally suitable for HTS.
Materials and methods
Chemicals and buffers
Mouse monoclonal ADP antibody (3 mg mL−1), monoclonal AMP/GMP antibody (1.2 mg mL−1), ADP Alexa Fluor® 633 tracer (400 nM), AMP/GMP Alexa Fluor® 633 tracer (800 nM) and ADP (5 mM) were provided by BellBrook Laboratories (Madison, WI).34,35 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) was obtained from AppliChem (Darmstadt, Germany), Tris(hydroxymethyl)aminomethane (Tris) from Carl Roth GmbH (Karlsruhe, Germany). ATP (100 mM), Brij® L23, calcium chloride, dimethyl sulfoxide (DMSO) and ethylenediaminetetraacetic acid (EDTA) were purchased from Sigma-Aldrich (Steinheim, Germany). Sodium 1-amino-4-(1-naphthylamino)-9,10-dioxo-9,10-dihydroanthracene-2-sulfonate (PSB-06126) was synthesized as previously described.19 The assay buffer consisted of 5 mM CaCl2 and 80 mM Tris with the pH value adjusted to 7.4. The detection reagent buffer consisted of 200 mM HEPES, 400 mM EDTA and 0.2% Brij® L23 with the pH value adjusted to 7.5.Instruments
FP measurement was performed on a BMG PheraStar FS plate reader (BMG Labtech GmbH, Ortenberg, Germany) at an excitation wavelength of 590 nm and an emission wavelength of 675 nm (50 nm bandwidth). The free tracer reference was set to 20 mP. Black 384-well flat bottom small volume microplates with a non-binding surface from Greiner Bio-One GmbH (Frickenhausen, Germany) were used.Transfection and membrane preparations of human NTPDases
COS-7 cells were transiently transfected with a plasmid encoding either human NTPDase1, human NTPDase2, human NTPDase3, or human NTPDase8. Cell membranes were prepared as described elsewhere.36,37 Briefly, COS-7 cells (in 15 cm dishes) were transfected with an expression vector (pcDNA3) incorporating the cDNA that encodes each ecto-nucleotidase using Lipofectamine (Invitrogen) and harvested 40–72 h later. For the preparation of protein extracts, transfected cells were washed three times with 95 mM NaCl and 45 mM Tris, pH 7.5, at 4 °C, collected by scraping using the same buffer supplemented with 0.1 mM PMSF and washed twice by centrifugation (300 × g, 10 min, 4 °C). The cells were then resuspended in harvesting buffer supplemented with 10 mg mL−1 aprotinin and sonicated. Nucleus and cellular debris were discarded after another centrifugation (300 × g, 10 min, 4 °C) and the resulting supernatant (hereinafter called protein extract) was aliquoted and stored at −80 °C until use. Protein concentration was estimated by a Bradford microplate assay using bovine serum albumin as a standard.38Preparation of standard solutions and detection reagents
Nucleotide solutions at concentrations equal to 2.5× final substrate concentration of 20 μM ATP or 10 μM ADP in assay buffer were prepared. A 10 mM stock solution of PSB-06126 (sodium 1-amino-4-(1-naphthylamino)-9,10-dioxo-9,10-dihydroanthracene-2-sulfonate) in DMSO was prepared. It was further diluted to a 1 mM solution in 10% aq. DMSO, and an appropriate serial dilution in 10% aq. DMSO was prepared. An in-house drug library (http://www.pharma.uni-bonn.de/www/pharmchem1-en/mueller-laboratory/compound-library) consisting of 438 diverse compounds addressing many different targets in 10% aq. DMSO (100 μM) was prepared in 96-well plates, each well containing a single compound. The ADP detection reagent was prepared by adding ADP antibody (final concentration 23 μg mL−1), ADP Alexa 633 tracer (final concentration 4 nM) and the detection reagent buffer (final concentration of 20 mM HEPES, 40 mM EDTA and 0.02% Brij-35, pH 7.5) to distilled water. The AMP/GMP detection reagent was prepared by adding AMP/GMP antibody (final concentration 8 μg mL−1), AMP/GMP Alexa 633 tracer (final concentration 8 nM) and the detection reagent buffer (final concentration of 20 mM HEPES, 40 mM EDTA and 0.02% Brij-35, pH 7.5) to distilled water.Enzyme fluorescence polarization assay procedure
Substrate solutions, either 4 μL ATP (50 μM) or ADP (25 μM) and 2 μL of 10% aq. DMSO, or inhibitor solution, respectively, were added to a well of a 384-well microplate (final concentrations of substrate and DMSO were 20 μM ATP, or 10 μM ADP, and 2% DMSO). The final DMSO concentration was 2% to facilitate screening of compounds that possess low water-solubility. The reaction was initiated by adding 4 μL of the appropriate enzyme suspension in assay buffer to each test well. The plate was incubated for 10 min at 37 °C. To stop the reaction 10 μL of the appropriate detection reagent containing optimal antibody and tracer concentration were added to each well. The FP signals were read on the plate reader after equilibration for 1 h (ADP assay) or 2 h (AMP assay) at room temperature (with orbital shaking).Antibody optimization
Due to the linear correlation between ATP concentration and ADP antibody concentration the quantity of ADP antibody required for enzyme reactions can be determined using the equation y = mx + b, where x = ATP concentration (μM) in the 10 μL enzyme reaction, y = ADP antibody concentration (μg mL−1) in the detection reagent, m (slope) = 1.08, and b (y-intercept) = 1.0.34To determine the optimal antibody concentration for using the AMP assay by working under initial velocity conditions an antibody titration was performed in the presence of an initial substrate concentration of 10 μM ADP (0% conversion) and additionally in the presence of a 9:1 ratio of substrate:product (9 μM ADP:1 μM AMP) which mimics 10% conversion.30 Twelve different concentrations of antibody ranging from 0.061 to 125 μg mL−1 were used in combination with 8 nM of AMP tracer. The appropriate detection reagent (10 μL) was added to 10 μL of nucleotide solution in a well of a 384-well plate (in duplicates). The assay was performed as described above and the results were plotted as mP versus log antibody concentration using sigmoidal dose-response (variable slope) curve fitting (GraphPad Prism 4, GraphPad Software Inc., San Diego, CA, USA).
Standard curves
Standard curves were determined using decreasing amounts of the substrate ATP or ADP and increasing amounts of the product ADP or AMP. The nucleotide mixtures represented 0%, 0.5%, 1%, 2%, 3%, 5%, 7.5%, 10%, 12.5%, 15%, 20%, 50% and 100% of substrate conversion. To 10 μL of the nucleotide substrate–product solution 10 μL of the appropriate detection reagent were added and the plates were incubated as described above before fluorescence polarization measurements. The polarization values (mP) were plotted against ADP or AMP concentrations, respectively (half-logarithmic representation).Enzyme titrations
For determination of the optimal enzyme concentration, titrations were performed in the presence of 20 μM ATP (human NTPDase2, human NTPDase3 or human NTPDase8) or 10 μM ADP (human NTPDase1). Therefore different enzyme concentrations were incubated with the substrate and 10% DMSO (final concentration 2%) for 10 min at 37 °C in a 384-well microplate. The assays were further performed as described above and the results were fitted to a non-linear regression curve using GraphPad Prism. To determine the optimal enzyme concentration, the obtained polarization values for different enzyme concentrations were inserted into the appropriate standard curves and the amount of formed product was calculated. The enzyme concentrations that lead to ≤10% conversion of substrate were used for further experiments.Assay validation
The validation of the assay performance for each enzyme was quantified by calculation of the Z′-factor using the formula of Zhang et al.:39where σ+ and σ− are the standard deviations and μ+ and μ− are the mean values of the positive and negative controls, respectively. A series of negative and positive controls was measured. For each, positive and negative controls, 16 wells were analyzed. For the positive controls 4 μL of the appropriate NTPDase were added to 2 μL of 10% DMSO solution and 4 μL of substrate solution. After the enzymatic reaction time 10 μL of the detection reagent was added. For the negative controls 4 μL of the appropriate enzyme solution was added to 2 μL 10% DMSO solution and 4 μL of substrate solution in a well which already contained 10 μL of the detection reagent (enzyme reaction was stopped immediately). The assay was performed as described above in three separate experiments and the Z′-factors were calculated.
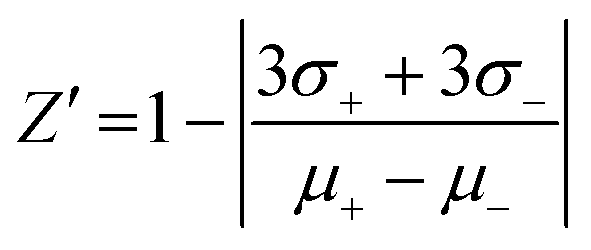
The plate was incubated for 10 min at 37 °C. To stop the reaction 10 μL of the appropriate detection reagent containing optimal antibody and tracer concentration were added to each well.
Enzyme inhibition assays
A solution of the NTPDase inhibitor PSB-06126 (2 μL in 10% aq. DMSO) was added to 4 μL ATP solution in a well of a 384-well plate. To start the enzymatic reaction 4 μL of human NTPDase3 suspension were added. The enzymatic reaction was stopped as described above. Three separate inhibition experiments were performed, each in duplicates. The concentration–inhibition curve was fitted using GraphPad Prism 4, the IC50 value was determined and the Ki value was calculated by using the Cheng–Prusoff equation.40Screening of a drug library
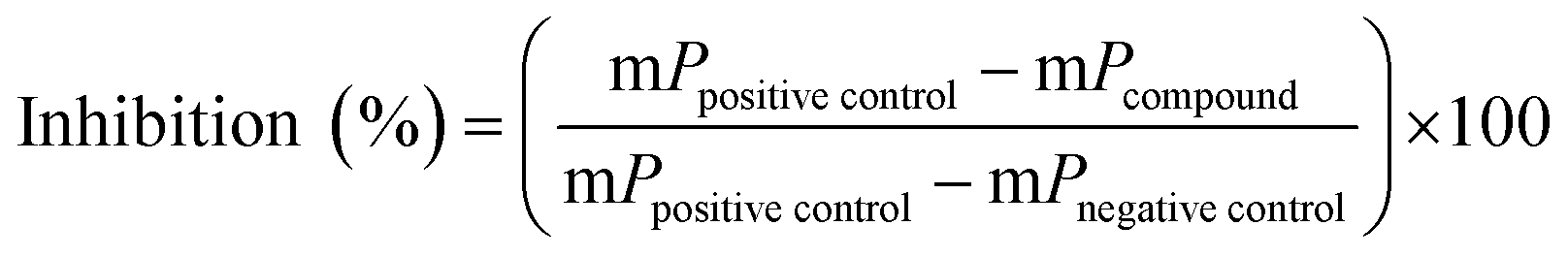
For screening of a small drug library (438 compounds) 4 μL of human NTPDase3 suspension were added to a well of a 384-well plate containing 2 μL compound solution in 10% aq. DMSO (100 μM) and 4 μL substrate solution, resulting in a final concentration of 20 μM test compound (2% DMSO) and 20 μM ATP. Positive controls containing only 10% DMSO (no inhibitor) and negative controls containing inactivated enzyme by addition of the detection reagent at the beginning of the 37 °C incubation period, were included. The enzymatic reaction was stopped as described above, and after fluorescence polarization measurements the percent inhibition was calculated using the formula below:where mP is milli polarization value.
The Z′-factor was determined according to the equation described above.
Results and discussion
Enzyme fluorescence polarization assay
All NTPDase screening assays described so far show many drawbacks and are not ideally suited for HTS campaigns. In the present study we report on the establishment of a new assay procedure for the detection of NTPDase activity using a fluorescence polarization methodology. Fluorescence polarization is based on the assessment that molecule size affects the polarization of fluorescence emission.41 When a fluorescent molecule is excited with plane-polarized light, the intensity of the emitted light can be monitored in vertical and horizontal planes to the excitation plane and the resulting polarization value (P) can be calculated using the following formula:where Ivertical is the fluorescence intensity measured in the vertical and Ihorizontal the fluorescence intensity in the horizontal plane. The polarization value is often expressed in millipolarization (mP) values and is low when a fluorescence molecule is attached to a small structure, and high when attached to a large molecule based on their different rotation rates during the excited state.28,41,42 This fact provides the basis for the newly established NTPDase screening assays using ATP or ADP as a substrate. The detection reagent consists of an Alexa Fluor 633-labeled ADP or AMP bound to an ADP or AMP antibody, respectively, leading to the measurement of high mP values (bound tracer = large molecule).34,35 During the enzymatic reaction unlabeled product ADP when using ATP as a substrate, for human NTPDase2, -3 and -8, or AMP when using ADP as a substrate, for human NTPDase1, is generated and displaces the tracer from the antibody leading to a decrease in fluorescence polarization (free tracer = small molecule).34,35 The choice of an Alexa Fluor 633 tracer in the far red wavelength range allows to overcome interference of test compounds due to autofluorescence and/or light scatter.43 A general procedure for the NTPDase fluorescence polarization assays is shown in Fig. 2. In a first step the enzymatic reaction is started in a 384-well plate with a final volume of 10 μL in each test well. In a second step the reaction is stopped by addition of the appropriate detection reagent. After the equilibration time (third step) of 1 h (ADP assay) or 2 h (AMP assay) the polarization signal can be measured. The procedure shows that these newly developed FP-based assays easily allow the direct quantification of the enzymatic reaction products ADP or AMP in a 384-well format. The possibility to directly monitor ADP and AMP formation makes these assays also well suitable for the screening of a variety of enzymes including other nucleotide-metabolizing enzymes, as well as kinases and phosphodiesterases. Only low amounts of enzyme suspension, substrate and test compound are required, which indicates the suitability of the assays for the screening of large compound libraries where enzyme preparation and/or compounds are valuable. Stopping of the enzymatic reaction by the addition of the EDTA-containing detection reagent is more gentle than the addition of sulfuric acid (e.g. in the malachite green assay) or by heating of the reaction mixture for denaturation of the protein (e.g. in CE-assays), and therefore less background signals occur in the presented assay procedure. Moreover, only short equilibration times of the samples and the detection reagent are required, which is an advantage for HTS in comparison to more time-consuming analytical methods (e.g. CE- or HPLC-assays). In order to achieve a sensitive, robust and reproducible method for the screening of NTPDases, several parameters had to be carefully optimized.
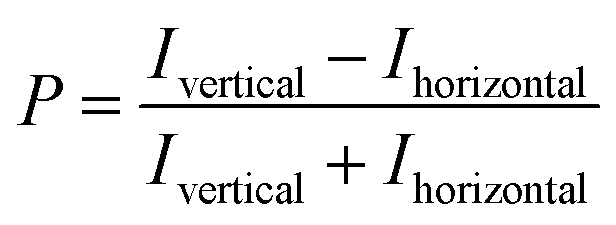
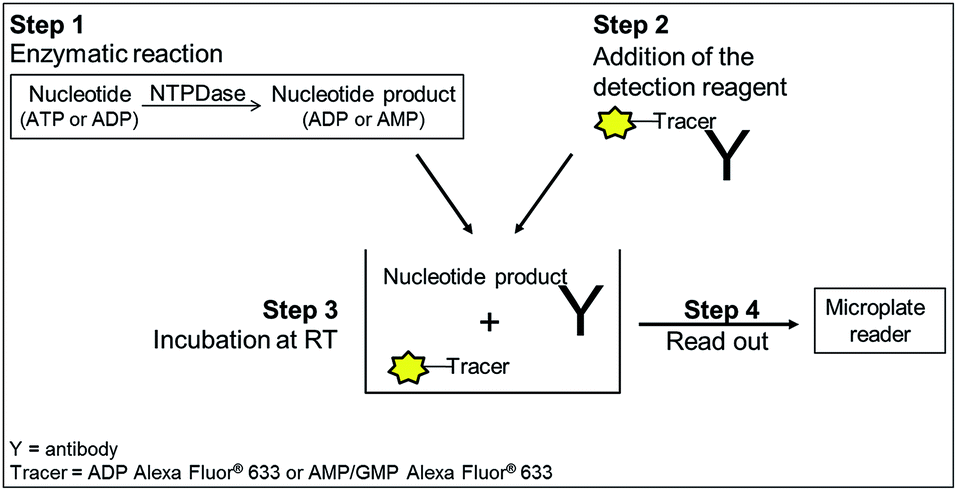 | ||
| Fig. 2 General procedure for NTPDase fluorescence polarization assays. Step 1: incubation of enzyme and substrate; step 2: detection reagent containing tracer and antibody is added; step 3: equilibration for 1 h (ADP assay) or 2 h (AMP assay) at room temperature. The tracer is displaced by the nucleotide generated during the enzymatic reaction; step 4: read-out of the microplate. The displaced tracer can rotate faster (small molecule), which leads to a decrease in fluorescence polarization. | ||
Optimal antibody concentrations
The antibody concentration is the main variable controlling the total polarization shift and the assay window in the novel fluorescence polarization assays.30 Therefore optimal antibody concentrations for using the ADP and AMP assays have to be determined in the presence of the relevant substrate. The substrate concentrations of ATP and ADP were set at 20 μM and 10 μM, respectively. These low substrate concentrations were chosen, as they are below the described KM values of all NTPDases. An optimal ADP antibody concentration of 23 μg mL−1 was calculated for a substrate concentration of 20 μM ATP. Using this antibody concentration a substrate conversion of 10% (initial velocity conditions) resulted in an adequate assay window (shift of close to 159 mP).In order to determine the optimal antibody concentration for the AMP assay an antibody titration was performed in the presence of 0% conversion of substrate (10 μM ADP), and 10% conversion of substrate (9 μM ADP and 1 μM AMP), respectively. The 10% conversion rate of substrate simulates the amount of product formation expected in the enzyme assay. The measured polarization values were plotted against different antibody concentrations resulting in sigmoidal titration curves (Fig. 3A). The difference between the polarization values (ΔmP) of both curves was also plotted against the antibody concentration. An antibody concentration of 8 μg mL−1 resulted in a maximal difference and was used for further experiments (Fig. 3B). A substrate conversion of 10% resulted in a shift of close to 176 mP and thus leads to an adequate assay window.
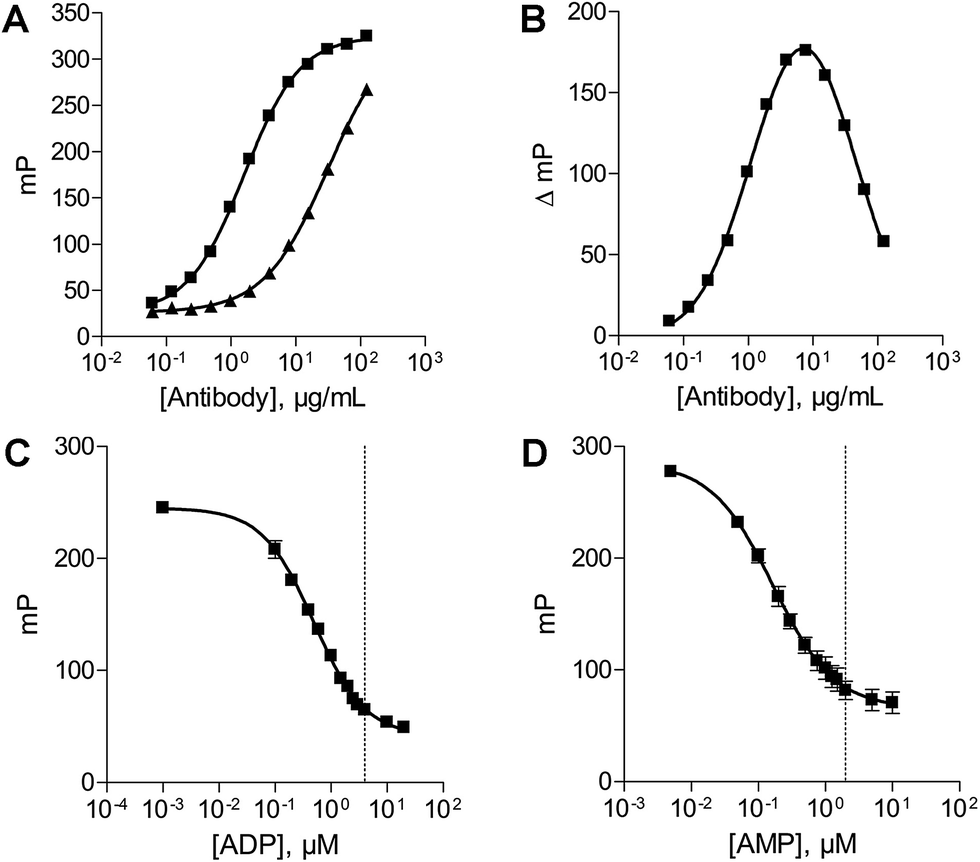 | ||
| Fig. 3 Optimization of the assay parameters. (A) Antibody titration curve in the presence of 10 μM ADP (■) and 9 μM ADP/1 μM AMP (▲). (B) Difference plot of the two antibody titration curves; the maximal difference was determined to be 8 μg mL−1. (C) ATP/ADP standard curve (20 μM); dotted line: 20% conversion of substrate. (D) ADP/AMP standard curve (10 μM); dotted line: 20% conversion of substrate. | ||
By using the optimal antibody concentration the assays provide a very sensitive method for detecting enzyme activity working under initial velocity conditions with a good detection range. It is possible to use the assays for every substrate concentration of interest between 0.1 μM–1000 μM for ATP and 1 μM–1000 μM for ADP, because the optimal antibody concentration can easily be adapted either by calculating (ADP assay) or by determining it by titration (AMP assay).34,35 The possibility to work with low substrate concentrations below the KM values of NTPDases simplifies the identification of competitive inhibitors in screening campaigns and will allow the screening of compounds with moderate solubility. Moreover, it will allow fragment-based screening, an emerging strategy in drug discovery, by which useful starting points for lead structure identification can be discovered from a library of low molecular weight compounds.44
Standard curves
After determination of the optimal antibody concentrations, standard curves were generated to show a correlation between product formation and FP signal. Based on these curves quantification of the enzymatic reaction products ADP (for the substrate ATP) and AMP (for the substrate ADP) can be performed. Two standard curves for each initial substrate concentration of 20 μM ATP or 10 μM ADP, which mimic the enzyme reactions by decreasing the amounts of substrate and increasing the amounts of product, were prepared. A linear relationship could be shown for substrate conversions up to 20% (dotted line) (Fig. 3C and D), which allows for the calculation of product formation from polarization values. Only a low substrate conversion is required for an adequate polarization shift, which allows working under initial velocity conditions and demonstrates the high sensitivity of these assays.Enzyme titration
In order to determine the optimal enzyme concentrations upon working under initial velocity conditions (<10% substrate conversion) enzyme titrations were performed at the selected substrate concentrations of 20 μM ATP (human NTPDase2, human NTPDase3 and human NTPDase8), or 10 μM ADP (human NTPDase1), respectively. The polarization values (mP) were plotted against the enzyme concentration (Fig. 4). The amount of nucleotide product for each enzyme concentration was calculated by using the standard curves and was further fitted against enzyme concentrations. A linear increase in product formation in relation to increasing enzyme concentrations was observed (Fig. 5). Enzyme concentrations which led to <10% conversion (dotted lines) of 20 μM ATP were determined to be lower than 0.5 ng μL−1 (NTPDase2), 0.5 ng μL−1 (NTPDase3), and 5.0 ng μL−1 (NTPDase8), and for 10 μM ADP to be lower than 1.0 ng μL−1 (NTPDase1). For each enzyme a polarization change of at least 130 mP was observed during the linear phase of the reaction, which reflects an adequate assay window.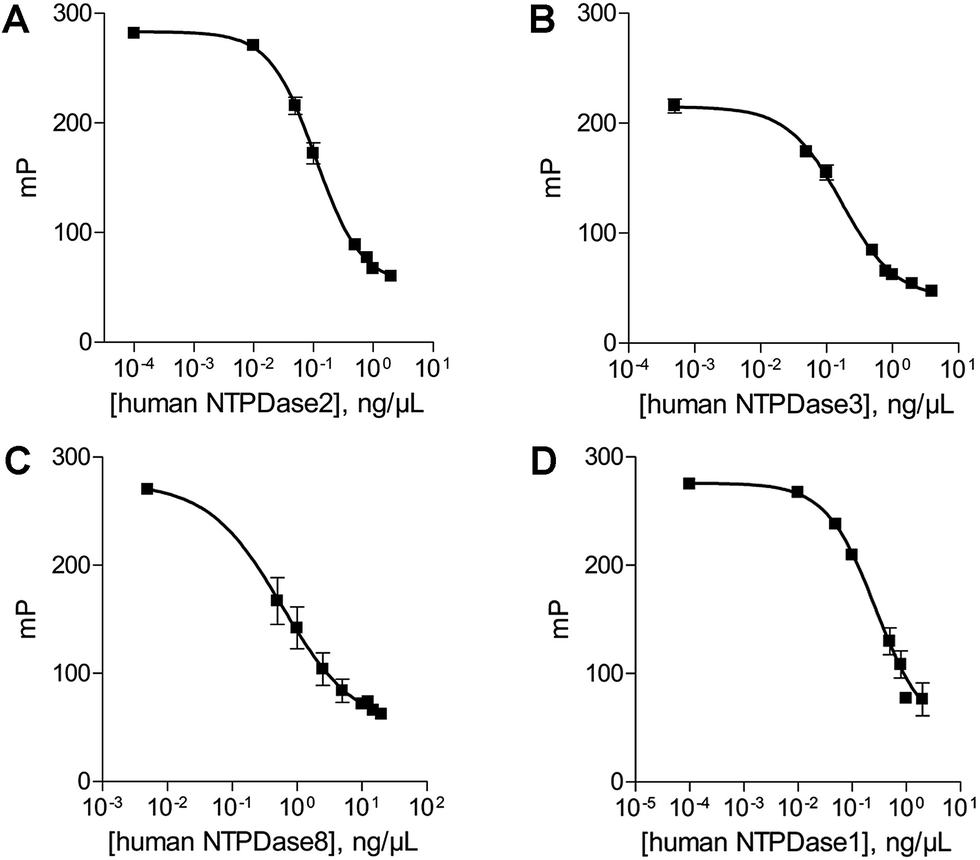 | ||
| Fig. 4 Enzyme titration curves in the presence of 20 μM ATP ((A) human NTPDase2; (B) human NTPDase3; (C) human NTPDase8) and 10 μM ADP ((D) human NTPDase1). | ||
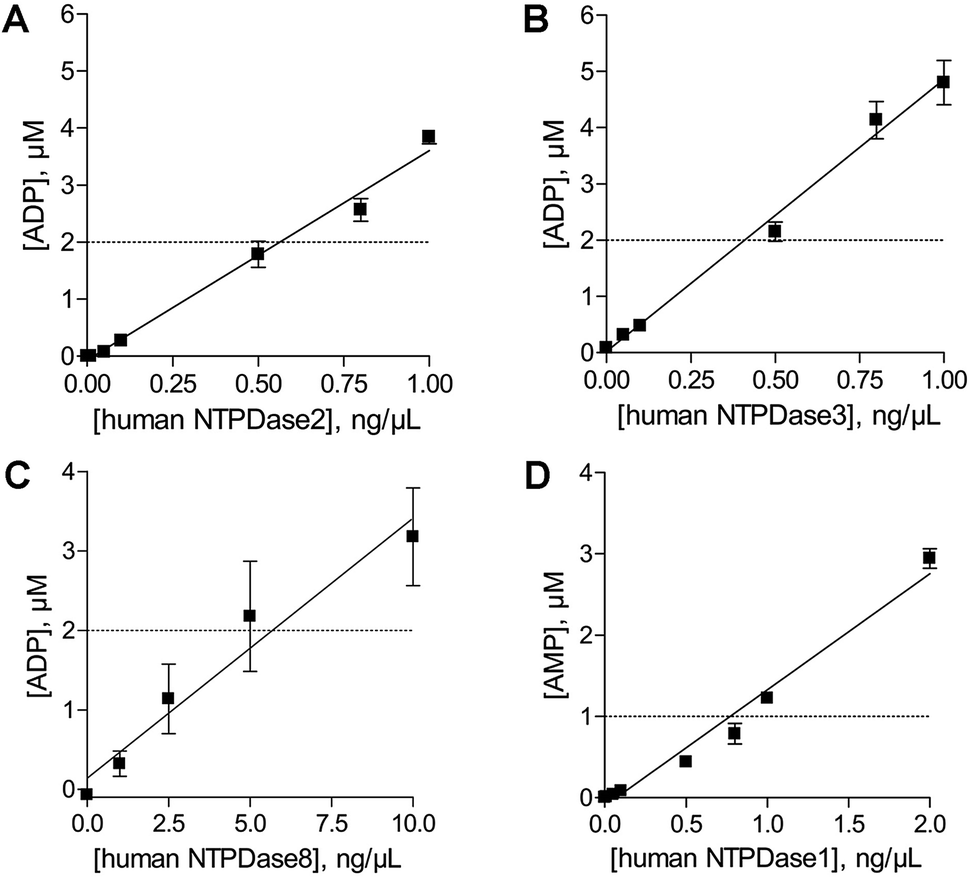 | ||
| Fig. 5 The amount of product generated during the enzymatic reaction was plotted against the enzyme concentration; dotted line: 10% product formation of ADP ((A) human NTPDase2; (B) human NTPDase3; (C) human NTPDase8) and AMP ((D) human NTPDase1). | ||
Assay validation
After optimization of the assay parameters we evaluated the assays with regard to their suitability for HTS. The Z′-factors were calculated for assay validation according to the formula published by Zhang and coworkers.39 The Z′-factor is a characteristic statistical parameter, which is suitable for assay quality assessment. It takes full account of the variability in the measurements (positive and negative controls) and the signal dynamic range. To decide if an assay is suitable for HTS without further optimization, a Z′-factor of greater than 0.5 is required.39 The Z′-factors determined for the NTPDase assays measuring ADP were 0.77 (for NTPDase2), 0.70 (for NTPDase3), and 0.76 (for NTPDase8). For the assay measuring AMP a Z′-factor of 0.71 (for NTPDase1) was obtained. Thus, the assays show excellent robustness and reproducibility and are therefore well suitable for high-throughput screening.Enzyme inhibition assays
For further validation of the FP assays enzyme inhibition studies were exemplarily performed with the FP-based NTPDase3 assay using the anthraquinone derivative PSB-06126 as a standard E-NTPDase inhibitor (Fig. 6).19 Anthraquinone derivatives with similarity to reactive blue 2 (RB-2) have been shown to be moderately potent inhibitors of NTPDases. PSB-06126 was reported to inhibit rat NTPDase3 at low micromolar concentrations.19 The inhibition curve determined by the new FPIA-based assay is shown in Fig. 6. An IC50 value of 7.76 ± 0.88 μM (n = 3) was determined for PSB-06126 at human NTPDase3, and a Ki value of 4.39 ± 0.50 μM could be calculated according to the Cheng–Prusoff equation assuming a competitive mechanism of inhibition as previously described for PSB-06126 at the rat enzyme.19 For comparison, the CE-based assay had yielded a Ki value of 1.50 ± 0.10 μM for PSB-06126 at rat NTPDase3. These results show that the newly developed and optimized assay method is suitable for detecting enzyme inhibition and leads to reliable results, as the determined Ki value is well in agreement with the published value at the closely related rat enzyme.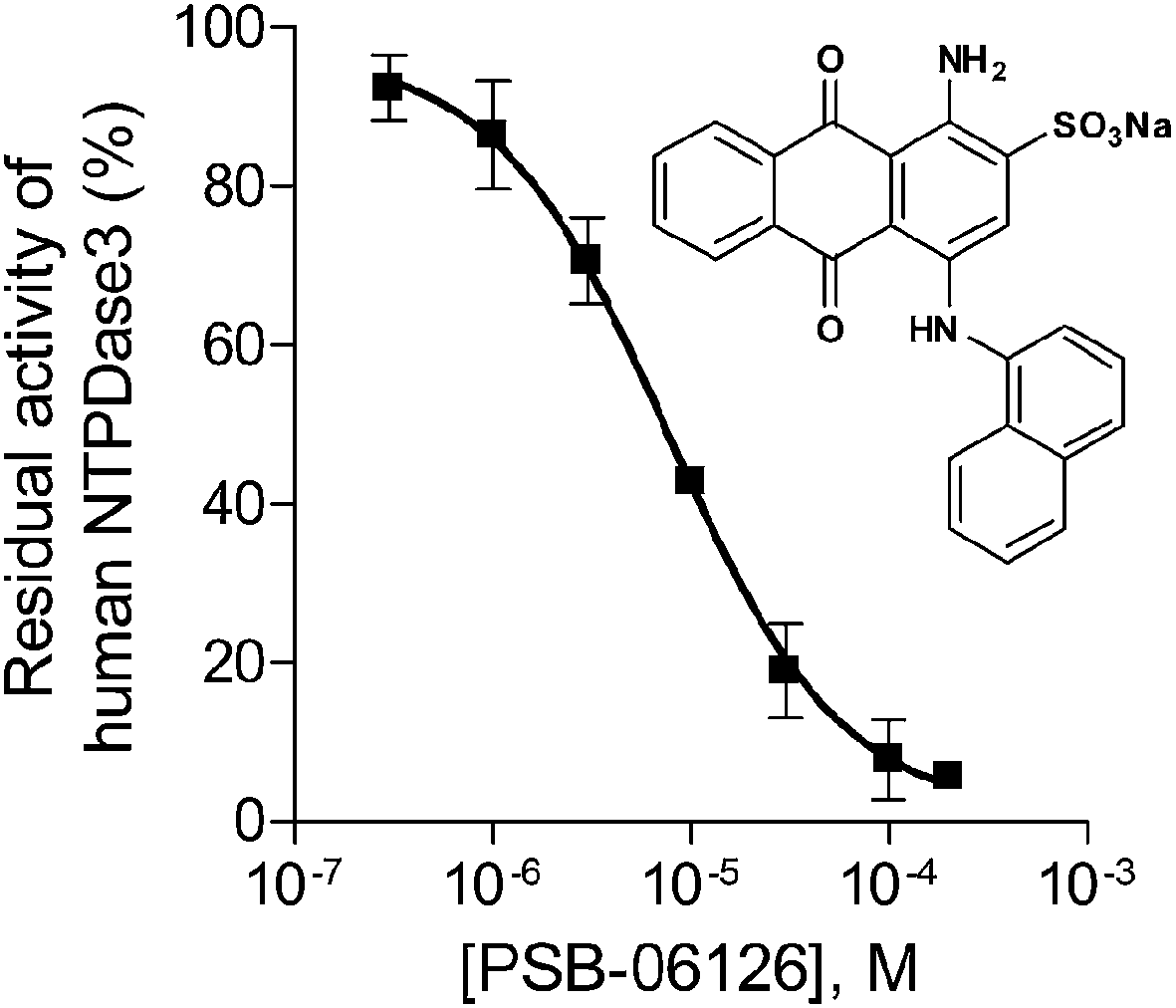 | ||
| Fig. 6 Concentration–inhibition curve of PSB-06126 at human NTPDase3 (IC50 = 7.76 ± 0.88 μM, n = 3) determined by the FPIA-based NTPDase3 assay using ATP (20 μM) as substrate. | ||
Screening of a drug library
Since the development of potent and selective NTPDase inhibitors has not been successful so far, HTS of compound libraries could be a promising approach for the identification of novel scaffolds. To this end, we used the newly established FP-based NTPDase3 assay for the screening of a small library consisting of 438 structurally diverse drugs. They were tested at a final concentration of 20 μM. Compounds that showed at least 30% inhibition at 20 μM concentration were considered as hit compounds. The results are shown in Fig. 7. Two compounds were identified as inhibitors of human NTPDase3 (0.5% hit rate) and could be confirmed in a second confirmatory screen (data not shown). These compounds may represent useful starting points for the development of novel NTPDase inhibitors. These results, along with the determined Z′-factor of 0.87, show that the FP-based assay is suitable for compound library screening and the identification of novel inhibitors. Advantageously, the assay is very specific due to the direct detection of the enzymatic reaction product. The new FP-assay is expected to produce less false-positive results in HTS, than typically obtained in the current standard assays.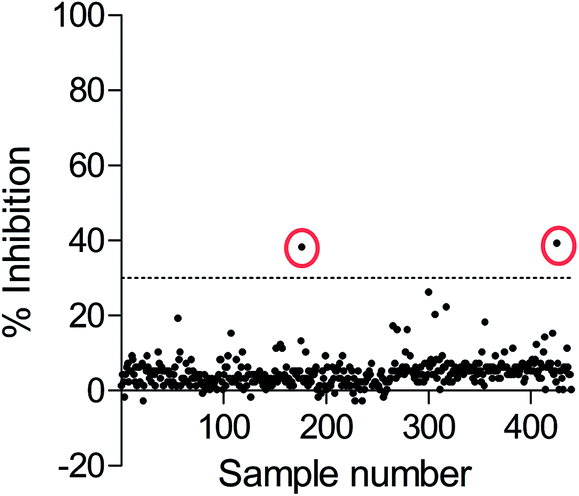 | ||
| Fig. 7 High-throughput screening results of FPIA-based NTPDase3 assay using ATP as a substrate. A small compound library consisting of 438 compounds was screened at 20 μM concentration, enzyme inhibition (%) was calculated for each compound; dotted line: hit threshold of 30% inhibition (Z′-factor: 0.87). Two hit compounds (encircled points) were identified showing significant inhibition of NTPDase3. | ||
Conclusions
NTPDases are of increasing interest as attractive therapeutic targets for interfering with purine receptor-mediated signaling pathways. Novel FP-based ADP and AMP assays for detecting enzyme activity using recombinant NTPDases were therefore successfully established and validated (Z′-factor ≥0.70). They are very fast and highly sensitive, allowing the usage of substrate concentrations as low as 0.1 μM ATP, or 1 μM ADP, respectively. Thus, they allow to perform screening with substrate concentrations around or even below the KM value of the NTPDases.34,35 This simplifies the identification of competitive inhibitors and enables fragment-based screening approaches. In a first exemplary screening campaign >400 drugs were investigated for their potential to inhibit human NTPDase3 (Z′-factor: 0.87) and two compounds were identified and confirmed as inhibitors (0.5% hit rate). This demonstrates the suitability of the assay for compound library screening and the identification of novel inhibitors. The new FP assays show significant advantages in comparison with previously reported and utilized assays for monitoring NTPDase reactions. A direct detection and quantification of the products ADP, or AMP, respectively, is feasible in a 384-well format and drawbacks of other assay methods are avoided. A far-red shifted fluorophor (Alexa 633) is used, which leads to low assay interference from compound fluorescence and light scatter and therefore gives an excellent specificity and performance in FP assays.43 Moreover, these assays can be used for detecting any ADP, AMP or GMP producing enzymes including other ecto-nucleotidases (e.g., APs and E-NPPs) and further nucleotide-metabolizing enzymes (e.g. soluble calcium-activated nucleotidase). They are therefore a promising technology for the screening of a variety of enzymes where low substrate concentrations have to be used and novel inhibitors are still lacking. A major advantage is that they allow the use of the natural substrates (like ATP or ADP) instead of artificial ones, which might produce results of questionable relevance. Finally, the low sample volume saves expensive enzyme preparations and test compounds. The newly developed FPIA assays will be highly useful for the identification and characterization of potent and selective NTPDase inhibitors which may have potential as drugs, e.g. for the treatment of cancer and immunodeficiency disorder, and as novel anti-bacterial therapeutics.Abbreviations
| Aq | Aqueous |
| CD | Cluster of differentiation |
| CE | Capillary electrophoresis |
| DMSO | Dimethyl sulfoxide |
| EDTA | Ethylenediaminetetraacetic acid |
| Ecto-5′-NT | Ecto-5′-nucleotidase |
| E-NTPDase | Ecto-nucleoside triphosphate diphosphohydrolase |
| FP | Fluorescence polarization |
| FPIA | Fluorescence polarization immunoassay |
| HTS | High-throughput screening |
| IC50 | Half maximal inhibitory concentration |
| K i | Inhibition constant |
| K M | Michaelis constant |
| mP | Milli polarization value |
| P | Polarization value |
| RB-2 | Reactive Blue 2 |
| TLC | Thin-layer chromatography |
| Tris | Tris(hydroxymethyl)aminomethane. |
Notes and references
- G. G. Yegutkin, Biochim. Biophys. Acta, 2008, 1783, 673–694 CrossRef CAS PubMed.
- H. Zimmermann, M. Zebisch and N. Sträter, Purinergic Signalling, 2012, 8, 437–502 CrossRef CAS PubMed.
- J. P. Vivian, P. Riedmaier, H. Ge, J. Le Nours, F. M. Sansom, M. C. J. Wilce, E. Byres, M. Dias, J. W. Schmidberger, P. J. Cowan, A. J. F. D'Apice, E. L. Hartland, J. Rossjohn and T. Beddoe, Structure, 2010, 18, 228–238 CrossRef CAS PubMed.
- S. C. Robson, J. Sévigny and H. Zimmermann, Purinergic Signalling, 2006, 2, 409–430 CrossRef CAS PubMed.
- H. Zimmermann, Naunyn-Schmiedeberg's Arch. Pharmacol., 2000, 362, 299–309 CrossRef CAS.
- F. Kukulski, S. A. Lévesque, E. G. Lavoie, J. Lecka, F. Bigonnesse, A. F. Knowles, S. C. Robson, T. L. Kirley and J. Sévigny, Purinergic Signalling, 2005, 1, 193–204 CrossRef CAS PubMed.
- P. Heine, N. Braun, A. Heilbronn and H. Zimmermann, Eur. J. Biochem., 1999, 262, 102–107 CrossRef CAS.
- T. Vorhoff, H. Zimmermann, J. Pelletier, J. Sévigny and N. Braun, Purinergic Signalling, 2005, 1, 259–270 CrossRef CAS PubMed.
- E. G. Lavoie, F. Kukulski, S. A. Lévesque, J. Lecka and J. Sévigny, Biochem. Pharmacol., 2004, 67, 1917–1926 CrossRef CAS PubMed.
- M. Fausther, J. Lecka, F. Kukulski, S. A. Lévesque, J. Pelletier, H. Zimmermann, J. A. Dranoff and J. Sévigny, Am. J. Physiol.: Gastrointest. Liver Physiol., 2007, 292, 785–795 CrossRef PubMed.
- A. F. Knowles and C. Li, Biochemistry, 2006, 45, 7323–7333 CrossRef CAS PubMed.
- M. Al-Rashida and J. Iqbal, Med. Res. Rev., 2013, 34, 703–743 CrossRef PubMed.
- J. Stagg and M. J. Smyth, Oncogene, 2010, 29, 5346–5358 CrossRef CAS PubMed.
- L. Wang, S. Tang, Y. Wang, S. Xu, J. Yu, X. Zhi, Z. Ou, J. Yang, P. Zhou and Z. Shao, Clin. Exp. Metastasis, 2013, 5, 671–680 CrossRef PubMed.
- J. Spychala, Pharmacol. Ther., 2000, 87, 161–173 CrossRef CAS.
- L. S. Bergamin, E. Braganhol, R. F. Zanin, M. I. A. Edelweiss and A. M. O. Battastini, J. Biomed. Biotechnol., 2012, 2012, 1–10 CrossRef PubMed.
- F. M. Sansom, H. J. Newton, S. Crikis, N. P. Cianciotto, P. J. Cowan, A. J. F. D'Apice and E. L. Hartland, Cell. Microbiol., 2007, 9, 1922–1935 CrossRef CAS PubMed.
- F. M. Sansom, P. Riedmaier, H. J. Newton, M. A. Dunstone, C. E. Müller, H. Stephan, E. Byres, T. Beddoe, J. Rossjohn, P. J. Cowan, A. J. F. D'Apice, S. C. Robson and E. L. Hartland, J. Biol. Chem., 2008, 283, 12909–12918 CrossRef CAS PubMed.
- Y. Baqi, S. Weyler, J. Iqbal, H. Zimmermann and C. E. Müller, Purinergic Signalling, 2009, 5, 91–106 CrossRef CAS PubMed.
- I. Koichi and U. I. Michio, Clin. Chim. Acta, 1965, 14, 361–366 Search PubMed.
- A. A. Baykov, O. A. Evtushenko and S. M. Avaeva, Anal. Biochem., 1988, 171, 266–270 CrossRef CAS.
- Life Technologies online, http://www.lifetechnologies.com/de/de/home/life-science/cell-analysis/labeling-chemistry/fluorescence-spectraviewer.html#product=M689, accessed 3 July 2014.
- E. B. Cogan, G. B. Birrell and O. H. Griffith, Anal. Biochem., 1999, 271, 29–35 CrossRef CAS PubMed.
- J. Feng, Y. Chen, J. Pu, X. Yang, C. Zhang, S. Zhu, Y. Zhao, Y. Yuan, H. Yuan and F. Liao, Anal. Biochem., 2011, 409, 144–149 CrossRef CAS PubMed.
- J. Iqbal, P. Vollmayer, N. Braun, H. Zimmermann and C. E. Müller, Purinergic Signalling, 2005, 1, 349–358 CrossRef CAS PubMed.
- J. Iqbal, PhD thesis, University of Bonn, 2005.
- M. Helenius, S. Jalkanen and G. Yegutkin, Biochim. Biophys. Acta, 2012, 1823, 1967–1975 CrossRef CAS PubMed.
- R. G. Lowery and K. Kleman-Leyer, Expert Opin. Ther. Targets, 2006, 10, 179–190 CrossRef CAS PubMed.
- M. Koresawa and T. Okabe, Assay Drug Dev. Technol., 2004, 2, 153–160 CrossRef CAS PubMed.
- M. Staeben, K. M. Kleman-Leyer, A. L. Kopp, T. A. Westermeyer and R. G. Lowery, Assay Drug Dev. Technol., 2010, 8, 344–355 CrossRef PubMed.
- M. J. Vazquez, B. Rodriguez, C. Zapatero and D. G. Tew, Anal. Biochem., 2003, 320, 292–298 CrossRef CAS.
- K. M. Kleman-Leyer, T. A. Klink, A. L. Kopp, A. Thane, M. D. Koeff, B. R. Larson and T. J. Worzella, Assay Drug Dev. Technol., 2009, 7, 56–67 CrossRef CAS PubMed.
- M. Rowlands, C. McAndrew, C. Prodromou, L. Pearl, A. Kalusa, K. Jones, P. Workman and W. Aherne, J. Biomol. Screening, 2010, 15, 279–286 CrossRef CAS PubMed.
- BellBrook Labs: Transcreener ADP2 FP assay, Technical Manual, 2009, pp. 1–12 Search PubMed.
- BellBrook Labs: Transcreener AMP2/GMP2 FP assay, Technical Manual, 2011, pp. 1–11 Search PubMed.
- J. Lecka, I. Gillerman, M. Fausther, M. Salem, M. N. Munkonda, J.-P. Brosseau, C. Cadot, M. Martín-Satué, P. D'Orléans-Juste, E. Rousseau, D. Poirier, B. Künzli, B. Fischer and J. Sévigny, Br. J. Pharmacol., 2013, 169, 179–196 CrossRef CAS PubMed.
- S. A. Lévesque, E. G. Lavoie, J. Lecka, F. Bigonnesse and J. Sévigny, Br. J. Pharmacol., 2007, 152, 141–150 CrossRef PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- J.-H. Zhang, T. D. Y. Chung and K. R. Oldenburg, J. Biomol. Screening, 1999, 4, 67–73 CrossRef PubMed.
- Y.-C. Cheng and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef CAS.
- P. Held and X. Amouretti, BioTek Application Note, 2006, 1–12 Search PubMed.
- J. C. Owicki, J. Biomol. Screening, 2000, 5, 297–306 CrossRef CAS PubMed.
- K. L. Vedvik, H. C. Eliason, R. L. Hoffman, J. R. Gibson, K. R. Kupcho, R. L. Somberg and K. W. Vogel, Assay Drug Dev. Technol., 2004, 2, 193–203 CrossRef CAS PubMed.
- M. N. Schulz and R. E. Hubbard, Curr. Opin. Pharmacol., 2009, 9, 615–621 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |