
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revealing highly unbalanced energy barriers in the extension and contraction of the muscle-like motion of a [c2]daisy chain†
Yan-Ling
Zhao
*a,
Rui-Qin
Zhang
b,
Christian
Minot
ac,
Klaus
Hermann
d and
Michel A.
Van Hove
a
aInstitute of Computational and Theoretical Studies & Department of Physics, Hong Kong Baptist University, Hong Kong, China. E-mail: ylzhao2008@gmail.com
bDepartment of Physics and Materials Science, City University of Hong Kong, Hong Kong, China
cLaboratoire de Chimie Théorique, Université Pierre & Marie Curie – Paris 6, CNRS, UMR7616, case 137, 4 place Jussieu 75252 Paris Cedex 05, France
dInorganic Chemistry Department, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany
First published on 19th May 2015
Abstract
Nanoscale muscle-like materials have aroused great interest as they may provide controllable mechanical operations by artificial actuations. Molecular designs to achieve the desired motion at the macroscopic scale in experiments require atomic level understanding. By systematic quantum chemical and molecular dynamics calculations we reveal that the length change is not only due to the linear telescoping from the dibenzo[24]crown-8 recognition at two docking stations but also the folding/unfolding of two bulky stoppers. The extension and contraction processes of a [c2]daisy chain under acidic vs. basic conditions are exothermic but need to cross very different energy barriers, being at least double the height under acidic compared to basic conditions, hindering balanced cyclic motions at moderate excitation. Our result suggests that to realize the desired muscle-like motion one should adopt sufficiently high external excitation, using for example reasonably high temperature and further optimizing the solution used.
Introduction
Biomimetic research harnesses nature's creations to guide us in developing new valuable products. Hence, muscle-like materials are envisaged to provide controllable mechanical operations in a variety of environments, from benign to hostile. In accordance with the bottom-up principle, design and synthesis at the molecular level are fundamentally conducive to creating precise molecular machines and then intentionally amplifying their action to the macro-scale. Chemists have synthesized several muscle-like molecular shuttles, where one functional group reciprocates between two fixed “stations” along the axis under external stimuli such as a chemical environment,1–5 electrochemical redox reactions,1,6–13 or illumination.8,14–19 Notably, Sauvage et al.20 first synthesized a doubly threaded rotaxane capable of contraction and stretching under the action of a chemical stimulus due to exchanging Cu+/Zn2+ in solution. This opens a research arena to synthesize complex molecular shuttles in functional analogy with natural muscles.Recently, the Giuseppone21 group has successfully polymerized doubly threaded rotaxanes and in principle can thereby magnify the elongation/contraction of the individual units by a factor of ∼3000. The Stoddart22 group highlighted this work afterwards. As shown in Fig. 1a, each half of the rotaxane polymer unit consists mainly of five components, (1) a rod-shaped backbone, (2) a dibenzo[24]crown-8 (DB24C8) ring-like ether, (3) a docking station that can vary between a secondary dialkylammonium ion (NH2+) in a low pH environment and a neutral amino group (NH) in high pH, (4) a triazolium cation docking station, and (5) a terpyridine end group. The length of each unit varies dynamically as the binding affinity of DB24C8 to the NH2+/NH docking station changes with pH: with a solution of trifluoroacetic acid, the DB24C8 ring preferentially forms hydrogen bonds with NH2+, cf.Fig. 1b;23–27 in contrast, under a basic solution of sodium hydroxide, the DB24C8 ring prefers to combine with the triazolium cation docking station, as the NH2+ is neutralized to NH, cf.Fig. 1c. Therefore, the pH of the environment determines which of these two docking stations is favored, allowing alternation between contracted and extended states by cycling between high and low pH, which also provides the needed energy. For polymerization,21cf.Fig. 1d, two terpyridine groups of two rotaxanes are 6-fold coordinated to a single Fe2+ ion (originating from FeCl2 present in the solution), so as to connect the units head to tail and form a snake-like linear polymer. As revealed by light scattering and small-angle neutron scattering measurements,21 the unit lengths in the extended and contracted forms are found to be ca. 5.4 and 3.2 nm, respectively. The 2.2 nm length difference in each unit is amplified in a linear polymer of about 3000 units to cause a stretch from about 9400 to about 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 nm, thus hopefully realizing the muscle-like motion in a quasi-macroscopic regime.
900 nm, thus hopefully realizing the muscle-like motion in a quasi-macroscopic regime.
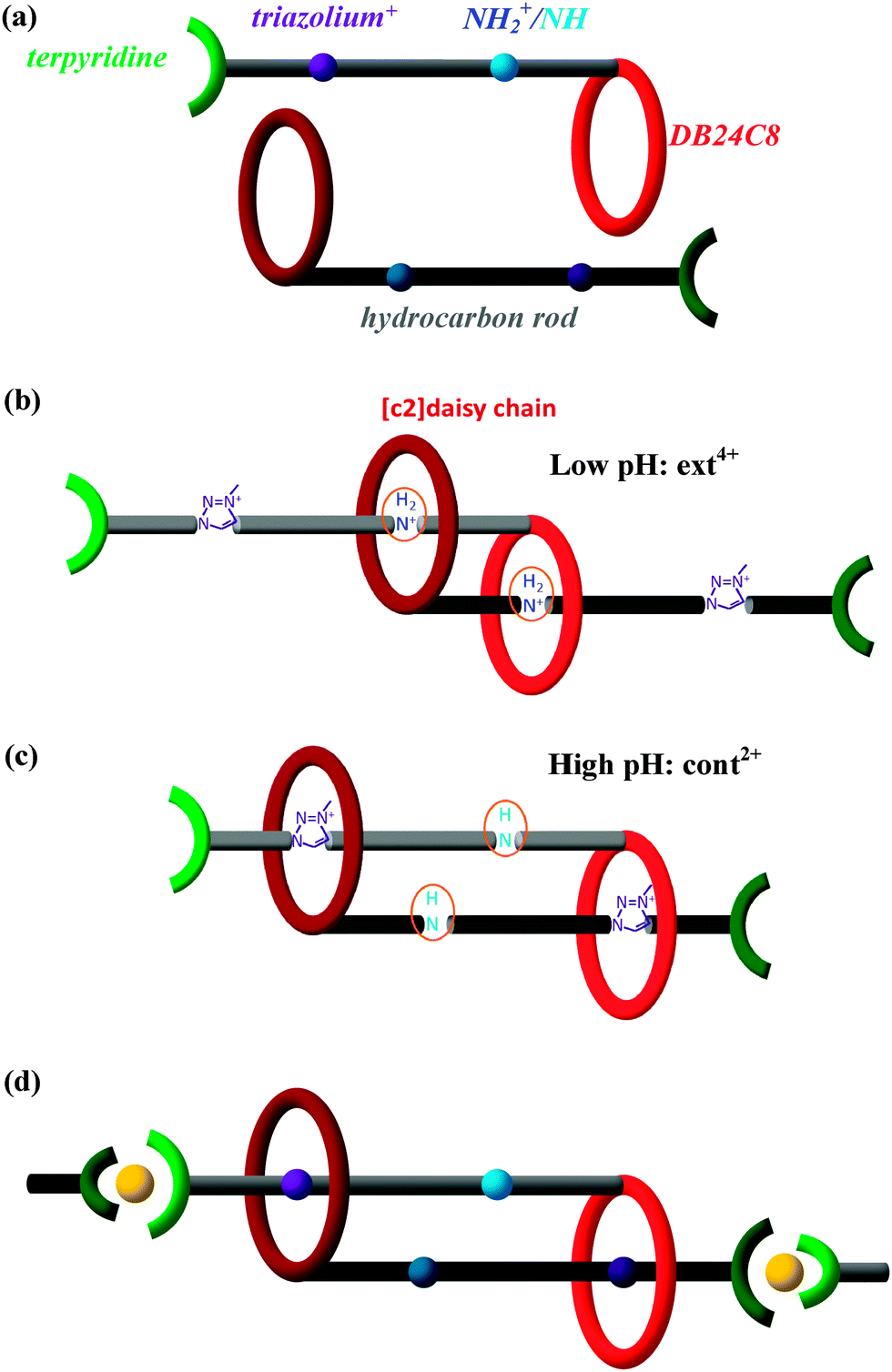 | ||
| Fig. 1 Schematic representations of the muscle-like [c2]daisy chain polymer. (a) Each half of the doubly threaded rotaxane polymer unit consists of a hydrocarbon rod with two docking stations, a crown ether ring DB24C8, and a terpyridine end group; (b) two identical halves are mutually threaded into a [c2]daisy chain, docked at the NH2+ station (in low pH) in the extended state and (c) at the triazolium+ station (in high pH) in the contracted state; (d) polymerized [c2]daisy chains are linked by Fe2+ (yellow balls), held by terpyridine groups, and docked in the contracted state. | ||
However, many challenging problems still exist in going from the laboratory setting to practical molecular machines. The study of the structural evolution presenting a macroscopic muscle-like motion is very demanding. By means of mass spectrometry, 1H NMR, UV-Vis spectra, and scattering techniques, experimental studies have successfully identified the synthesized functional groups, tracked the configuration variations under the external stimuli, and extracted the polymer size and shape. With quantum mechanical calculations, performed for the first time for this system, we herein add deeper insight into the mechanisms of such a muscle-like material at an atomic level which can help design future molecular machines. Our calculations contribute useful insight by presenting the equilibrated end configurations as well intermediate configurations of the rotaxane unit of the polymer obtained by Giuseppone et al.,21 and analyze detailed potential energy profiles as well as structural evolution during contraction and stretching, thus aiming to identify the functioning mechanisms and to provide important thermodynamic data. Solvent effects are taken into account to show the impact of the environment on the motion.
Models and computational details
Generally, a rotaxane is a dumbbell-like molecule that is threaded through a macrocycle ring which can move along the molecule's shaft without being able to escape due to bulky end groups. In this work, each half of the doubly threaded rotaxane provides the other one with a crown ether ring, allowing mutual threading which affords the freedom of linear contraction and stretching while preventing dethreading. Such a type of rotaxane possesses a reciprocal symmetry and is often called a [c2]daisy chain, where “c” stands for cyclic and “2” stands for a two-membered chain (as opposed to, for example, an [a3]daisy chain for an acyclic three-membered chain).28The initial model is built manually from the structure shown in Fig. 1 of ref. 22, in agreement with the molecular formula reported in the supporting information of ref. 21. The acidic environment of the [c2]daisy chain is simulated in the present study by adding two protons near the nitrogen of its amino groups, leading to an extended structure in acid. For the basic environment of the [c2]daisy chain the two protons of NH2+ are assumed to be neutralized by OH− ions and, thus, are left out of the simulation which results in the contracted structure in base. Hereafter, the extended and contracted forms are respectively designated as ext4+ and cont2+. The configuration ext4+ (C116H134O16N14) carries four positive charges localized at two NH2+ and two triazolium cations, while the two positive charges of cont2+ (C116H132O16N14) localize at two triazolium cations.
The [c2]daisy chains are self-assembled groups of molecules held together by weak bonds, mainly hydrogen bonds, van der Waals (vdW) interactions, electrostatic interactions, and/or coupling by π–π stacking. At present, many theoretical approaches include long-range dispersion forces to model such weak interactions.29–32 In this work, we chose the self-consistent-charge density-functional tight-binding (SCC-DFTB) method33,34 to carry out most calculations where dispersion contributions are included via Lennard-Jones potentials to describe the universal force field parameters. The SCC-DFTB method uses linear combinations of frozen atomic orbitals to represent electrons that are delocalized over the entire system. Corresponding interaction integrals for pairs of atomic orbitals are calculated separately and used when evaluating self-consistent molecular orbitals of a given system. The SCC-DFTB method has proven to be both computationally efficient for very large systems and accurate when compared with standard DFT results for electronic and geometric configurations as well as energies. Therefore, it is extensively employed to deal with large clusters containing hundreds of atoms, and particularly suitable for those consisting mainly of C, H, O, and N atoms.35–39
To investigate the solvent effect on the [c2]daisy chain, we perform SCC-DFTB-D-based molecular dynamics (SCC-DFTB-D/MD) simulations for a period of 1 ps. The canonical ensemble is adopted with an Anderson thermostat in an NVT model for a temperature of 300 K. The time step is set at 1 fs for the integration of the equations of motion by a Verlet velocity algorithm. Periodic boundary conditions are used with a unit cell defined by a = 6.6 nm, b = 3.3 nm, c = 2.7 nm, and α = β = γ = 90°. After the [c2]daisy chain was placed in the center of the box the remaining space is filled with 413 acetonitrile (CH3CN) molecules, which are randomly distributed and automatically generated with the help of the Gromacs package40 by checking overlaps of Coulomb and vdW radii.
In order to obtain more accurate binding energies, energy profiles, dipole moments and electrostatic potential (ESP) distributions, single-point calculations on the [c2]daisy chain supramolecules are carried out by using the ωB97XD/6-31G** level of theory, which also considers the dispersion correction and is included in the G09 package.41 The SMD solvation model42 is adopted as we describe the energy profiles in solution. In the experiment, the cationic [c2]daisy chain was synthesized in an ionic liquid, where stoichiometric PF6− counterions accompany the supramolecules to neutralize the charge in the environment of organic solvents. Thus, the role of solvation is considered for binding with PF6− counterions. Optimization and frequency calculations on smaller species such as PF6−, CH3CN, and chloroform (CHCl3), and PF6−(CH3CN)n (n = 1–8) and PF6−(CHCl3)n (n = 1–8) complexes are performed at the same calculation level.
Results and discussion
1. Equilibria and intermediates
We here analyze in detail the contraction/extension cycle of the [c2]daisy chain. When the equilibrated ext4+ is exposed to an alkaline solution (cf. the schematic diagram in Fig. 2), two protons (one from each of the two NH2+ groups) rapidly leave, leading to an intermediate which we call deproton-ext2+. This intermediate then gradually evolves to the final equilibrated form of cont2+ in basic solution. When cont2+ encounters an acidic environment, two protons attach to two neutral NH groups, generating the proton-cont4+ intermediate, which then evolves back into the equilibrated form of ext4+ in acid. This cycle repeats as the solution is switched back and forth between basic and acidic conditions. The two equilibration processes of this cycle are:| Contraction in base: deproton-ext2+ → cont2+ |
| Stretching in acid: proton-cont4+ → ext4+ |
 | ||
| Fig. 2 The cyclic process of acid-induced extension and base-promoted contraction through equilibrium and intermediate structures, from SCC-DFTB-D optimizations in the vacuum. The central schematic shows the cycle in a simplified energy diagram, with equilibrated forms detailed below and intermediates above. Gray, white, red, and blue atom colors represent C, H, O, and N, respectively. | ||
An extensive conformation search for the favorable intermediates and equilibria was carried out by SCC-DFTB-D optimizations in the vacuum without any position constraints. It resulted in the two equilibrated forms and two intermediates that are shown in Fig. 2 to illustrate their cyclic transformations. Fig. S1 (cf. ESI†) shows the geometric details of the two equilibrium structures more clearly, identifying those atoms that are essential for the formation of equilibrium geometries. The configuration ext4+ has a long thin shape after complete relaxation. Its central part is dominated by a four-level π–π stacking of four phenol rings with separations of 1.047 nm between the most distant rings and 0.324 nm between the two central parallel-displaced rings; it also exhibits four hydrogen bonds between NH2+ and DB24C8 of type N–H⋯O with H⋯O distances of 0.209 and 0.207 nm, and of type N+–H⋯O with H⋯O distances of 0.181 and 0.181 nm. To validate the SCC-DFTB-D results, we have also optimized the DB24C8 and NH2+ interacting part of ext4+ by ωB97XD/6-31G** (with the triazolium parts and two stoppers removed to reduce the computational cost). It turns out that the central interlayer spacing is about 0.316 nm, close to the 0.324 nm found by SCC-DFTB-D. Additionally, as compared with the interlayer distance (0.35 to 0.40 nm) of π–π stacking between two benzenes in previous DFT studies,29–32 this shows that four hydrogen bonds between NH2+ and DB24C8 and more layered benzene rings will help strengthen the group in the central part of ext4+. The configuration cont2+ presents a short thick shape with the triazolium cations staying outside the two DB24C8 rings and perpendicular to the dibenzo planes. Five C–H⋯O hydrogen bonds are found on one side with H⋯O distances from 0.217 to 0.245 nm, due to the hydrogen atoms from triazolium and hydrocarbons near the crown ether oxygen.
To evaluate the amount of telescoping which takes place between the two forms, we first consider the distance between two oxygen atoms (O1 and O2, labeled in Fig. S1a and f, ESI†) of the respective crown ether rings: the O1–O2 distances are 0.91 nm for ext4+ and 1.70 nm for cont2+. A difference of 0.79 nm is much smaller than the experimental value 2.2 nm, which suggests that the telescoping between two threads only accounts for a part of the length change in the full [c2]daisy chain. Thus, other contributions affecting its length change must exist.
If we examine the individual left and right halves of one unit in Fig. S1c, d, h and i (ESI†), we clearly observe the planar rigidity of the bulky stoppers, each composed of a terpyridine group together with its adjacent ethinyl and phenyl species. In ext4+, these bulky stoppers are slightly folded away from the long axis of the [c2]daisy chain, while in cont2+, they are strongly folded back around the crown ether rings. As shown in Fig. S1e and j (ESI†), the dihedral angles for the chain of atoms C1–C2–C3–N4, which are highlighted by enlarged balls, are 139.6° and 85.0° for ext4+ and cont2+, respectively. This folding greatly decreases the total length of the contracted form: we measure the distances between two N atoms of the central pyridines at the two ends (which are used for polymerization) as 3.23 nm for cont2+ but 4.82 nm for ext4+. We thus see that the folding of the rigid end groups actually contributes substantially to the expansion/contraction, in addition to the mutual telescoping of the two linear chains.
As shown in Fig. 2, the structure of the intermediate deproton-ext2+ is similar to that of ext4+, while the other intermediate proton-cont4+ has a relatively large distortion as compared with cont2+. To confirm the stability of the [c2]daisy chain in these four supramolecules, we have calculated the frozen binding energy (ΔEb) between the two halves. Here “frozen” means that the optimized geometries of the paired halves are kept unchanged when they are separated. The frozen binding energy is calculated as: ΔEb = Etot([c2]daisy chain) − [Etot(left half) + Etot(right half)]. The single point total energy of the [c2]daisy chain is calculated with the basis set superposition error (BSSE) correction at the level of ωB97XD/6-31G**. As listed in Table 1, the two more negative ΔEb values (−67.33 and −110.97 kcal mol−1) reflect the thermodynamic stability of the equilibrated structures ext4+ and cont2+. The two less negative ΔEb values (−13.83 and −40.70 kcal mol−1) reasonably imply the relative instability of the intermediates proton-cont4+ and deproton-ext2+ as starting points towards the equilibrium states during the muscle-like motion. cont2+ and deproton-ext2+ are more stable than ext4+ and proton-cont4+, respectively. We ascribe this difference to the crowding into a smaller space of the two NH2+ groups of ext4+ and proton-cont4+: their two extra positive charges generate more Coulomb repulsion and higher interior strain energy. Additionally, the ten C–H⋯O hydrogen bonds in cont2+ further improve their stability, even though the C–H⋯O hydrogen bond is usually weaker than the N–H⋯O hydrogen bond.43–45
| Items | μ (Debye) | BSSE-ΔEb (kcal mol−1) |
|---|---|---|
| ext4+ | 2.50 | −67.33 |
| Proton-cont4+ | 5.70 | −13.83 |
| cont2+ | 1.35 | −110.97 |
| Deproton-ext2+ | 1.42 | −40.70 |
| PF6− | 0 | — |
| CH3CN | 3.90 (3.9247) | — |
| CHCl3 | 1.30 (1.0147) | — |
| PF6−(CH3CN) | 0.59 | −10.44 |
| PF6−(CH3CN)2 | 0.00 | −19.81 |
| PF6−(CH3CN)3 | 1.69 | −27.13 |
| PF6−(CH3CN)4 | 0.78 | −33.80 |
| PF6−(CH3CN)5 | 2.27 | −38.67 |
| PF6−(CH3CN)6 | 0.00 | −44.05 |
| PF6−(CH3CN)7 | 2.99 | −51.89 |
| PF6−(CH3CN)8 | 0.02 | −57.96 |
| PF6−(CHCl3) | 5.71 | −10.67 |
| PF6−(CHCl3)2 | 0.063 | −20.26 |
| PF6−(CHCl3)3 | 3.59 | −27.64 |
| PF6−(CHCl3)4 | 1.14 | −35.81 |
| PF6−(CHCl3)5 | 1.75 | −42.33 |
| PF6−(CHCl3)6 | 0.81 | −49.02 |
| PF6−(CHCl3)7 | 0.33 | −54.65 |
| PF6−(CHCl3)8 | 0.86 | −61.88 |
2. Energy profiles and structural evolutions in the vacuum
In this section, we analyze the detailed potential energy profiles during two muscle-like movements and track the structural evolution during the back-and-forth sliding of the DB24C8 rings between the docking stations provided by the NH2+/NH and triazolium cations. We first study the situation in the vacuum by SCC-DFTB-D constrained geometry optimizations. We progressively slide one half of an isolated [c2]daisy chain relative to the other by imposing and freezing a sequence of distances between two pairs of O atoms in the two DB24C8 rings: O1 and O2, as well as O3 and O4 (which are in principle equivalent by symmetry to O1 and O2, respectively, see Fig. S1b, ESI†). At each O1–O2 distance, which is varied in steps of about 0.05 nm, we optimize the positions of all other atoms and obtain the total energy by SCC-DFTB-D calculations. This allows us to track the total energy and structural change as a function of the O1–O2 distance, which represents the ring–ring distance, as shown in Fig. 3. Note that increasing the O1–O2 distance corresponds to contracting the [c2]daisy chain, and vice versa. It should be mentioned that the present constrained search is approximate and cannot completely exclude the existence of alternative equilibration paths. However, it can provide a reasonable qualitative thermodynamic description of the muscle-like motion.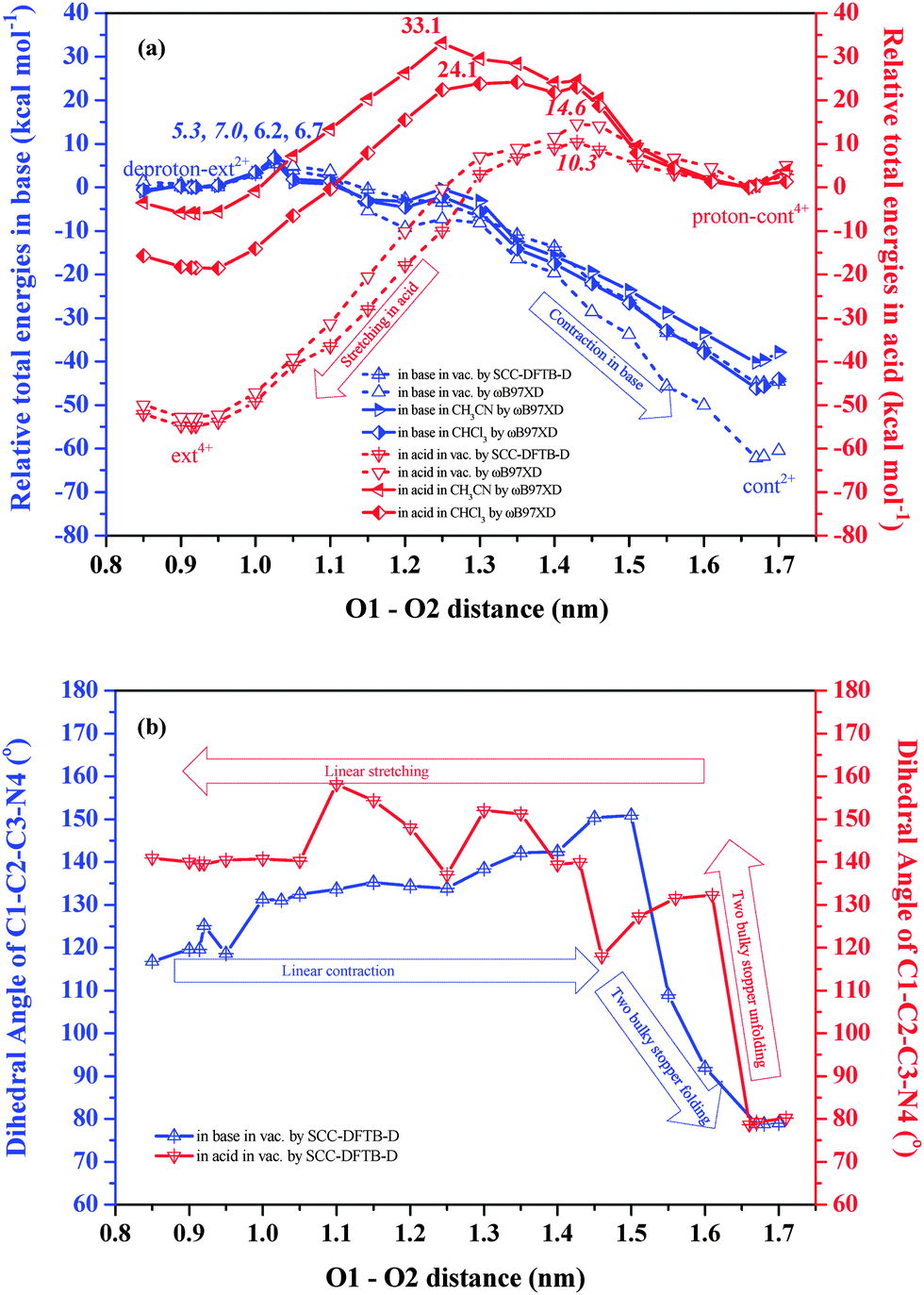 | ||
| Fig. 3 (a) Relative total energy profiles in the vacuum by SCC-DFTB-D constrained geometry optimizations and by ωB97XD/6-31G** single point calculations and in solutions by ωB97XD/6-31G** single point calculations with the consideration of the SMD solvent model of CH3CN/CHCl3, as labeled in the graph (numbers are barrier energies, italicized in vacuum, otherwise in solution), and (b) evolution of the dihedral angle between the backbone (rod) and the rigid bulky stopper by SCC-DFTB-D constrained geometry optimizations in the vacuum as a function of the O1–O2 distance (note that the overall length of the [c2]daisy chain decreases when the O1–O2 distance increases, and vice versa). The contraction of the [c2]daisy chain is shown in blue, while expansion is shown in red, following the arrows' directions. | ||
On the basis of the constrained geometries, we also perform single point energy calculations at the level of ωB97XD/6-31G** to obtain an energy profile comparison between the supramolecule in acid and in base, and between two methods. The energy data are shown in Tables S1 and S2 (ESI†). Fig. 3a displays energy profiles for the stretching in acid (red dashed curve, right to left) and contraction in base (blue dashed curve, left to right) in the vacuum, starting from the intermediates discussed earlier. We find by ωB97XD that, in the vacuum, the contraction must surmount a barrier of 7.0 kcal mol−1 before descending to a net total energy of −62.2 kcal mol−1, while the stretching experiences a barrier of 14.6 kcal mol−1 and a net total energy decrease of −52.8 kcal mol−1. We note that both barriers are comparable to the thermal energy at room temperature (0.6 kcal mol−1) and can thus be overcome by thermal fluctuations. The barrier for stretching is about twice that for contraction. Such an asymmetry implies different kinetic constants and different response velocities under external acid vs. base stimuli: stretching should be significantly slower than contraction. Merely from the viewpoint of conformational changes without temperature and solvent effects, the large total energy decrease (−62.2 and −52.8 kcal mol−1) from the intermediates to the equilibrated structures, following relatively small barriers, is very suitable for muscle-like motion: while thermal energy can overcome these barriers and trigger the equilibration after a pH change, the motion can then not easily be reversed before the pH itself is reversed. Moreover, we find that the stretching barrier (10.3 kcal mol−1) by SCC-DFTB-D is also two times that for contraction (5.3 kcal mol−1), which supports the validity of total energies predicted by this semi-empirical method.
An animation (Movie S1) showing our detailed model of the full contraction and elongation cycle of the [c2]daisy chain is available in the ESI.† The relative total energies and dihedral angles are listed in Tables S1 and S2 of the ESI.† Selected frames are shown in Fig. S2 (ESI†). Before the contraction process, the O1–O2 distance of deproton-ext2+ is about 0.91 nm. As the O1–O2 distance increases, the total energy of the conformation rises until reaching the barrier at 1.03 nm where the transition state is reached (see Frame 5b in Fig. S2, ESI†). The barrier is due to breaking the four-layered π–π stacking in the central part of the [c2]daisy chain. The geometry changes little from deproton-ext2+ to the barrier. In contrast, the initial stretching process from cont2+ (see Fig. S1f, ESI†) has already shown large structural changes in going to the intermediate (see Frame 1a). The backbones of the two chains (rods in Fig. 1) arch away from each other due to the Coulomb repulsion between two NH2+ ions. As a result, the transition state (see Frame 7a) appears at a relatively larger O1–O2 distance of 1.43 nm. The geometry at the barrier is quite different from the starting structure proton-cont4+ and already close to that of ext4+. The energy barrier caused by structural deformation is now mainly due to the DB24C8 rings breaking loosely from the triazolium cations. Hence, the stretching in acid should be more difficult and slower than the contraction in base.
One key reason for different barriers in contraction vs. extension is the folding of two bulky stoppers occurring in the last phase of contraction, which contrasts with the unfolding in the first phase of stretching. This change can be described by the dihedral angle between the backbone and the bulky stopper within each half, as sketched in Fig. S1e and j (ESI†). We measure this dihedral angle for the chain of atoms C1–C2–C3–N4 to track the folding and unfolding of the bulky stoppers as a function of the O1–O2 distance. As shown in Fig. 3b, for the contraction in base, when the O1–O2 distance grows toward 1.50 nm, the dihedral angle gradually increases from ∼120° to ∼150°. The configuration change in this process originates mainly from the [c2]daisy chain linear contraction. When the O1–O2 distance exceeds 1.50 nm, the dihedral angle rapidly decreases to ∼80°, reflecting the folding of the two bulky stoppers until the final cont2+ is produced. For the stretching in acid, the dihedral angle at first rapidly increases from ∼80° to ∼130° as the O1–O2 distance is reduced from 1.67 nm to 1.61 nm, indicating that the two bulky stoppers are quickly unfolding. Afterwards, the dihedral angle steps up and down, until by 1.05 nm its value stabilizes at ∼140°. In particular, in the 1.61–1.46 nm range, we find that the molecule is trying to encapsulate the NH2+ into the DB24C8 ring (see Frame 6a). Accordingly, due to the DB24C8 covering NH2+, the large deformation generates a higher barrier for stretching than contraction. After 1.43 nm, the process corresponds to the smooth stretching of the [c2]daisy chain until ext4+ is formed. In this phase, the hydrogen bonds of N–H⋯O/ N+–H⋯O and π–π stacking among phenol rings are formed, which also impacts the dihedral angle between the backbone and the bulky stopper.
3. Solvent effects
Experimentally, the muscle-like polymer was synthesized in a solution of CH3CN and CHCl3 with a mixing ratio of 1:1.21 Therefore, we have also evaluated the [c2]daisy chain thermal energy data in an SMD solvation model by ωB97XD/6-31G**. The results are included in Fig. 3a (full lines). Clearly, the stretching barriers in both solutions (33.1 and 24.1 kcal mol−1 in CH3CN and CHCl3) are much higher than in the vacuum (14.6 kcal mol−1) and at the same time the reaction heats in solution (−6.0 and −18.6 kcal mol−1 in CH3CN and CHCl3) become much smaller than in the vacuum (−52.8 kcal mol−1). This clearly means that the solvents will significantly hinder the stretching movement in acid. In contrast, the solvent effect on the contraction in base is rather small with energy profiles similar to the vacuum in the initial part of the path but becoming somewhat different in the final stage. Altogether, in solvents the contraction barriers for a single [c2]daisy chain are found to be much lower than those for stretching, analogous to the results in the vacuum which confirms that stretching will be slower than contraction. The acid-promoted elongation of the polymer may thus also occur during the cyclic muscle-like motion.
To further clarify the solvent effect on the contraction and stretching processes, we first perform the SCC-DFTB-D/MD simulations by two equilibrium configurations and two intermediate states in an environment of 413 molecules of CH3CN. Here we only discuss CH3CN as a typical model of the solution environment. After 1 ps, the four configurations remain almost the same as in the vacuum, as compared visually in Fig. 4. This means that the [c2]daisy chain in these four states is well stabilized in the organic solvents. As shown in Table S3 (ESI†), in the final 100 fs, the root mean square deviation (RMSD) around room temperature is 3.9 to 5.5 K and the RMSD of the potential energy is 25 to 31 kcal mol−1. We assume that the system has reached equilibrium at this stage. According to the average potential energies calculated in the last 100 fs, the two reaction heats in solution are −32.0 kcal mol−1 for the contraction and −12.1 kcal mol−1 for the stretching, as listed in Table 2. Compared with the results in the vacuum via full atom relaxations and constrained geometry optimization by SCC-DFTB-D, it is found that the energy differences in the solution are significantly reduced, especially for the stretching process.
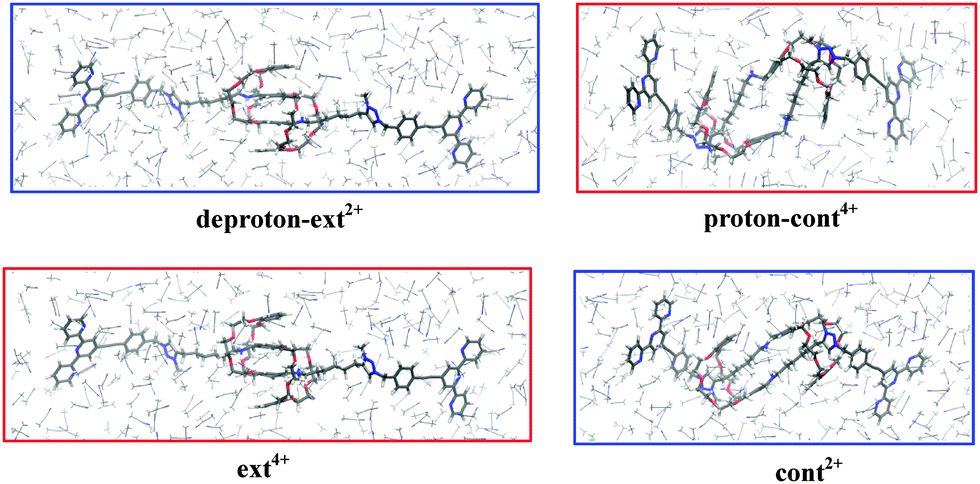 | ||
| Fig. 4 SCC-DFTB-D/MD frames of two equilibria and two intermediates at 1 ps in an environment of 413 CH3CN molecules. | ||
| Muscle-like motion | ΔE (full opt. in vac.) | ΔE (four O fixed opt. in vac.) | ΔE (NVT in sol.) |
|---|---|---|---|
| Contraction in base | −46.8 | −45.1 | −32.0 |
| Stretching in acid | −49.9 | −54.9 | −12.1 |
We believe that the dominant effect resulting in energy differences in acid as compared to base solution is due to unequal electrostatic interactions of the cationic [c2]daisy chain with polar solvents. An opposite example indirectly justifies this speculation. As reported in a recent study,46 the free-energy landscape of a similar but neutral [c2]daisy chain rotaxane shows nearly equal energy barriers for both contraction and extension processes, with values of 13.65 ± 0.04 kcal mol−1 derived from 1H NMR at 228 K in a solution of CDCl3. Therefore, we have calculated and compared corresponding dipole moments, μ, of four [c2]daisy chains. As shown in Table 1, the μ values are in the order cont2+ < deproton-ext2+ < ext4+ < proton-cont4+. The μ of proton-cont4+ is rather high. Thus, we may expect that the potential energy of proton-cont4+ will be dramatically decreased in solution so as to reduce the reaction heat and simultaneously increase the energy barrier during the stretching process. Fig. 5a–d display the ESP distributions of the four supramolecules on the vdW surface, defined at an electron density of 0.001 e Bohr−3. Ranging from −125.5 to +125.5 kcal mol−1, a stronger ESP distribution (dark blue) is found from proton-cont4+ to ext4+ and a milder spread (mostly light blue) is observed from deproton-ext2+ to cont2+. This implies again that the solvent effects during the stretching process will be stronger than during contraction. Additionally, the two bulky stoppers of these supramolecules are found to have smaller ESP values than the central sections. Although the ESP in the center is strongly impacted by solvents, the two ends still have greater opportunities to undergo a polymerization reaction with Fe2+ cations.
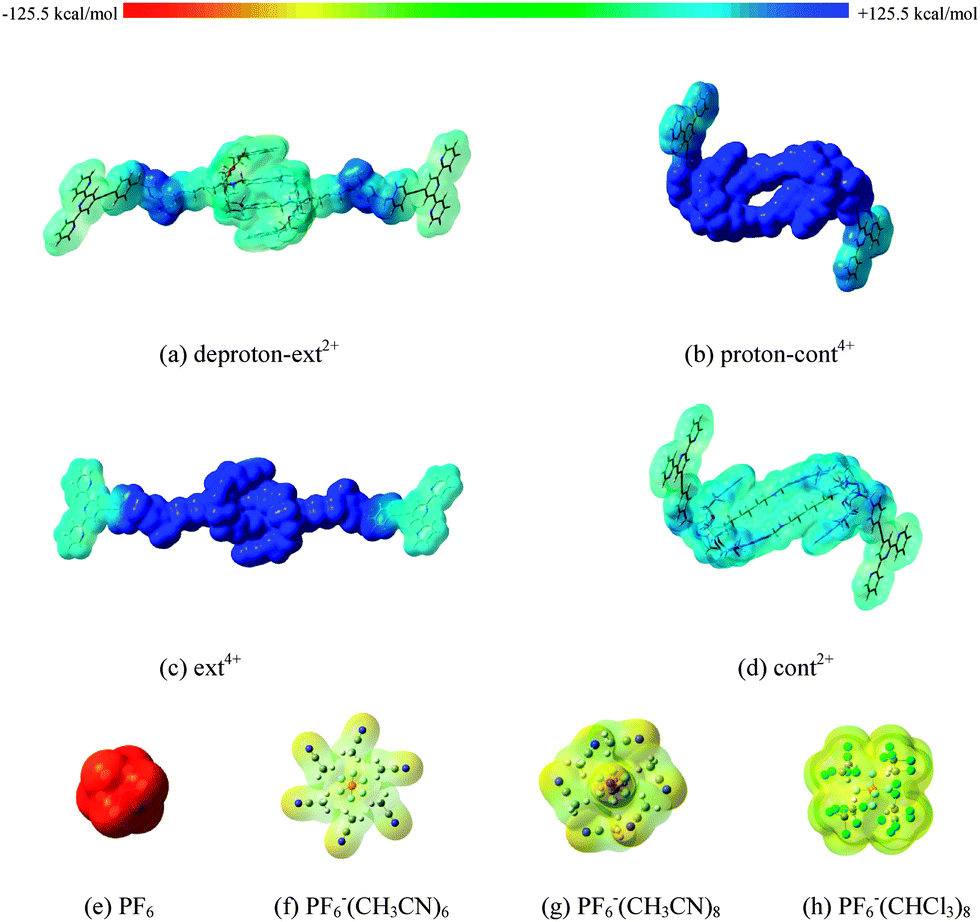 | ||
| Fig. 5 Color-coded electrostatic potential (ESP) distributions on vdW surfaces defined at an electron density of 0.001 e Bohr−3 (isovalue = 0.001) ranging from −125.5 to +125.5 kcal mol−1 of four supramolecules, PF6−, PF6−(CH3CN)6, PF6−(CH3CN)8 and PF6−(CHCl3)8, calculated by ωB97XD/6-31G**. The color bar at the top refers to a linear scale. | ||
Last, we have to consider the effect of solvation on the PF6− counterions that are present experimentally in the solvent to neutralize the charges of the [c2]daisy chains. In fact, we find that flexible combinations between small ions like PF6− and solvent molecules can easily occur, i.e., solvents could reduce the direct contact between PF6− and [c2]daisy chain cations. At the level of ωB97XD/6-31G**, stable configurations of PF6−(CH3CN)n or PF6−(CHCl3)n (n = 1–8) complexes are found, see Fig. S3 (ESI†), which are confirmed to have no imaginary frequencies. They belong to weak C–H⋯F hydrogen bonding complexes, where two or three H⋯F distances within 0.30 nm are formed between each solvent molecule and PF6−. As listed in Table 1, the BSSE-ΔEb values in these complexes can reach up to −62 kcal mol−1, which indicate favorable aggregations between PF6− and solvents. More importantly, the ESP of PF6− can be significantly reduced after binding with CH3CN or CHCl3 molecules, judging from the changes illustrated in Fig. 5e–h. Without solvents, a direct contact of PF6− will strongly distort the cationic [c2]daisy chain, especially the extended form, and likely result in losing the muscle-like function, according to our tests on the direct interaction between the [c2]daisy chain and PF6− by PM6 semi-empirical optimizations. Our cations in the [c2]daisy chain are not expected to exhibit a strong Coulomb interaction with PF6−. The solvents provide a soft negative ESP environment for supramolecules to avoid strong electrostatic interactions between the [c2]daisy chain and surrounding PF6− and keep the [c2]daisy chain in a working state during the muscle-like motion.
4. Polymer forms
Finally, we consider briefly the polymerized version of the [c2]daisy chain (see Fig. 6) where the terpyridine end groups of individual [c2]daisy chains are mutually linked via Fe2+ ions in a periodic fashion. The rotaxanes in the polymers are repeated in a zigzag chain allowing contraction and elongation of the individual units to be multiplied by the number of units in the polymer. With optimization by SCC-DFTB-D of geometry and repeat cell, the optimum unit lengths for the extended and contracted polymers are found to be 5.38 and 3.70 nm, respectively. The calculated extended unit length fits the experimental one (5.4 nm) well, while the contracted unit length is somewhat larger than in the experiment (3.2 nm).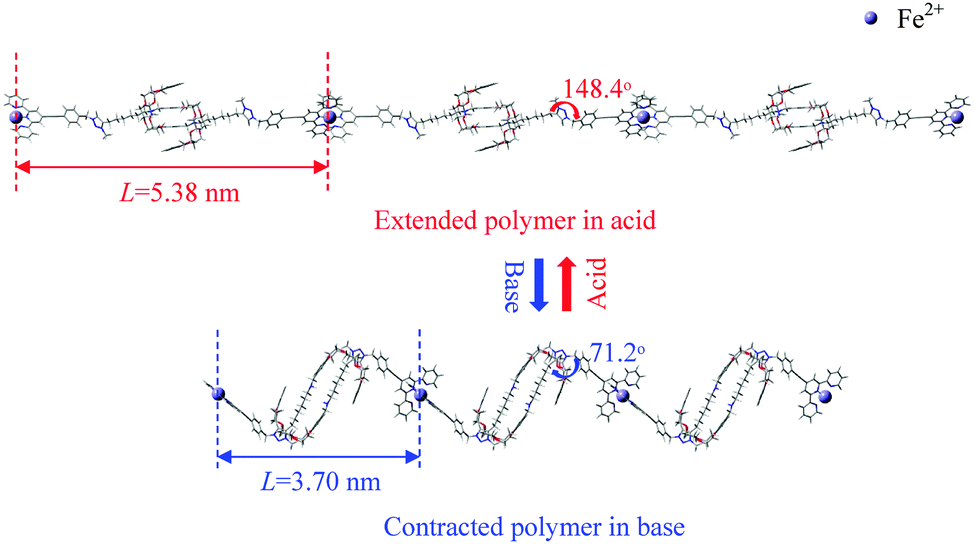 | ||
| Fig. 6 The extended vs. contracted polymer (i.e. Fe2+-linked [c2]daisy chains) confirmed by SCC-DFTB-D geometry and cell optimizations. The repeat length of the polymer unit L and dihedral angle C1–C2–C3–N4 are labeled in red and blue, resp. | ||
In our case, the dihedral angles of C1–C2–C3–N4 in the two polymerized forms are 148.4° and 71.2°, respectively. Compared with the single [c2]daisy chains (having dihedral angles of 139.6° and 85.0°, respectively), Fe2+ unfolds the two bulky stoppers somewhat in the extended form and folds them more in the contracted form due to the 6-fold coordination reaction. Although the muscle-like motion of the polymer will be impacted by the folding/unfolding of the bulky stoppers to different extents, which flexibly determines the unit length, the pathway of the contraction and stretching in the polymer is expected to be in principle consistent with the monomer, as the geometry of the [c2]daisy chain in the polymer is basically the same as in a single unit. Thus, we believe that our muscle-like motion simulated by a single [c2]daisy chain gives a realistic and informative understanding of the detailed motion.
A final remark on the energetics of this muscle-like system is in order: it is not a perpetual motion machine since we need to repeatedly inject acid or base with the concomitant accumulation of waste products and consumption of new energy. In a natural muscle also, it is the chemical energy that is being converted into mechanical energy which is not accounted for by the present simulation.
Summary and remarks
In summary, at the atomic level, we have given a detailed description of how a highly promising rotaxane executes muscle-like mechanical motion under acidic vs. basic conditions. We determine its equilibrated and intermediate forms and find that the change in length comes from two main mechanisms: the back-and-forth telescoping of the DB24C8 rings between NH2+ and triazolium ionic docking sites, as proposed in the literature, and also, to a significant extent, the folding vs. unfolding of two bulky stoppers; π–π stacking and multiple hydrogen bonds as well contribute to the stabilization of the energetics of the process. We present activated exothermic processes for both the expansion and the contraction by evaluating the corresponding energy barriers.The stretching is found to be more difficult and slower than the contraction as determined by the highly unequal energy barriers and reaction heats in the vacuum and in solution, electrostatic potential distributions, and dipole moment comparison. The difference occurs in the particular protonated contraction intermediate, which undergoes a large conformation distortion and has a strong interaction with solvents due to its large dipole moment. In addition, by studying weak hydrogen bonding complexes between PF6− and CH3CN/CHCl3, we conclude that solvation plays an important role in effectively reducing the strong electrostatic interactions between [c2]daisy chain cations and PF6− counterions and provides a soft polar environment for the muscle-like motion of the [c2]daisy chain polymer. Our understanding can be helpful to enable the desired muscle-like motion at a larger length scale in experiments with sufficiently high external excitation using for example reasonably high temperature and further optimized solution.
Acknowledgements
We thank the High Performance Cluster Computing Centre, Hong Kong Baptist University, which receives funding from the Research Grants Council, University Grants Committee of the HKSAR and HKBU. This work is supported in part by project no. 9041650 of the Research Grants Council of HKSAR. MAVH and YLZ were supported in part by the HKBU Strategic Development Fund. CM acknowledges support from HKBU's Sabbatical Visitor scheme and partial support from the HKBU Strategic Development Fund. KH acknowledges support from HKBU's Distinguished Scholar scheme and partial support from the HKBU Strategic Development Fund.References
- R. A. Bissell, E. Córdova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133–137 CrossRef CAS PubMed.
- M.-V. Martínez-Díaz, N. Spencer and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1997, 36, 1904–1907 CrossRef PubMed.
- P. R. Ashton, R. Ballardini, V. Balzani, I. Baxter, A. Credi, M. C. T. Fyfe, M. T. Gandolfi, M. Gómez-López, M.-V. Martínez-Díaz, A. Piersanti, N. Spencer, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 11932–11942 CrossRef CAS.
- P. G. Clark, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 13631–13633 CrossRef CAS PubMed.
- Z. Zhang, C. Han, G. Yu and F. Huang, Chem. Sci., 2012, 3, 3026–3031 RSC.
- P. Gaviña and J.-P. Sauvage, Tetrahedron Lett., 1997, 38, 3521–3524 CrossRef.
- L. Raehm, J.-M. Kern and J.-P. Sauvage, Chem. – Eur. J., 1999, 5, 3310–3317 CrossRef CAS.
- N. Armaroli, V. Balzani, J.-P. Collin, P. Gaviña, J.-P. Sauvage and B. Ventura, J. Am. Chem. Soc., 1999, 121, 4397–4408 CrossRef CAS.
- R. Ballardini, V. Balzani, W. Dehaen, A. E. Dell'Erba, F. M. Raymo, J. F. Stoddart and M. Venturi, Eur. J. Org. Chem., 2000, 591–602 CrossRef CAS.
- J.-P. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS PubMed.
- Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, B. H. Northrop, H.-R. Tseng, J. O. Jeppesen, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. D. Solares, W. A. Goddard, C.-M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745–9759 CrossRef CAS PubMed.
- S. Saha, A. H. Flood, J. F. Stoddart, S. Impellizzeri, S. Silvi, M. Venturi and A. Credi, J. Am. Chem. Soc., 2007, 129, 12159–12171 CrossRef CAS PubMed.
- G. Fioravanti, N. Haraszkiewicz, E. R. Kay, S. M. Mendoza, C. Bruno, M. Marcaccio, P. G. Wiering, F. Paolucci, P. Rudolf, A. M. Brouwer and D. A. Leigh, J. Am. Chem. Soc., 2008, 130, 2593–2601 CrossRef CAS PubMed.
- A. C. Benniston and A. Harriman, Angew. Chem., Int. Ed. Engl., 1993, 32, 1459–1461 CrossRef PubMed.
- A. C. Benniston, A. Harriman and V. M. Lynch, Tetrahedron Lett., 1994, 35, 1473–1476 CrossRef CAS.
- H. Murakami, A. Kawabuchi, K. Kotoo, M. Kunitake and N. Nakashima, J. Am. Chem. Soc., 1997, 119, 7605–7606 CrossRef CAS.
- P. R. Ashton, R. Ballardini, V. Balzani, A. Credi, K. R. Dress, E. Ishow, C. J. Kleverlaan, O. Kocian, J. A. Preece, N. Spencer, J. F. Stoddart, M. Venturi and S. Wenger, Chem. – Eur. J., 2000, 6, 3558–3574 CrossRef CAS.
- A. Harada, Acc. Chem. Res., 2001, 34, 456–464 CrossRef CAS PubMed.
- R. E. Dawson, S. F. Lincoln and C. J. Easton, Chem. Commun., 2008, 3980–3982 RSC.
- M. C. Jiménez, C. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284–3287 CrossRef.
- G. Du, E. Moulin, N. Jouault, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2012, 51, 12504–12508 CrossRef CAS PubMed.
- C. J. Bruns and J. F. Stoddart, Nat. Nanotechnol., 2013, 8, 9–10 CrossRef CAS PubMed.
- J. Wu, K. C.-F. Leung, D. Benítez, J.-Y. Han, S. J. Cantrill, L. Fang and J. F. Stoddart, Angew. Chem., Int. Ed., 2008, 47, 7470–7474 CrossRef CAS PubMed.
- L. Fang, M. Hmadeh, J. Wu, M. A. Olson, J. M. Spruell, A. Trabolsi, Y.-W. Yang, M. Elhabiri, A.-M. Albrecht-Gary and J. F. Stoddart, J. Am. Chem. Soc., 2009, 131, 7126–7134 CrossRef CAS PubMed.
- C.-J. Chuang, W.-S. Li, C.-C. Lai, Y.-H. Liu, S.-M. Peng, I. Chao and S.-H. Chiu, Org. Lett., 2009, 11, 385–388 CrossRef CAS PubMed.
- Y. Jiang, J.-B. Guo and C.-F. Chen, Org. Lett., 2010, 12, 4248–4251 CrossRef CAS PubMed.
- M. Hmadeh, L. Fang, A. Trabolsi, M. Elhabiri, A.-M. Albrecht-Gary and J. F. Stoddart, J. Mater. Chem., 2010, 20, 3422–3430 RSC.
- P. R. Ashton, I. Baxter, S. J. Cantrill, M. C. T. Fyfe, P. T. Glink, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 1294–1297 CrossRef CAS.
- M. O. Sinnokrot and C. D. Sherrill, J. Phys. Chem. A, 2004, 108, 10200–10207 CrossRef CAS.
- M. P. Waller, A. Robertazzi, J. A. Platts, D. E. Hibbs and P. A. Williams, J. Comput. Chem., 2006, 27, 491–504 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- R. M. Parrish and C. D. Sherrill, J. Am. Chem. Soc., 2014, 136, 17386–17389 CrossRef CAS PubMed.
- G. Seifert, J. Phys. Chem. A, 2007, 111, 5609–5613 CrossRef CAS PubMed.
- B. Aradi, B. Hourahine and T. Frauenheim, J. Phys. Chem. A, 2007, 111, 5678–5684 CrossRef CAS PubMed.
- D. Porezag, T. Frauenheim, T. Köhler, G. Seifert and R. Kaschner, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 12947–12957 CrossRef CAS.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- T. Frauenheim, G. Seifert, M. Elstner, Z. Hajnal, G. Jungnickel, D. Porezag, S. Suhai and R. Scholz, Phys. Status Solidi B, 2000, 217, 41–62 CrossRef CAS.
- M. Elstner, T. Frauenheim, E. Kaxiras, G. Seifert and S. Suhai, Phys. Status Solidi B, 2000, 217, 357–376 CrossRef CAS.
- T. Frauenheim, G. Seifert, M. Elstner, T. Niehaus, C. Köhler, M. Amkreutz, M. Sternberg, Z. Hajnal, A. D. Carlo and S. Suhai, J. Phys.: Condens. Matter, 2002, 14, 3015–3047 CrossRef CAS.
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model., 2001, 7, 306–317 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- G. R. Desiraju and B. N. Murty, Chem. Phys. Lett., 1987, 139, 360–361 CrossRef CAS.
- F. H. Allen, J. A. K. Howard, V. J. Hoy, G. R. Desiraju, D. S. Reddy and C. C. Wilson, J. Am. Chem. Soc., 1996, 118, 4081–4084 CrossRef CAS.
- G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441–449 CrossRef CAS PubMed.
- C. J. Bruns, J. Li, M. Frasconi, S. T. Schneebeli, J. Iehl, H.-P. J. de Rouville, S. I. Stupp, G. A. Voth and J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 1953–1958 CrossRef CAS PubMed.
- R. D. Nelson, Jr., D. R. Lide and A. A. Maryott, NSRDS-NBS 10, 1967.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp00315f |
| This journal is © the Owner Societies 2015 |