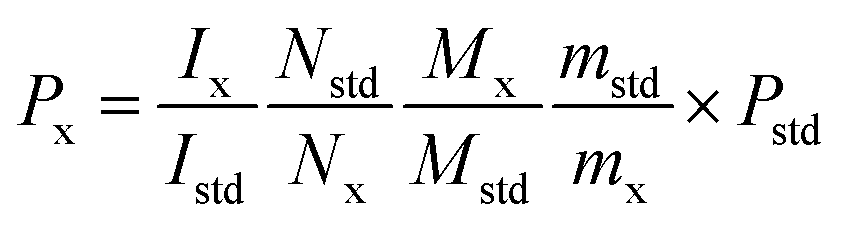DOI:
10.1039/C4AN01681E
(Paper)
Analyst, 2015,
140, 280-286
Quantitative analysis of sitagliptin using the 19F-NMR method: a universal technique for fluorinated compound detection
Received
13th September 2014
, Accepted 18th October 2014
First published on 20th October 2014
Abstract
To expand the application scope of nuclear magnetic resonance (NMR) technology in quantitative analysis of pharmaceutical ingredients, 19F nuclear magnetic resonance (19F-NMR) spectroscopy has been employed as a simple, rapid, and reproducible approach for the detection of a fluorine-containing model drug, sitagliptin phosphate monohydrate (STG). ciprofloxacin (Cipro) has been used as the internal standard (IS). Influential factors, including the relaxation delay time (d1) and pulse angle, impacting the accuracy and precision of spectral data are systematically optimized. Method validation has been carried out in terms of precision and intermediate precision, linearity, limit of detection (LOD) and limit of quantification (LOQ), robustness, and stability. To validate the reliability and feasibility of the 19F-NMR technology in quantitative analysis of pharmaceutical analytes, the assay result has been compared with that of 1H-NMR. The statistical F-test and student t-test at 95% confidence level indicate that there is no significant difference between these two methods. Due to the advantages of 19F-NMR, such as higher resolution and suitability for biological samples, it can be used as a universal technology for the quantitative analysis of other fluorine-containing pharmaceuticals and analytes.
Introduction
The quantitative analysis of active pharmaceutical ingredients (API) is one of the most important issues associated with both previously approved drugs and newly developed molecular entities.1 This issue could not be solved without the assistance of modern analytical technologies, including a variety of capacity and instrumental analysis methods. Traditional titration is known for its accuracy but has poor specificity. Modern instrumental analysis, such as chromatography, spectroscopy, and their hyphenated techniques, makes the quantitative studies rapid, sensitive, and accurate.2 However, an authentic standard substance is always required for the quantitative analysis by these instrumental methods. Therefore, new reliable quantitative analysis technologies which do not require standard substances are of significance and are a challenge for the development of new drugs.
In recent years, quantitative nuclear magnetic resonance (QNMR) spectroscopy has been considered as a feasible approach to confirm the chemical structure and simultaneously determine the content of molecular entities.3 To achieve qualitative and quantitative determination of pharmaceuticals in one step, 1H quantitative NMR (qHNMR) needs no specific standard references and consumes a small amount of organic solvent when compared with other instrumental methods. This technique has recently been one of the most routinely used approaches for the quantitative analysis of new compounds due to its intrinsic advantages, including higher sensitivity and natural abundance when compared to the other nuclei (e.g., 31P and 13C).4,5 However, qHNMR also has its problems and limitations. Since most organic compounds contain several proton atoms in their chemical structure, (1) almost all the proton signals appear in a relative narrow chemical shift range (0–20 ppm), and (2) the mutual coupling effect between protons causes a typical proton signal to split into multiple peaks. These phenomena make the 1H-NMR spectra complicated. For some compounds, it is difficult to select a separated and qualified signal for quantitative analysis. Conversely, other compounds do not contain protons, such as perfluoro-quinoline and perfluorooctane sulfonic acid, making qHNMR not suitable for assessing the content of these analytes.
Fortunately, fluorine is a common atom in the chemical structure of pharmaceuticals, and commercially available fluorine-substituted compounds have been estimated to be about 20%.6 Thus, 19F quantitative NMR could be a complementary method for the QNMR approach. The main advantages associated with 19F NMR include a wide chemical shift range (about 400 ppm for organics) and high resolution as the signals do not overlap with each other. This makes the selection of a qualified signal easier for quantitative analysis when compared to qHNMR. Furthermore, without the interference from the water peak and less interference from endogenous or exogenous backgrounds, 19F-NMR is more suitable for the quantitative analysis of compounds in a biological background and commercial formulations than 1H-NMR.7 Based on these attributes, the 19F-NMR method has been applied to identification of structures,8–11 monitoring of the reaction process,12,13 and quantitative analysis of fluorine-substituted drug molecules.7,14–18
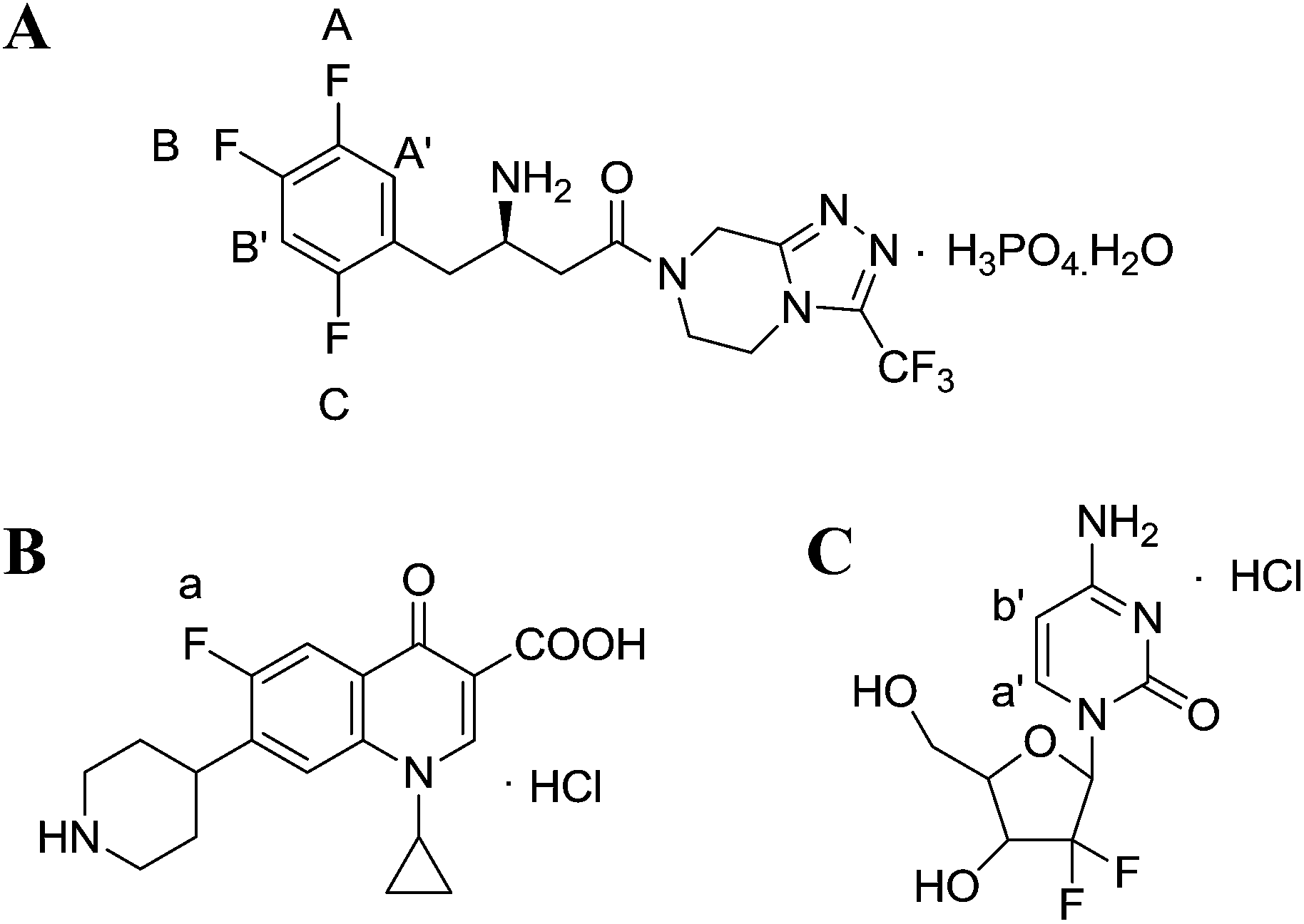
However, the studies on the research methodology of 19F-NMR for the quantitative analysis of fluorinated pharmaceuticals have not been reported as well as its comparison with the 1H-NMR method. In this study, 19F-NMR is employed to detect the absolute content of sitagliptin (STG, [(2R)-1-(2,4,5-trifluorophenyl)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4] triazolo [4,3-a]pyrazin-7(8H)-yl] butan-2-amine]phosphate monohydrate) (Fig. 1A), a newly reported dipeptidyl peptidase IV (DPPIV) inhibitor that can be administrated orally for the treatment of type II diabetes.19 Influential factors, such as relax delay time (d1) and pulse angle, affecting the accuracy and precision of the spectral results are systematically studied and optimized. The reliability and feasibility of the proposed 19F-NMR technology have been compared with 1H-NMR.
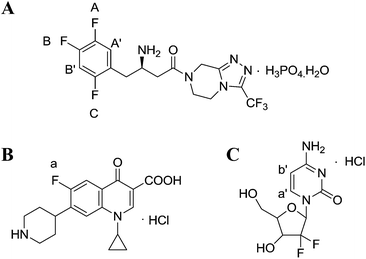 |
| | Fig. 1 Chemical structures of (A) STG, (B) ciprofloxacin (Cipro), and (C) gemcitabine hydrochloride. | |
Experimental section
Materials and reagents
Analytical or HPLC grade chemicals were used throughout. STG was provided by Changzhou pharmaceutical factory. Ciprofloxacin (Cipro, 130451-201203, 84.2%) and gemcitabine hydrochloride (G0G083, 99.7%, USP standards) were purchased from the National Institute for the Control of Pharmaceutical and Biological Products; DMSO-d6 (99.9% atom%D) and D2O (99.9% atom%D) were purchased from Sigma-Aldrich.
Apparatus
19F-NMR and 1H-NMR experiments were performed by using a Bruker AV300 MHz and an AV500 MHz FT-NMR spectrometer equipped with a 5 mm quattro nucleus probe (QNP), respectively. The BT125D analytical balance (Sartorius) was used for weighing samples and the DZF-vacuum oven was used for drying the substances.
Experiment conditions
All 19F-NMR spectra were recorded in the range of δF −20.0 to −130.0 ppm (CF3COOH as reference, δ = −76.2 ppm)20 with a center frequency (O1P) at −98.6 ppm. The NMR probe was maintained at 313 K during the whole experiment. A d1 of 20 s was adopted to ensure full T1 relaxation, and a pulse angle of 90° was used to provide the maximum signal to noise ratio (S/N). 1H-NMR spectra were documented in the range of δH 0.0–11.2 ppm (TMS as reference, δ = 0.0 ppm) with O1P at 4.9 ppm. Signals of benzene were selected for quantitative analysis with gemcitabine hydrochloride as the internal standard (IS) for 1H NMR. The NMR probe was maintained at 303 K during the whole experiment. Manual shimming was acquired to get a symmetrical magnetic field and data were processed using TOPSPIN 2.1 software with manual baseline, phase correction and integration.
Effect of d1 between two scans
One of the main advantages of QNMR is that the same NMR signal response of a compound could be obtained by optimizing instrument parameters. An appropriate S/N, (for 19F-NMR, it is 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13) of the selected quantitative peak is needed to allow an accurate and precise outcome. The d1 is an important parameter affecting the S/N as when d1 is at least 5 times of T1, nearly 99% of excited nuclei can relax to the ground state. Consequently, in this part S/N was tested by varying d1 of five replicates of 10.0 mg STG and 7.0 mg Cipro dissolved in selected solutions at 2 s, 5 s, 10 s, 20 s, and 40 s.
:13) of the selected quantitative peak is needed to allow an accurate and precise outcome. The d1 is an important parameter affecting the S/N as when d1 is at least 5 times of T1, nearly 99% of excited nuclei can relax to the ground state. Consequently, in this part S/N was tested by varying d1 of five replicates of 10.0 mg STG and 7.0 mg Cipro dissolved in selected solutions at 2 s, 5 s, 10 s, 20 s, and 40 s.
Effect of pulse angle
A pulse angle of 30–90° is routinely used for the quantitative test. Lesser pulse angle was more popular as more excited nuclei can relax to the ground state at the same time, although a 90° pulse provides the maximum signal intensity, usually. However, to achieve good accuracy and precision, the pulse angle should be calibrated considering that samples have different physicochemical properties. Herein, we also test the effect of pulse angle on S/N of quantitative peaks by varying the pulse angle of five replicates of 10 mg STG and 7.0 mg IS dissolved in selected solutions at 10°, 20°, 30°, 45°, 50°, 60°, 70°, 80°, and 90°.
Analysis of bulk pure samples
For 19F-NMR analysis, 10.0 mg STG and 7.0 mg Cipro were weighed accurately and transferred to stoppered tubes. Then, 0.25 mL DMSO-d6, 0.25 mL DMSO, and 0.1 mL redistilled water with two drops of appropriate diluted hydrochloric acid (HCl:H2O = 1:3) were added and the solution was heated until complete dissolution. Then the clear solution was transferred to NMR tubes for later detection. For the 1H-NMR test, accurately weighed 10.0 mg STG and 6.0 mg gemcitabine hydrochloride were transferred to stoppered tubes. After, 0.5 mL DMSO-d6 was added with 0.1 mL D2O to avoid the interference of active protons. Solution was mixed thoroughly on the vortex mixer till complete dissolution and transferred to NMR tubes. Acquisition was performed under conditions afore-mentioned and the results were calculated according to eqn (1).| | 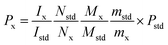 | (1) |
where Ix and Istd are the areas of selected signals for STG and IS; Nx and Nstd are the fluorine or proton number of STG and IS for quantitative analysis; Mx and Mstd are the molar mass (Mr) of STG and IS, mx and mstd are the milligram amounts of STG and IS, Pstd and Px are the absolute content of IS and STG, respectively.
Method validation
The developed 19F-NMR approach was validated according to International Conference on Harmonization (ICH) guidelines,21 which includes precision and intermediate precision, linearity, LOD and LOQ, stability, and robustness.
Results and discussion
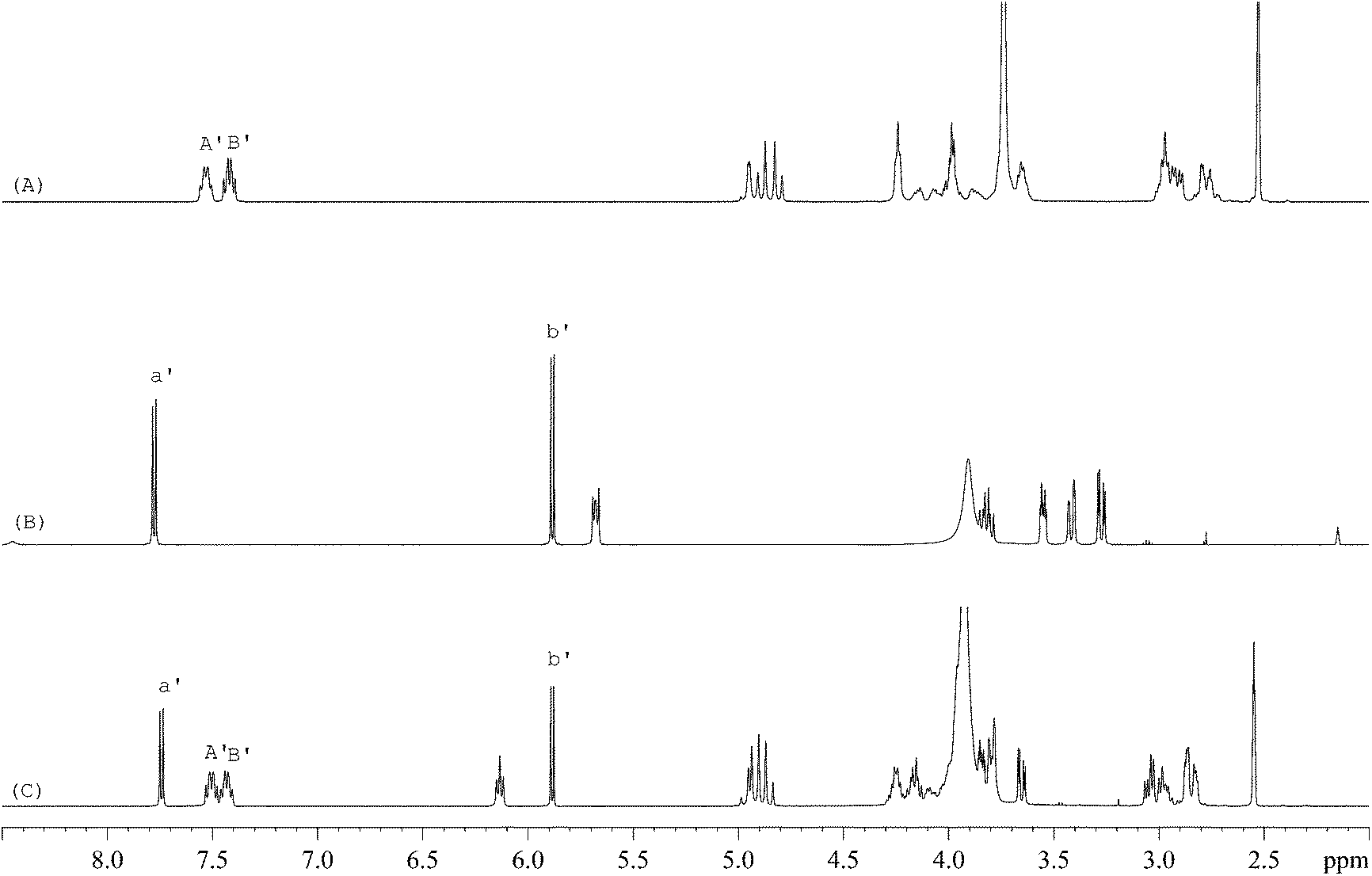
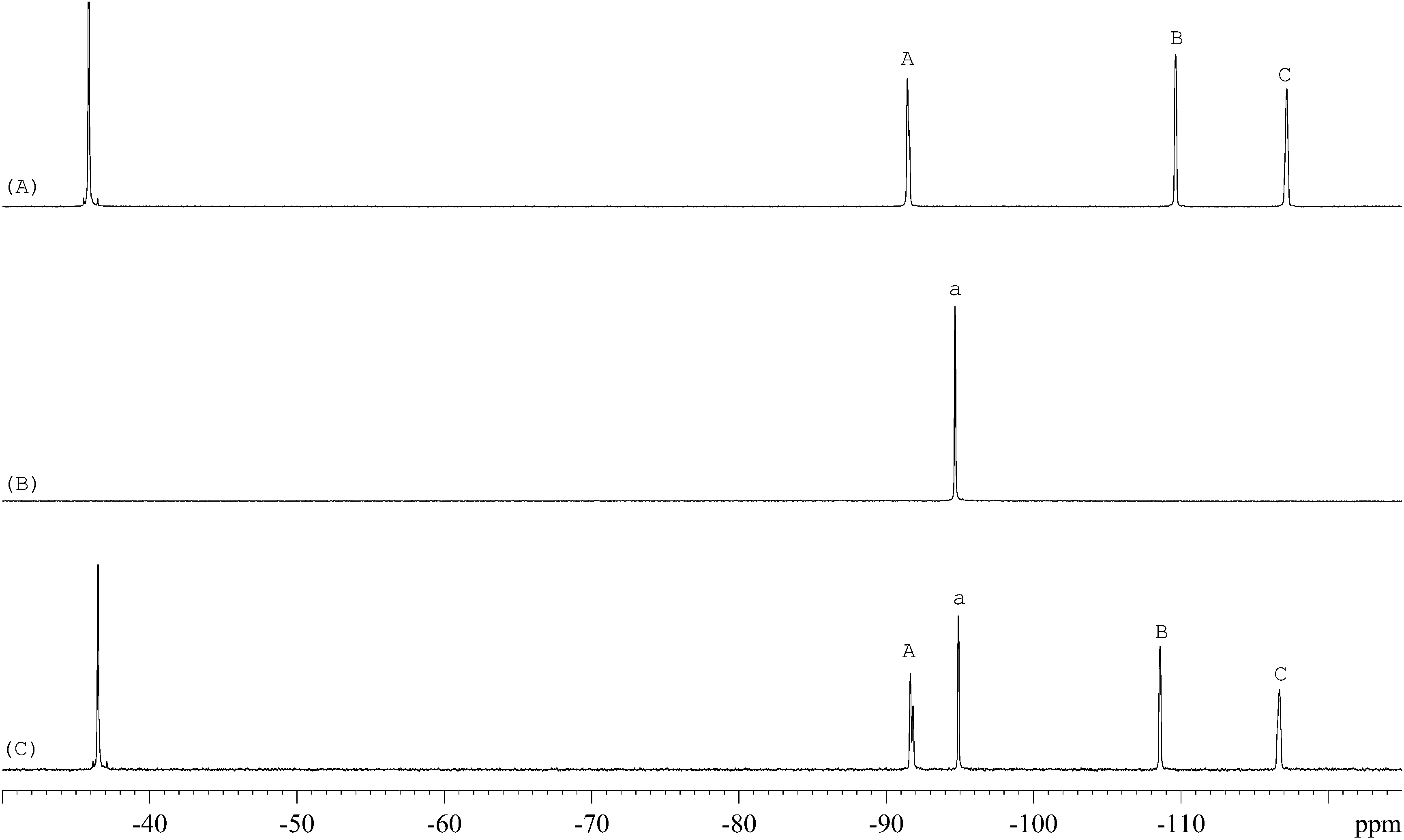
The 1H-NMR spectra of STG, its IS (gemcitabine hydrochloride), and their mixture are shown in Fig. 2. 19F-NMR spectra of these samples are shown in Fig. 3, but the IS here is Cipro. Compared to 1H-NMR, 19F-NMR spectra showed more simple and clear resonance signals, peaks A, B, and C (Fig. 3A) have been chosen as the quantitative signals for determination. Before the quantitative analysis of STG, a variety of influential factors have been optimized.
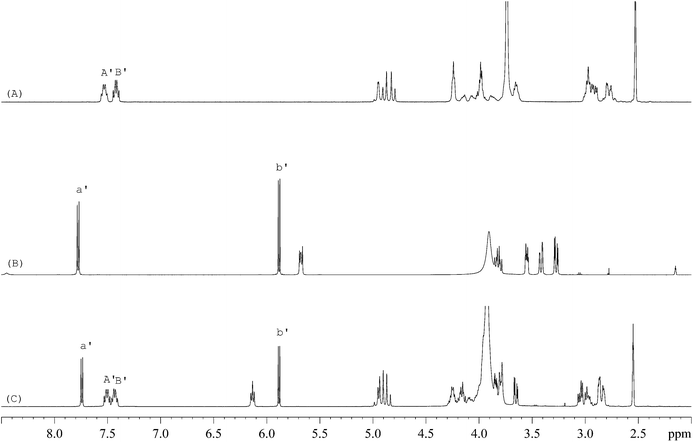 |
| | Fig. 2
1H-NMR spectra of (A) STG, (B) gemcitabine hydrochloride, and (C) their mixed solutions in 0.6 mL DMSO-d6 + D2O (5:1, v/v) (marked signals were selected for quantitative analysis). | |
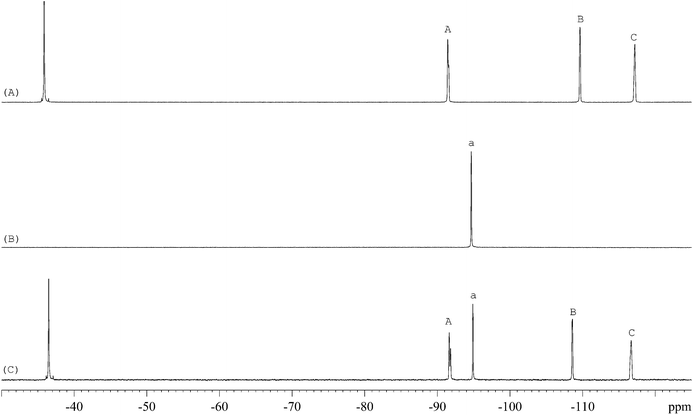 |
| | Fig. 3
19F-NMR spectra of (A) STG, (B) Cipro, and (C) their mixed solutions in 0.6 mL DMSO-d6 + DMSO + H2O (2.5:2.5:1, v/v) with 2 drops of diluted HCl (marked signals were selected for quantitative analysis). | |
Solvent selection for 19F-NMR detection
One of the most important principles for quantitative analysis is that the target material must be well dissolved in the selected solvent. In our primary test, either STG or Cipro is soluble in 0.6 mL DMSO-d6 + D2O (5:1, v/v). Considering that a little portion of non-deuterated solvent will not affect the fluorine signal in the final spectrum, for reducing the cost, the solvent 0.6 mL DMSO-d6 + DMSO + H2O (2.5:2.5:1, v/v) has been selected. However, the suspension appears when both STG and Cipro are dissolved in this solvent, which could be due to the acid–base neutralization making it harder for the conjugate to dissolve. Considering that acidification would increase the solubility of Cipro, HCl was added into the mixture. After several trials, 2 drops of appropriate diluted HCl (HCl:H2O = 1:3) were added into the mixed solution. After the addition, the mixed solution was found to be clear. Thereby, 0.25 mL DMSO-d6, 0.25 mL DMSO and 0.1 mL H2O with 2 drops of diluted HCl were selected as the ideal solvents for 19F-NMR experiment.
Selection of an appropriate IS
Selection of an appropriate IS is of significant importance in QNMR experiment and the options are limited as they should conform to several specific requirements such as: (i) high purity; (ii) good stability and solubility in the selected solvent; (iii) equivalent molecular weight to target compound; (ii) sharp and separated signal in the NMR spectra.22 Cipro (Fig. 1B) is soluble and stable in the selected solution. Its single 19F peak is located at δF = −94.7 ppm and its Mr is 367.8 g mol−1. These physicochemical parameters meet above criteria and therefore it is selected as IS in this study.
Selection of quantitative signals
The 19F-NMR spectra of STG, Cipro, and their mixture are shown in Fig. 3. The single peak of Cipro appears at −94.7 ppm. Signals of STG are located at −91.5, −108.5, and −116.6 ppm. These quantitative signals are marked as a, A, B, and C in Fig. 3, and are well separated in the spectra, demonstrating no interference between the two substances and good specificity of this method.
Effect of d1
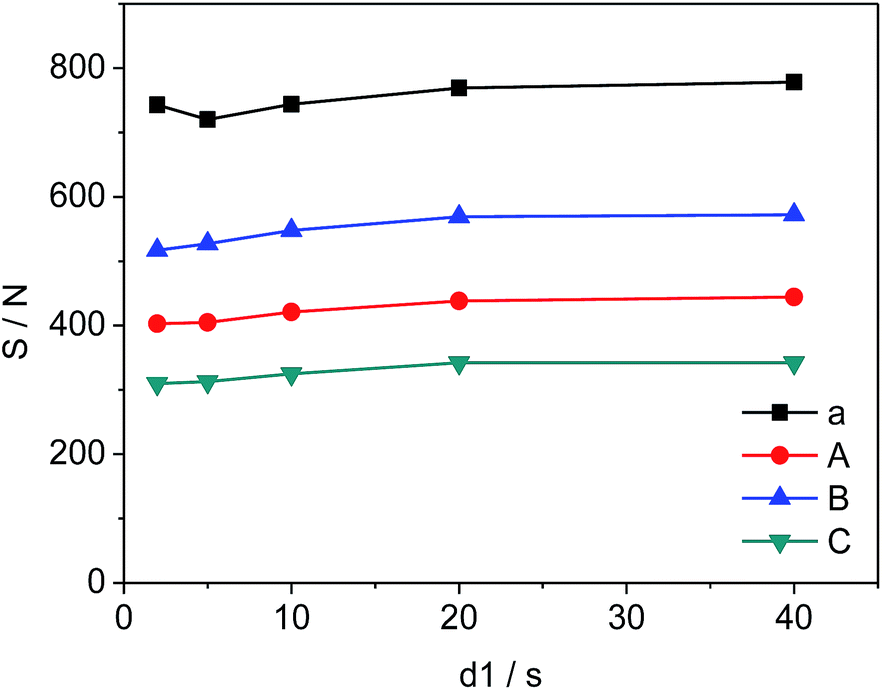
The changes in d1, the relaxation delay time, during the acquisition time could affect S/N of the selected quantitative signals and consequently influence the precision and accuracy of 19F-NMR results. Fig. 4 shows the effect of different d1 on the quantitative signals of the target drug and Cipro. The S/N ratio of selected signals changes from 720 to 778 (a), 403 to 444 (A), 517 to 572 (B), and 310 to 342 (C) as d1 increases from 2 s to 20 s respectively. However, S/N remains nearly unchanged in the interval of 20 s to 40 s indicating that S/N is saturated when d1 = 20 s. To improve the accuracy and shorten test time simultaneously, all 19F-NMR experiments in this work were performed at d1 = 20 s.
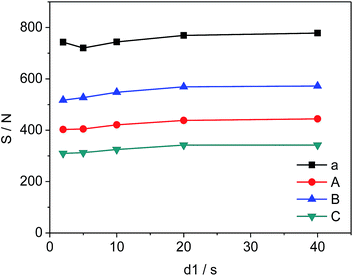 |
| | Fig. 4 Effects of d1 on S/N of selected signals (a, A, B and C are shown in Fig. 3, respectively). | |
Effect of pulse angle
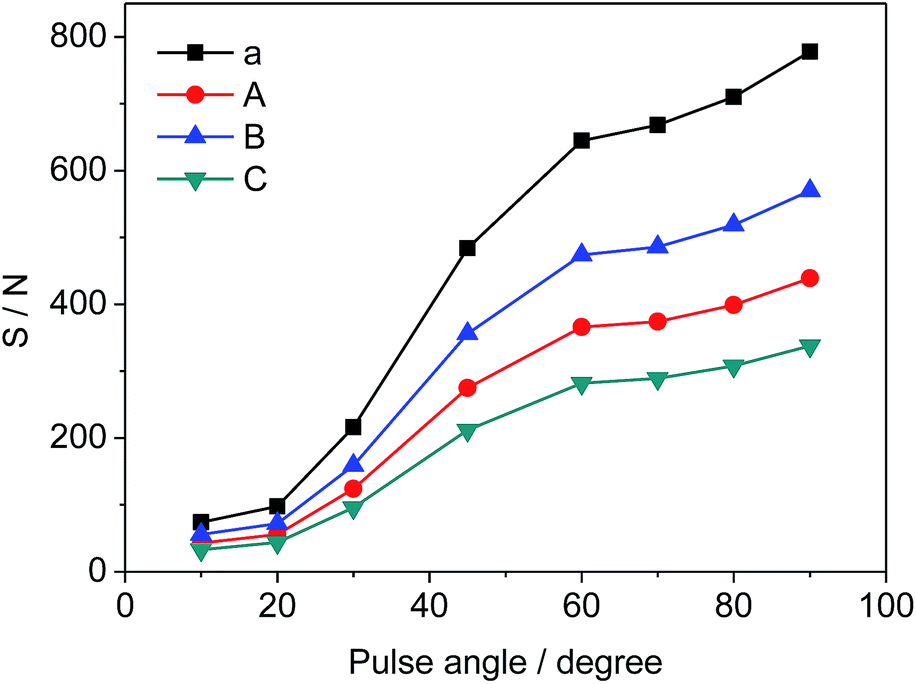
Different pulse angles also have significant influence on S/N of selected signals, which can be deduced from the results in Fig. 5. S/N values vary from 74 to 778 (a), 43 to 439 (A), 56 to 570 (B), and 33 to 338 (C), respectively as the pulse angle increases from 10° to 90°. Since the pulse angle of 90° provides the maximum S/N and signal intensity for STG, it is effective and adopted for the measurement of 19F-NMR spectra.
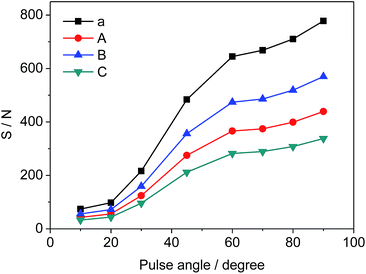 |
| | Fig. 5 Effects of pulse angle on S/N of selected signals (a, A, B and C as shown in Fig. 3, respectively). | |
Method validation
Precision and intermediate precision
To investigate the precision and intermediate precision of the proposed method, we performed recovery studies on two different days with the same concentration: 10.0 mg STG + 7.0 mg Cipro. Precision was accessed by six preparation processes (n = 5) in one day and intermediate precision was evaluated on two different days. The mean content of drug in % assay and RSDs were tabulated (Table 1). The average content of the drug was found to be 99.3% and 99.3% and RSDs were 0.74% and 0.82% for precision and intermediate precision analysis, respectively. These results demonstrated high precision and accuracy for 19F-NMR, able to determine the content of fluorine-containing drugs.
Table 1 Precision and intermediate precision of the quantitative test (n = 6)
| No. |
Precision |
Intermediate precision |
| Taken (mg) |
Found (mg) |
Assay (%) |
Mean (%) |
RSD (%) |
Taken (mg) |
Found (mg) |
Assay (%) |
Mean (%) |
RSD (%) |
| 1 |
10.31 |
10.28 |
99.7 |
99.3 |
0.74 |
10.32 |
10.26 |
99.6 |
99.3 |
0.82 |
| 2 |
10.68 |
10.49 |
98.2 |
10.59 |
10.50 |
98.2 |
| 3 |
10.17 |
10.18 |
100.1 |
10.32 |
10.24 |
100.6 |
| 4 |
10.07 |
10.06 |
99.9 |
10.31 |
10.18 |
101.1 |
| 5 |
9.91 |
9.83 |
99.2 |
9.91 |
9.90 |
99.5 |
| 6 |
10.11 |
9.98 |
98.7 |
10.10 |
10.04 |
99.9 |
Linearity
Linearity of the proposed 19F-NMR method was validated by preparing seven different concentrated samples, their amounts ranging from 75% to 140% (n = 5). The linearity results are shown in Fig. 6, which gave the regression equation of Y = 0.983X + 0.015 with a correlation coefficient R2 = 0.9998, an indication of good linearity.
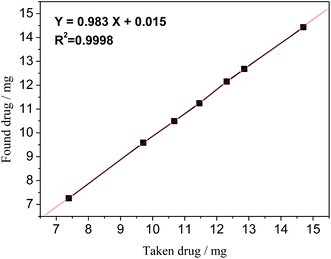 |
| | Fig. 6 Linearity curve of STG of taken vs. found amount of drug. | |
LOD & LOQ
The LOD and LOQ are defined as three-times and ten-times the standard deviation (SD) of the least tested concentration divided by the slope of the linearity curve. They were calculated to be 0.014 mg and 0.046 mg per 0.6 mL according to the regression equation obtained in the linearity analysis, respectively.23
Stability
The stability test of STG was conducted at 0, 6, 48, 120, and 168 h by measuring replicates (n = 5) of an aliquot solution at refrigeration temperature (273–281 K) and compared with initial values, which are tabulated in Table 2. It illustrated that the stability results offered by this recommended approach was satisfactory with RSD values ranging from 0.16 to 0.81 and relative error (RE) values ranging from −1.53 to 0.63, respectively.
Table 2 Stability results of STG under storage conditions (n = 5)
| Time interval (h) |
Taken (mg) |
Found (mg) |
Assay (%) |
RSD (%) |
RE (%) |
| 0 |
14.29 |
14.07 |
98.4 |
0.47 |
−1.53 |
| 6 |
14.29 |
14.14 |
99.0 |
0.43 |
−1.05 |
| 48 |
14.29 |
14.38 |
100.6 |
0.16 |
0.63 |
| 120 |
14.29 |
14.22 |
99.5 |
0.58 |
−0.49 |
| 168 |
14.29 |
14.26 |
99.8 |
0.81 |
−0.21 |
Robustness
The robustness of this method was verified by varying three parameters independently: (i) the number of scans (48 ± 16); (ii) the temperature of the probe (298–313 K); (iii) the amount of IS (10.0 ± 1.0 mg).
The results obtained are summarized in Table 3 and demonstrated that: varying the number of scans, for example, 32, 48 and 64 did not influence the measured amount of the drug; a slight change in the amount of the IS at 9.0 mg, 10.0 mg, and 11.0 mg revealed no appreciable change in the detected content of the drug; different probe temperatures at 298 K, 303 K, 308 K and 313 K also gave similar results. Consequently, the 19F-NMR method is really robust in terms of these three parameters mentioned above and it should be selected as a universal procedure for other fluorine-substituted pharmaceutical analyses.
Table 3 Robustness study of STG
| Parameters |
Change |
Taken (mg) |
Found (mg) |
Assay (%) |
RE (%) |
| Number of scans |
32 |
14.81 |
14.51 |
98.0 |
−2.02 |
| 48 |
14.81 |
14.54 |
98.1 |
−1.82 |
| 64 |
14.81 |
14.53 |
98.2 |
−1.89 |
| IS (mg) |
9.31 |
14.19 |
13.97 |
98.5 |
−1.55 |
| 10.29 |
14.20 |
13.98 |
98.5 |
−1.55 |
| 11.39 |
14.69 |
14.51 |
98.8 |
−1.22 |
| Probe temperature (K) |
298 |
14.81 |
14.76 |
99.6 |
−0.33 |
| 303 |
14.81 |
14.67 |
99.0 |
−0.94 |
| 308 |
14.81 |
14.51 |
98.0 |
−2.02 |
| 313 |
14.81 |
14.67 |
99.0 |
−0.94 |
Comparison with the 1H-NMR method
To clarify the validity of the proposed 19F-NMR method, the results obtained in this approach were compared with 1H-NMR outcomes (Table 4). The statistical F-test and the student t-test at 95% confidence level were applied to detect whether there were significant differences between these two methods. Data shown in Table 4 illustrated that, at a confidence limit of 95%, there was no significant bias in precision between 19F-NMR and 1H-NMR methods. The calculated “F” value, 2.53, was smaller than the theoretical value 6.39. The t-test also showed that there was no marked difference between these two methods in terms of average content because the obtained “t” value is 0.54, less than t0.05,4 = 2.78. This result demonstrates that the proposed 19F-NMR method could be applied to the detection of STG and it can also be adopted as a universal technique for the analysis of other fluorinated substances.
Table 4 Comparison results of the 19F-NMR method with the 1H-NMR method
| No. |
19F-NMR |
1H-NMR |
| Taken (mg) |
Assay (%) |
Mean (%) |
RSD (%) |
Taken (mg) |
Assay (%) |
Mean (%) |
RSD (%) |
|
F-value in this table at P = 0.05 is 6.39.
t-value in this table at P = 0.95 is 2.78.
|
| 1 |
10.10 |
99.6 |
99.5 |
0.35 |
10.47 |
99.6 |
99.6 |
0.22 |
| 2 |
10.10 |
99.3 |
10.47 |
99.4 |
| 3 |
10.10 |
99.0 |
10.47 |
99.5 |
| 4 |
10.10 |
99.9 |
10.47 |
99.8 |
| 5 |
10.10 |
99.7 |
10.47 |
99.9 |
|
F-test at P = 0.95a |
2.53 |
|
t-test at P = 0.95b |
0.54 |
Conclusions
In summary, a quantitative 19F-NMR method for the determination of STG was studied and compared with the 1H-NMR method. Under the optimized conditions, 19F-NMR demonstrated advantages of high resolution, accuracy, detection speed, and reproducibility in the quantitative analysis of pharmaceuticals containing fluorine atoms. Therefore, this technology can be used as a supplementary method for qHNMR to broaden the application of QNMR, especially in the investigation process of drug research and development. At the same time, it is noted that, for the fluorinated samples containing some impurities that do not contain fluorine, the record on both 1H and 19F NMR spectra is necessary.
Acknowledgements
This work was financially supported by the Natural Science Foundation of China (31470916), the Program for New Century Excellent Talents in University (NCET-10-0816), A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, and the Open Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (SKLNMKF201307).
Notes and references
- R. M. Maggio, N. L. Calvo, S. E. Vignaduzzo and T. S. Kaufman, J. Pharm. Biomed. Anal., 2014, 101, 102–122 CrossRef CAS PubMed.
- S. Mahajan and I. P. Singh, Magn. Reson. Chem., 2013, 51, 76–81 CAS.
- C. Simmler, J. G. Napolitano, J. B. McAlpine, S. N. Chen and G. F. Pauli, Curr. Opin. Biotechnol., 2014, 25, 51–59 CrossRef CAS PubMed.
- U. Holzgrabe, Prog. Nucl. Magn. Reson. Spectrosc., 2010, 57, 229–240 CrossRef CAS PubMed.
- S. K. Bharti and R. Roy, Trends Anal. Chem., 2012, 35, 5–26 CrossRef CAS PubMed.
- Y. Amaoka, M. Nagatomo and M. Inoue, Org. Lett., 2013, 15, 2160–2163 CrossRef CAS PubMed.
- R. Martino, V. Gilard, F. Desmoulin and M. Malet-Martino, J. Pharm. Biomed. Anal., 2005, 38, 871–891 CrossRef CAS PubMed.
- R. K. Belter, J. Fluorine Chem., 2012, 137, 73–76 CrossRef CAS PubMed.
- M. A. Fox, G. Pattison, G. Sandford and A. S. Batsanov, J. Fluorine Chem., 2013, 155, 62–71 CrossRef CAS PubMed.
- C. Martineau, C. Legein, M. Body, O. Péron, B. Boulard and F. Fayon, J. Solid State Chem., 2013, 199, 326–333 CrossRef CAS PubMed.
- G. Arsenault, B. Chittim, J. Gu, A. McAlees, R. McCrindle and V. Robertson, Chemosphere, 2008, 73, S53–S59 CrossRef CAS PubMed.
- P. Wormald, B. Ameduri, R. K. Harris and P. Hazendonk, Polymer, 2008, 49, 3629–3638 CrossRef CAS PubMed.
- C. Langner, J. Meier-Haack, B. Voit and H. Komber, J. Fluorine Chem., 2013, 156, 314–321 CrossRef CAS PubMed.
- S. Trefi, V. Gilard, M. Malet-Martino and R. Martino, J. Pharm. Biomed. Anal., 2007, 44, 743–754 CrossRef CAS PubMed.
- A. Moghimi, I. Omrani, R. H. Khanmiri, R. Bahadorbeigi and M. Mahmoodi, Polym. Test., 2014, 33, 30–33 CrossRef CAS PubMed.
- A. Bock, M. Dubois, P. Bonnet, A. Hamwi, D. Avignant, L. Moch and B. Morel, J. Fluorine Chem., 2012, 134, 24–28 CrossRef CAS PubMed.
- N. Kitamura, K. Tanaka and Y. Chujo, Bioorg. Med. Chem. Lett., 2013, 23, 281–286 CrossRef CAS PubMed.
- V. Pourcelle, C. S. Le Duff, H. Freichels, C. Jérôme and J. Marchand-Brynaert, J. Fluorine Chem., 2012, 140, 62–69 CrossRef CAS PubMed.
- D. Q. Pham, A. Nogid and R. Plakogiannis, Am. J. Health-Syst. Pharm., 2008, 65, 521–531 CrossRef CAS PubMed.
-
W. R. Dolbier, Guide to fluorine NMR for organic chemists, John Wiley & Sons, 2009, p. 14 Search PubMed.
-
ICH Harmonized Tripartite Guidelines, Validation of Analytical Procedures: Text and Methodology Q2 (R1), 2005 Search PubMed.
- T. Rundlöf, M. Mathiasson, S. Bekiroglu, B. Hakkarainen, T. Bowden and T. Arvidsson, J. Pharm. Biomed. Anal., 2010, 52, 645–651 CrossRef PubMed.
- A. A. Salemand and H. A. Mossa, Talanta, 2012, 88, 104–114 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.