
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu(I) stabilizing crosslinked polyethyleneimine
Alireza
Movahedi
a,
Angelica
Lundin
a,
Nina
Kann
a,
Magnus
Nydén
*b and
Kasper
Moth-Poulsen
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-41296 Göteborg, Sweden. E-mail: kasper.moth-poulsen@chalmers.se; Tel: +46 31 772 3403
bIan Wark Research Institute, University of South Australia, Adelaide, SA 5001, Australia. E-mail: magnus.nyden@unisa.edu.au; Fax: +61 8 8302 3683; Tel: +61 8 8302 3203
First published on 18th June 2015
Abstract
Polyethyleneimine (PEI) is known for its metal-coordinating properties and in its crosslinked form has applications in different areas ranging from drug delivery to waste water treatment and recovery of trace metals. With the aim to regulate the coordination environment of Cu(I) and Cu(II) ions in marine coatings, we have prepared a triazole cross-linking agent with ‘soft’ coordination that can crosslink PEI via indirect reductive amination. We have shown that this triazole crosslinker not only increases the Cu(II) absorption capacity of the crosslinked PEI in comparison to the traditionally used glutaraldehyde-crosslinked PEI (PEI-GA), but also allows the crosslinked polymer network to stabilize the Cu(I) oxidation state more effectively. The Cu(II) uptake and Cu(I) stabilization of the polymer have been determined by elemental analysis and UV-vis spectroscopy. It was found that the triazole-crosslinked polymer (PEI-TA) could coordinate up to 12 wt% of Cu(II) before and 6 wt% Cu(II) after imine reduction.
1 Introduction
Trace metals such as Cu, Fe, and Zn are essential elements for most living organisms.1 At the same time, these metals are also hazardous to the environment and human health in higher concentrations.1–3 Due to their toxicity to marine species, Cu and Zn have been used in coatings to combat marine foulers that settle and grow on ship hulls.4,5 These foulers are a big challenge for marine transportation and increase fuel consumption and maintenance. However, the release of toxic substances into seawater is a major problem especially in harbours and other areas with limited water circulation. This has led to regulatory restrictions on the use and release of copper in some countries6,7 and there is a need to find environmentally friendly solutions to the problem of marine fouling.Both Cu(I) and Cu(II) are efficient anti-fouling agents.8,9 However, Cu(I) is unstable under aerobic conditions and rapidly oxidizes to Cu(II) in seawater.10,11 It is widely accepted that it is Cu(I) that passes through the biological membrane of organisms,12,13 and that the coordination environment of most biological cells favours Cu(I).14 Our working hypothesis is that by changing the coordinating characteristics of a coating, the redox potential for the Cu(I)/Cu(II) pair is attuned so that the concentration of Cu(I) increases, leading to a better antibiofouling efficiency.15 Maintaining such a window where the redox cycle of Cu(I)/Cu(II) occurs also allows the Cu ions to impose the most toxic effect.16 This work is one step towards developing a new type of ‘green’ coating that requires no toxic biocide additives, but instead makes use of the copper already present in seawater. The aim with this type of coating is to create a flux of copper at the coating interface; in effect, a permanent cyclic mechanism of copper uptake and release into and out of the coating. Of particular interest for this purpose are coordinating polymers that can bind to both Cu(II) and Cu(I) ions.
Tris[(benzyltriazolyl)methyl]amine (TBTA) (Fig. 1) is a tripodal ligand that has found applications in Cu(I)-catalysed reactions since its first introduction in 2004 by Fokin et al.17 Both the presence of triazole groups and the tripodal structure of this ligand, which can adapt to the different geometries required for Cu(I) and Cu(II) binding, have made it suitable for applications where coordination to both these ions is of importance. The calculated optimized structures of TBTA and its complexes with Cu(I) and Cu(II) are shown in Fig. 1, where the [Cu(I)2(TBTA)2] complex forms a distorted tetrahedral while the Cu(II) complex forms a distorted trigonal bipyramidal structure with the formula of [Cu(II)Cl(TBTA)]. The calculated structures are also in line with the reported crystal structures for these complexes.18 We have previously reported the synthesis and characterization of coordinating polymers functionalized by TBTA (poly(TBTA)) via a one-pot click reaction procedure.19 We demonstrated that a thin poly(TBTA) film can absorb up to 76 mg g−1 (7.1 wt%) of Cu(II) from an aqueous solution of copper. However, despite its performance, the multi-step synthesis process of poly(TBTA) can be a drawback for large-scale applications. Therefore, we here present alternative amine-based polymeric structures via a simpler synthetic route which possess a similar ‘soft’ coordinating character.
 | ||
| Fig. 1 (a) Tris[(benzyltriazol)methyl]amine (TBTA); (b) [Cu(I)2(TBTA)2]; and (c) [Cu(II)Cl(TBTA)], (b) and (c) are calculated at PBE/DNP(4.4). | ||
Polyethyleneimine (PEI) has a high affinity and selectivity for Cu ions.20–22 A common way to increase the stability of PEI on a surface is by chemical cross-linking using glutaraldehyde23,24 or epichlorohydrine.25 The nature of the cross-linker can alter the affinity and selectivity of PEI to metal ions. Moreover, its crosslinked form allowing reuse after regeneration,25,26 making it an interesting polymer for industrial uses such as waste water treatment.26 Also the selectivity can be changed by modifying the crosslinked structure with other coordinating groups.27,28 Considering the ability of PEI to bind to Cu(II), we hypothesise that PEI crosslinked with a triazole group will coordinate both Cu(II) and Cu(I) effectively. The crosslinker employed here consists of a triazole ring with aldehyde functionalities on the 1- and 4-positions. For convenience, we refer to the crosslinker as a triazole dialdehyde (TA). The cross-linked network of PEI-TA is also expected to be able to satisfy the different coordination geometries required for Cu(I) and Cu(II), i.e. 4-coordinated tetrahedral for Cu(I) and 5-coordinated trigonal bipyramidal for Cu(II). Glutaraldehyde (GA) was used as a reference crosslinker. It has already been shown that a cross-linked PEI-GA film can absorb copper from extremely dilute copper solutions.29,30 Our focus here is different since we aim to tune the affinity of the film towards a specific oxidation state of copper. To investigate the versatility of introducing triazole groups, we have also functionalized PEI-GA with a mixture of (benzyl)triazole carbaldehyde (BTA) and GA to achieve a Cu(I) stabilizing structure. To prevent further hydrolysis the imine bonds in the cross-linked polymers were reduced to amine-functionalities.31 The effect of this reduction on the copper uptake was also studied. Scheme 1 shows the reagents as well as the cross-linking reactions to prepare PEI-GA, PEI-TA, and PEI-GA-BT.
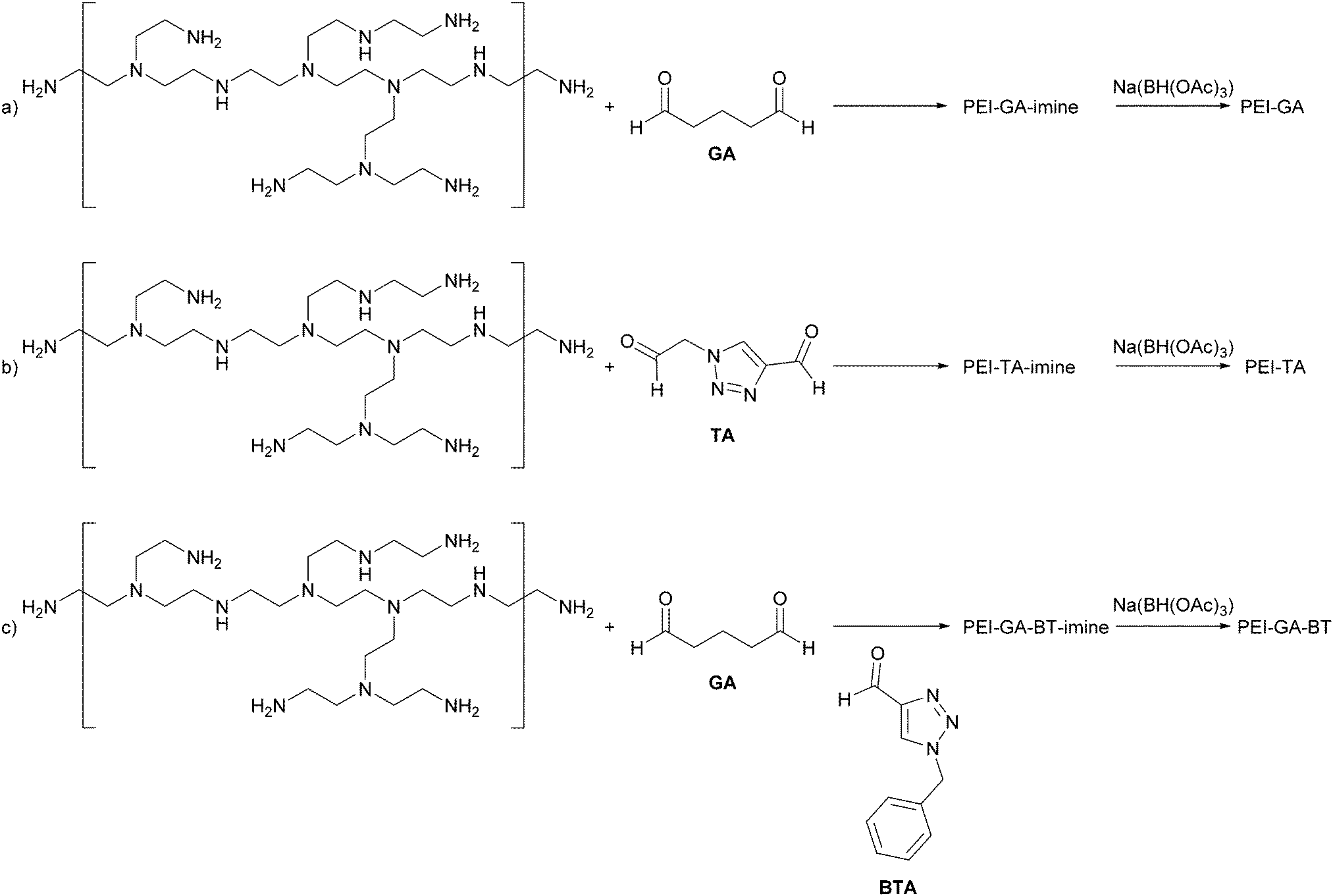 | ||
| Scheme 1 (a) Glutaraldehyde-crosslinked PEI (PEI-GA); (b) triazoledialdehyde-crosslinked PEI (PEI-TA); and (c) (benzyl)triazole-functionalized glutaraldehyde-crosslinked PEI (PEI-GA-BT). | ||
2 Experimental section
2.1 Materials and reagents
1-Benzyl-1H-1,2,3-triazole-4-carbaldehyde (BTA, Scheme 1) was prepared according to a previously reported procedure (Scheme 2).32 In this work, we have used a branched polyethyleneimine from Sigma Aldrich (average Mw ∼25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 by LS, average Mn ∼10000 by GPC, branched). Benzyl azide was purchased from Alfa Aesar. Remaining chemicals were purchased from Sigma-Aldrich.
000 by LS, average Mn ∼10000 by GPC, branched). Benzyl azide was purchased from Alfa Aesar. Remaining chemicals were purchased from Sigma-Aldrich.
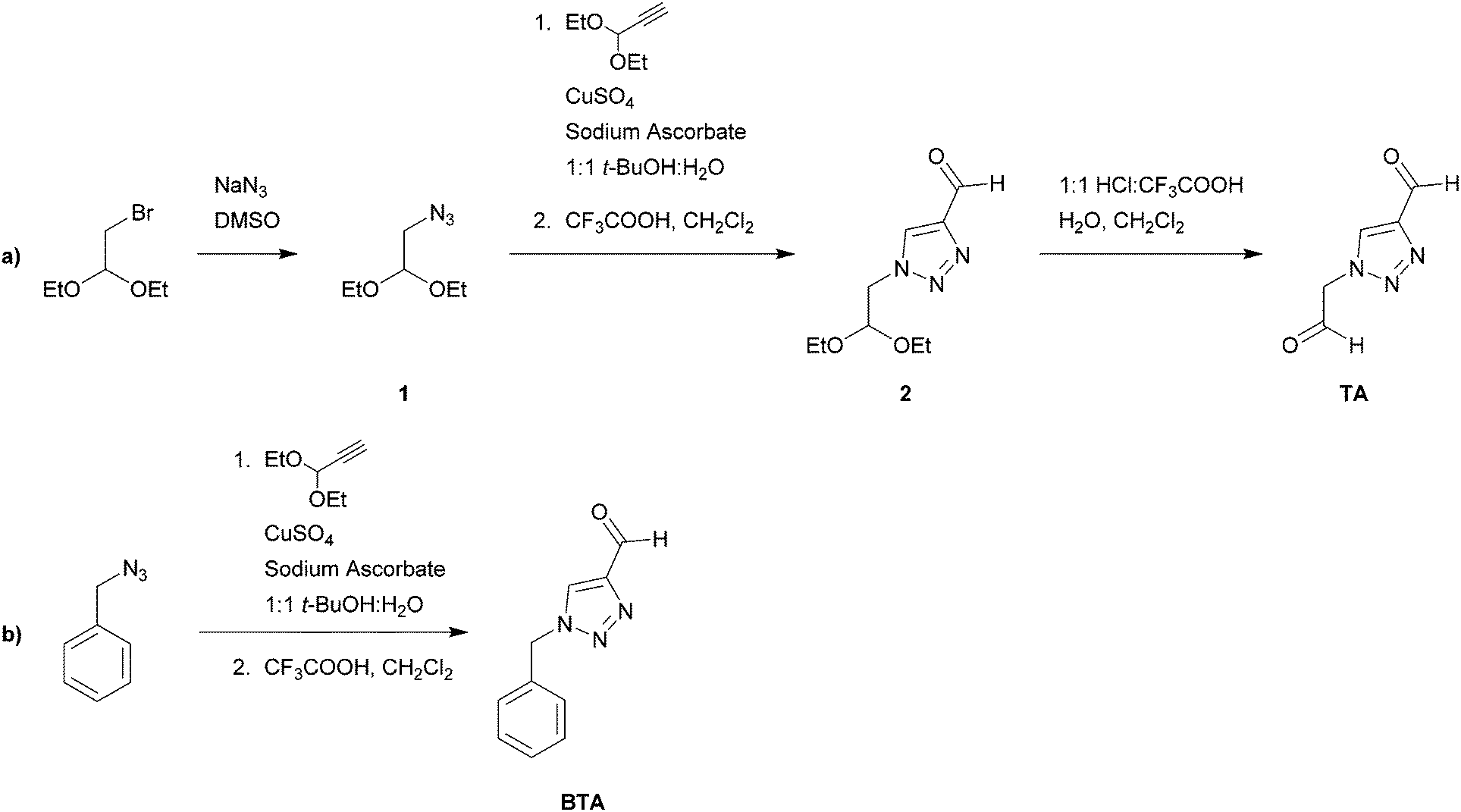 | ||
| Scheme 2 Synthetic procedures for (a) the TA crosslinker; (b) benzyl(triazole) aldehyde (BTA). | ||
2.2 Characterizations
FTIR-ATR spectra were recorded on a Perkin Elmer system Frontier (at a resolution of 2 cm−1 with 20 scans) The UV-vis measurements were carried out using a Perkin Elmer Lambda 900 UV/Vis/NIR spectrometer. Elemental analysis was carried out by Mikroanalytisches Laboratorium KOLBE (http://www.mikro-lab.de). Scanning electron microscopy (SEM) was performed with a Leo Ultra 55 FEG system equipped with an Oxford Inca EDS X-ray microanalysis system for energy dispersive X-ray spectroscopy (EDS) analysis.2.3 Synthesis
Scheme 2 shows the synthetic procedures for the crosslinker TA as well as the functional group BTA.:H2O (1:1, 15 mL), diethylpropargyl acetal (0.68 g, 5.00 mmol), sodium bicarbonate (0.10 g), CuSO4 1 M solution (0.4 mL, 10 mol%), and sodium ascorbate (0.30 g, 30 mol%) were added. The reaction mixture was stirred for 24 hours at 40 °C. Diethyl ether (50 mL) was added and the phases were separated. The organic phase was washed with saturated NaHCO3 (3 × 30 mL) and brine (2 × 50 mL), dried over MgSO4 and concentrated under vacuum to afford a yellow oil (0.89 g). This oil was then dissolved in dichloromethane (4 mL), followed by the addition of water (1 mL) and trifluoroacetic acid (TFA) (1 mL). The mixture was stirred for 12 hours. Diethyl ether (10 mL) was added and the organic phase was separated and washed with saturated NaHCO3 (3 × 40 mL) and brine (1 × 40 mL), dried over MgSO4, and concentrated under vacuum to give a yellow oil (0.30 g, 1.40 mmol, 33% over two steps). 1H NMR (400 MHz, CDCl3) δ 10.10 (s, 1H, CHO), 8.19 (s, 1H, H-triazole), 4.76 (t, 1H, O–CH–O), 4.49 (d, 2H, N–CH2–CH), 3.75–3.67 (m, 2H, O–CH2–CH3), 3.52–3.44 (m, 2H, O–CH2–CH3), 1.15 (t, 6H, –CH3), 13C NMR (400 MHz, CDCl3) δ 185.0, 147.7, 126.7, 100.1, 63.8, 52.9, 15.1. Elemental analysis: calc. for C9H15N3O3: C, 50.69; H, 7.09; N, 19.71; O, 22.51. Found: C, 50.48; H, 7.01; N, 19.42; O, 23.09.
:EDTA:sodium ascorbate solution (2 mM:10 mM:1 mM) for another 12 hours for the reduction and exchange between the copper loaded interface of polymer and the solution to proceed.
:EDTA:sodium ascorbate solution (2 mM:10 mM:1 mM) for additional 12 hours to allow time for equilibrium. The resulted suspensions were then centrifuged and 1 mL of the aqueous phase was diluted by adding 4 mL of deionized water just before the UV-vis absorbance measurements (250–800 nm).
3 Results and discussion
Upon the reaction of primary amines with aldehyde, imine bonds form while water is eliminated. To minimize the negative effect of the presence of initial water in the reaction environment, the initial cross-linking has been carried out in a relatively large volume of acetonitrile to dilute water. The miscibility of acetonitrile and water prevents the phase separation of this solution. Also, this method provide the flexibility to introduce other functional groups which may not be water soluble, but are still soluble in an organic solvent such as acetonitrile as BTA. Nevertheless, after initial formation of the crosslinked polymer particles, this latex solution is centrifuged and the particles were dried to remove water. This together with further reduction of the imine bonds to amine stabilizes the particle and prevents from hydrolysis.3.1 Elemental analysis of crosslinked polymers
The elemental analysis of the three crosslinked polymers in Table 1 shows that PEI-GA has 12.6 wt% of nitrogen while the value is 20.3 wt% for PEI-TA which has the triazole group as a part of its crosslinker. For the PEI-GA-BT structure, both the nitrogen and carbon content are higher than for PEI-GA due to the additional benzyl and triazole groups.| C (wt%) | H (wt%) | N (wt%) | |
|---|---|---|---|
| PEI-GA | 47.1 | 8.5 | 12.6 |
| PEI-TA | 34.0 | 7.3 | 20.3 |
| PEI-GA-BT | 49.8 | 7.2 | 15.5 |
3.2 IR spectroscopy
As presented in Fig. 2, the IR spectra for PEI-GA, PEI-TA, and PEI-GA-BT show almost the same pattern, mainly due to the similar vibrations of the C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds within the crosslinked structure of these polymers. This is especially true for PEI-GA and the functionalized PEI-GA-BT, where the main difference is found at 717 cm−1, that is the CC vibrations of the pendant benzyl group in compound BT. The remaining major signals originate from amine vibrations.
N bonds within the crosslinked structure of these polymers. This is especially true for PEI-GA and the functionalized PEI-GA-BT, where the main difference is found at 717 cm−1, that is the CC vibrations of the pendant benzyl group in compound BT. The remaining major signals originate from amine vibrations.
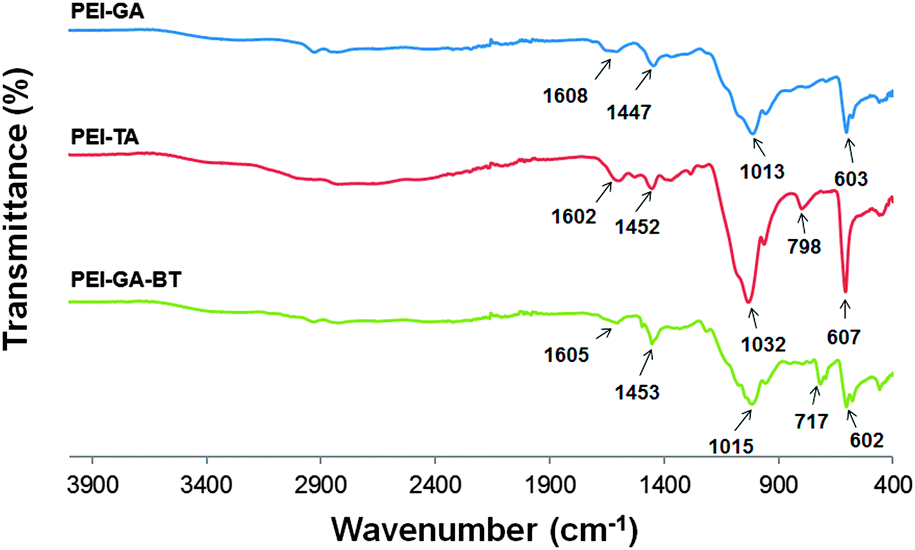 | ||
| Fig. 2 IR spectra for PEI-GA, PEI-GA-BT, and PEI-TA. | ||
3.3 Particle morphology for crosslinked polymers
Fig. 3 shows the scanning electron microscopy images of PEI-GA, PEI-TA, and PEI-GA-BT. The effect of the crosslinker on the surface morphology has been already shown for other crosslinking reactions and polymeric systems.37–39 During the crosslinking reaction and upon the formation of the cross-linked solid phase, a phase separation occurs.38 The dynamics of the phase separation, which has a determinative role on the morphology of the particles, can be influenced by the crosslinker. In Fig. 3, one can observe this difference in the morphology of PEI-GA and PEI-TA. While the PEI-GA structure is more compact, the structure of PEI-TA seems to be more porous. The structure of PEI-GA-BT more resembles PEI-GA, showing that the presence of the BT functional group does not have any strong effect on the final morphology of the particles. For the PEI-GA, there are a small number of spherical particles with varying sizes present. However, these microspheres are less frequent for PEI-GA-BT and absent in the case of PEI-TA. | ||
| Fig. 3 The morphology of the crosslinked polymers PEI-GA, PEI-TA, and PEI-GA-BT. | ||
3.4 Cu(II) absorption
For all the samples, the maximum copper uptake was measured by soaking the polymer in a 100 mM CuCl2 aqueous solution for 12 hours under vigorous shaking to assure that the equilibrium copper concentration was attained. Elemental analysis of the Cu(II) loaded polymers in Table 2 shows that reduction of the imine bonds reduces the capacity for copper uptake; PEI-GA absorbs 8.1 wt% Cu(II) and 4.5 wt% before and after reduction while for PEI-TA the corresponding values are 12.2 wt% and 5.8 wt% before and after the reduction step. PEI-GA-BT absorbs 3.5 wt% of Cu(II) before and 3.2 wt% after the reductive amination modification. The decrease in Cu(II) absorption upon reduction is probably correlated to the protonation of amine groups in the crosslinked structure. Although this effect can be minimized by optimizing the reduction process, it is ultimately a choice between the durability of the final structure and its copper uptake performance.| Absorption capacity for Cu(II) (wt%) | Absorption capacity for Cu(II) (wt%) | |
|---|---|---|
| Before imine reduction | After imine reduction | |
| PEI-GA | 8.1 | 4.5 |
| PEI-TA | 12.2 | 5.8 |
| PEI-GA-BT | 3.5 | 3.2 |
As it has been already pointed out, the higher capacity for the TA cross-linker is due to the presence of additional triazole functionalities, providing more binding sites for the copper ions. Also the more porous structure of PEI-TA, as compared to PEI-GA and PEI-GA-BT, provides an additional contribution to the higher Cu(II) absorption of this polymer as the Cu(II) absorption occurs mostly at the surface of the particle. Also, it can be seen from Fig. 3 that the PEI-GA-BT particles are on average larger than the PEI-GA particles, which explains the lower Cu(II) uptake of this polymer.
3.5 Absorption selectivity
The selectivity of the cross-linked polymers toward copper is important when a flux of copper at the interface is aimed for, otherwise the coordinating sites in the structure may be occupied by other competing metals ions. It has already been shown however, that a thin film of crosslinked PEI-GA can absorb copper from a mixture of metal ions with high selectivity.29 To investigate the selectivity of the crosslinked polymers PEI-GA, PEI-TA, and PEI-GA-BT upon metal coordination, the particles were soaked in 100 mM aqueous solutions of CuCl2, ZnCl2, and CuCl2/ZnCl2 and the metal content of these samples were measured by elemental analysis. The results are presented in Table 3. Both PEI-GA and PEI-TA were found to have a very low zinc absorption capacity after reaching equilibrium. PEI-GA and PEI-TA absorb less than 0.2 wt% when there is just ZnCl2 present in the solution. For the mixed solution of CuCl2/ZnCl2, almost no zinc was present in the samples, indicating the strong preference toward Cu(II) versus Zn(II) for both PEI-GA and PEI-TA. The ‘harder’ nature of Zn(II) compared to Cu(II) on one side, and the lack of hard oxygen donor groups in these systems may be a reason for the preference of copper over zinc.| Metal salt present in the solution | Absorption capacity for Cu(II) (wt%) | Absorption capacity for Zn(II) (wt%) | |
|---|---|---|---|
| PEI-GA | CuCl2 | 4.5 | — |
| ZnCl2 | — | 0.2 | |
| CuCl2/ZnCl2 | 4.5 | 0.0 | |
| PEI-TA | CuCl2 | 5.8 | — |
| ZnCl2 | — | 0.1 | |
| CuCl2/ZnCl2 | 6.3 | 0.0 | |
| PEI-GA-BT | CuCl2 | 3.2 | — |
| ZnCl2 | — | 0.7 | |
| CuCl2/ZnCl2 | 1.1 | 1.0 | |
For PEI-GA-BT however the selectivity for Cu(II) versus Zn(II) is not as good as the other two crosslinked system. PEI-GA-BT absorbs 0.7 wt% Zn(II) when there is only ZnCl2 is present and 1.0 wt% when both CuCl2 and ZnCl2 are present in the solution. The presence of ZnCl2 also affects the Cu(II) absorption capacity for the PEI-GA-BT crosslinked solution, reducing the Cu(II) content to 1.1 wt% which is less than half of the copper content of this polymer when there is just CuCl2 is present in the solution. It has already been shown that for a thin film of glutaraldehyde-crosslinked PEI, even though the dynamic of Zn(II) absorption is much faster at the initial absorption stage, the Cu(II) ions will eventually occupy the coordination sites in the crosslinked network.29,30 Our results from Table 3 indicates that although such a dominance by Cu(II) ions at equilibrium is the case for both PEI-GA and PEI-TA, the coordination sites will be distributed almost evenly between Zn(II) and Cu(II) for PEI-GA-BT at equilibrium.
3.6 Computational models of the triazolealdehyde (TA) and glutaraldehyde (GA) crosslinkers
The triazole monomer unit in PEI-TA can make a maximum of two coordinations to one Cu ion, but more than one unit can complex to one Cu ion to make a full coordination sphere. Fig. 4a shows a selection of probable binding geometries together with the corresponding complexation energies of the complexes of the model ‘monomers’ for PEI-GA and PEI-TA. It can be seen that the complexation energy for PEI-TA is higher than PEI-GA, i.e. Cu(I) reaches lower energy upon coordination to PEI-TA in comparison with PEI-GA. This is also in consistent with the experimental observations for the crosslinked system. Also, PEI-TA offers more possible coordination sites for Cu ions as opposed to PEI-GA (see Fig. 4b). Furthermore, and as it is shown in Fig. 4c, a lower energy is achieved when the second ‘PEI-TA monomer’ participates in the coordination sphere. This is a more realistic situation that one may also find in a crosslinked PEI-TA network.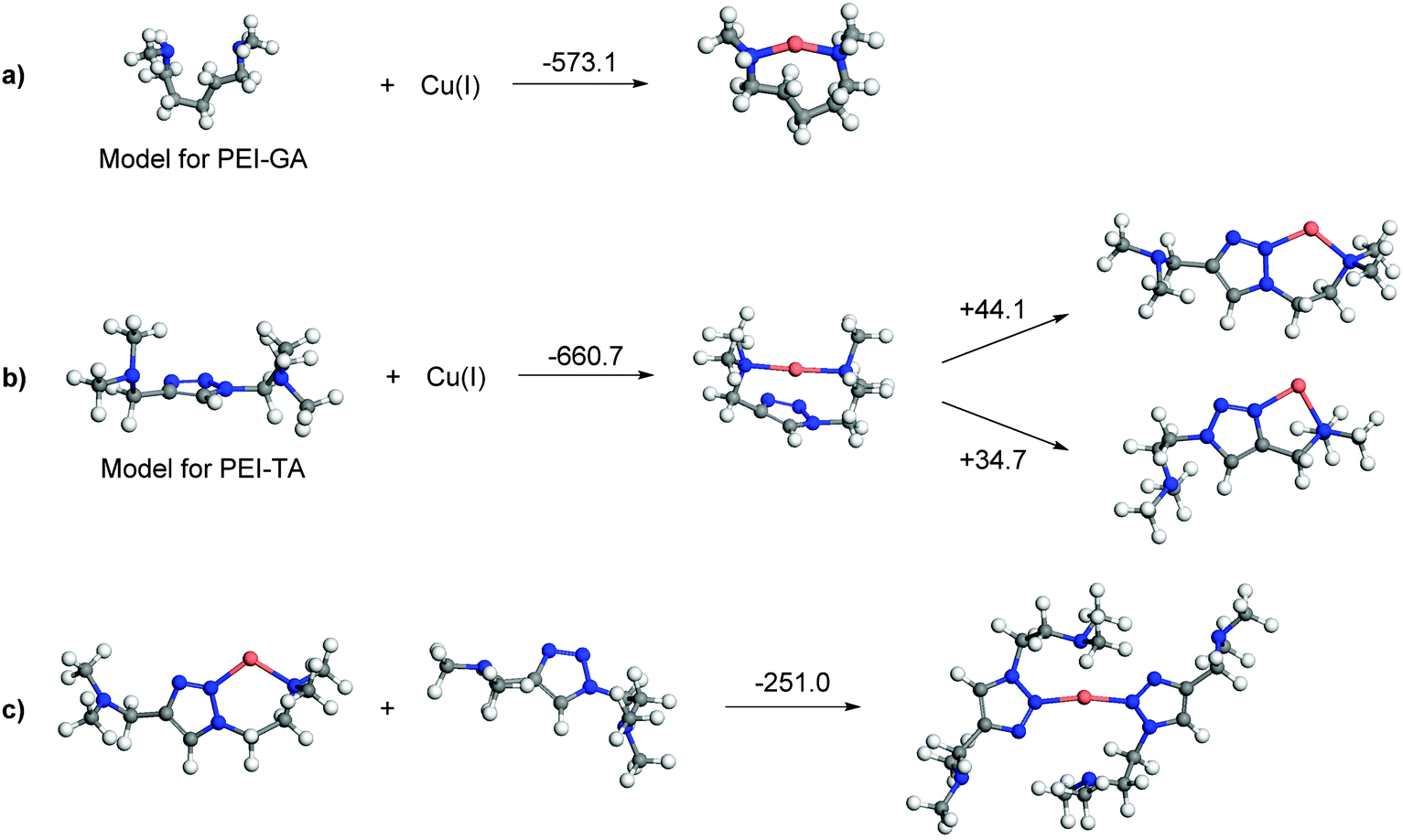 | ||
| Fig. 4 Complexation energies of Cu(I) in kJ mol−1 with (a) monomers of PEI-GA; (b) monomer of PEI-TA; and (c) dimer of PEI-TA. | ||
3.7 Cu(I) stabilization and exchange
In principle, Cu(I) can be produced in situ by the addition of a mild reducing agent such as sodium ascorbate. This procedure comes with a classic disappearance of the blue colour upon Cu(II) reduction. However, we have employed a titrating solution that not only contains sodium ascorbate, but also two more ligands that can coordinate strongly and specifically to Cu(I) or Cu(II). In this manner, we can also determine the ability of the polymer structures to retain Cu(I) upon competition with another strongly Cu(I) binding ligand bathocuproine (BC) (Fig. 5a). (In this work, we have used the water-soluble bathocuproinedisulfonic sodium salt (BCS) (Fig. 5b).) BCS shows a distinct red colour upon coordination to Cu(I) (absorbance wavelength at 488 nm). This has made BCS a useful compound for spectrophotometric determination of Cu(I).40 So, even though the quantitative measurement of Cu(I) is not feasible with this spectrophotometric method when a strong Cu(I)-coordinating ligand is present in the environment, the method can still provide valuable information for comparing different Cu-coordinating systems which has been prepared in the course of this work. Also, the strong Cu(I) preference of BCS can shift the Cu(II)⇌Cu(I) equilibrium in solution. To minimize this effect, one can use ethylenediaminetetraacetic (EDTA) (Fig. 5c) which is able to coordinates specifically and rapidly to Cu(II).40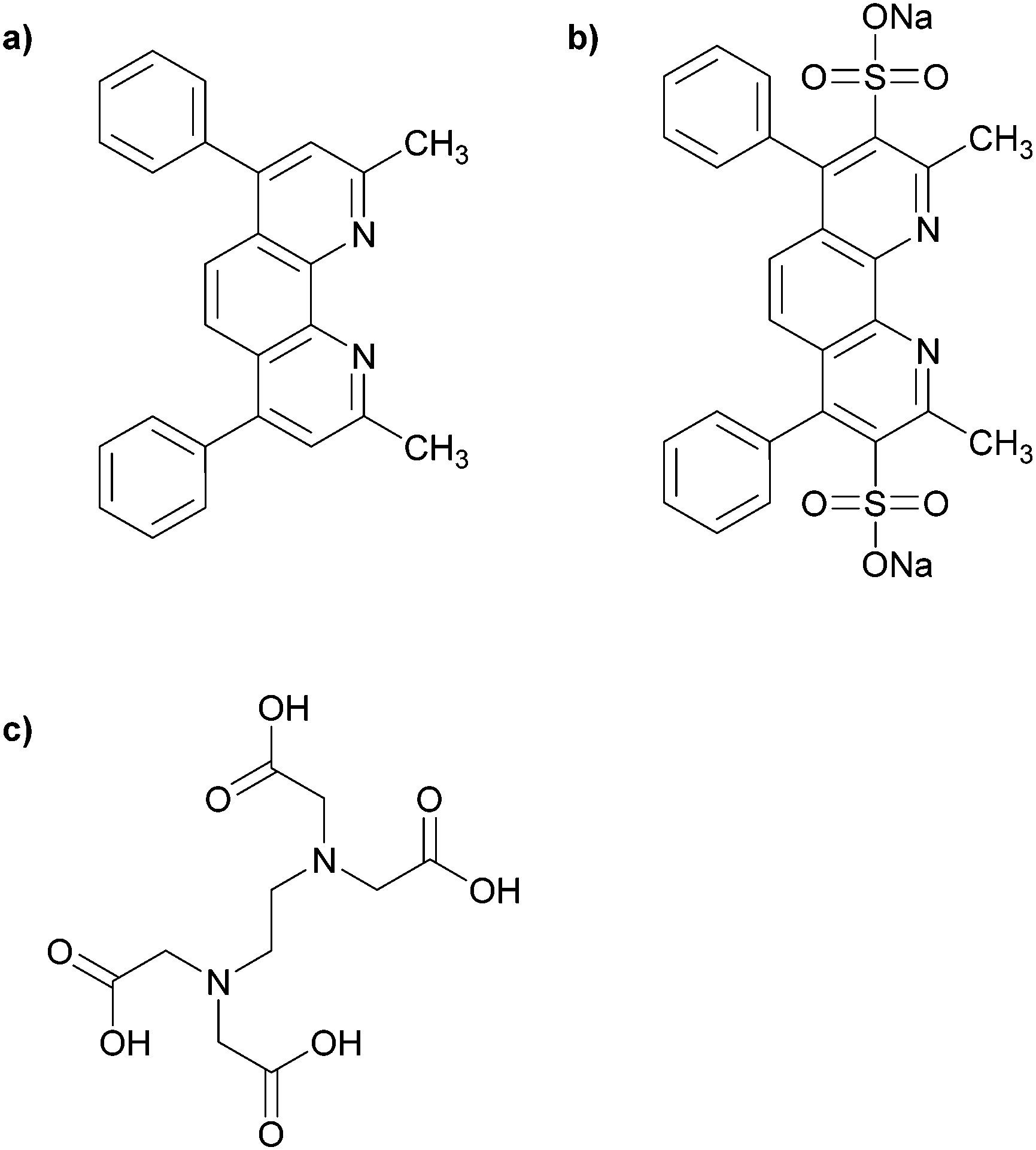 | ||
| Fig. 5 (a) Bathocuproine (BC); (b) bathocuproinedisulfonic sodium salt (BCS); (c) ethylenediaminetetraacetic acid (EDTA). | ||
A schematic representation of a system in which a Cu(II) loaded surface is treated with a solution of BCS:EDTA:sodium ascorbate is shown in Scheme 3. In such a system, there is a competition between different ligands in the solution and at the polymer-solution interface to coordinate to Cu ions. At equilibrium, a typical system is expected to contain a mixture of Cu(I) and Cu(II) coordinated to the different ligands both in solution and in the solid phase. For such a system, the conditional stability constants of the [Cu(ligand)] complexes as well as the [Cu(ligand 1)(ligand 2)] ternary complex system are important for the final equilibrium state of the system.
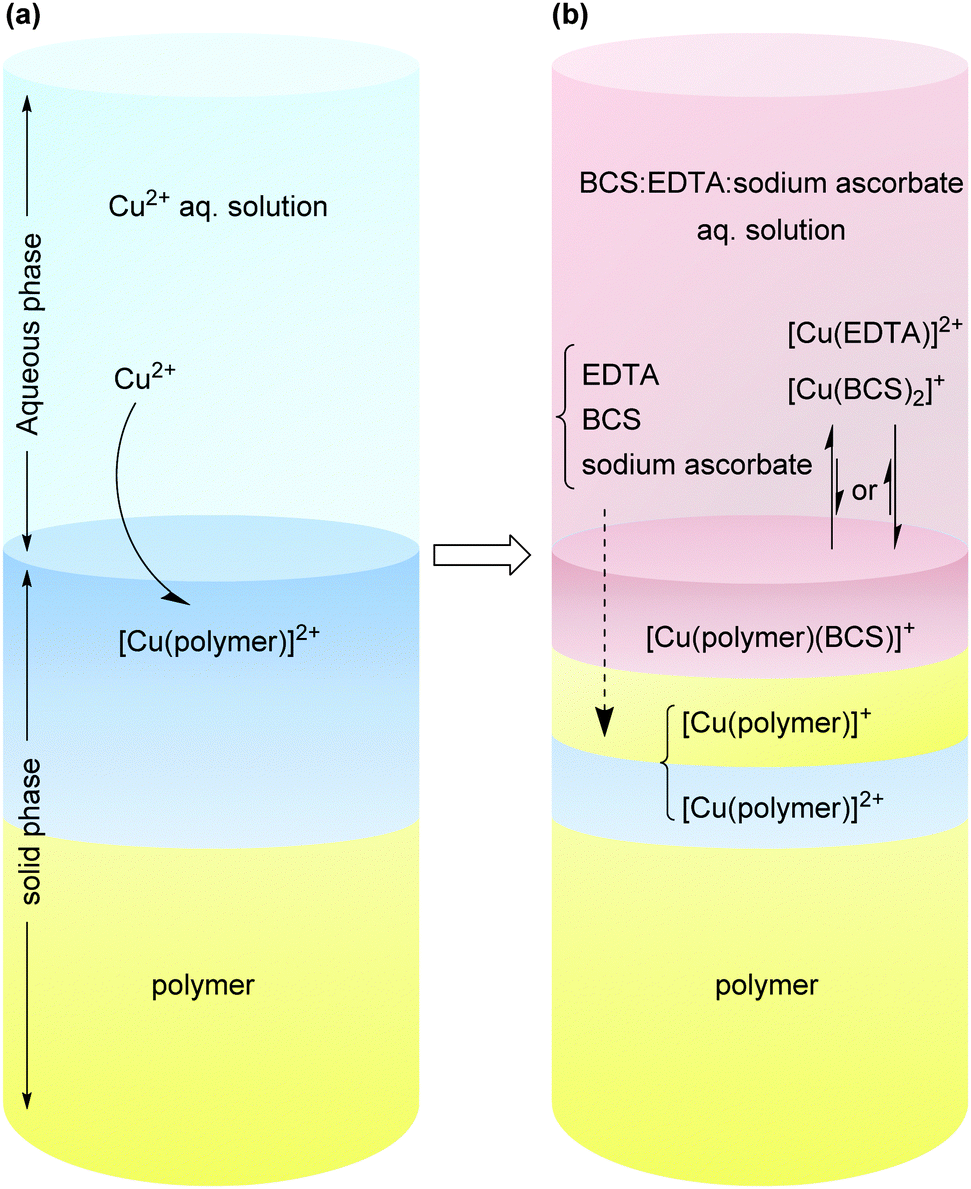 | ||
| Scheme 3 Schematic representation of (a) the Cu(II) absorption from an aqueous solution of Cu(II); (b) some of the important interactions between the titrating solution of BCS:EDTA:sodium ascorbate and a Cu-loaded surface. The structure–property of the polymer has a determinative influence on the [CuI(BCS)(polymer)]⇌[CuI(BCS)2] at equilibrium state. | ||
Fig. 6 shows copper soaked PEI-GA, PEI-TA, and PEI-GA-BT polymers before and after exposure to BCS:EDTA:sodium ascorbate aqueous solution. To mimic the conditions as in a typical coating, this experiment was carried out without shaking. The diffusion of sodium ascorbate causes a gradient of Cu(I) and Cu(II) across the polymer. For all these three polymers, the system follows Scheme 3 to different extents. However, the Cu(II) oxidation state gradient is mostly visible for PEI-TA where three different colour waves appear upon the diffusion of sodium ascorbate and the coordinating ligands, i.e. the red top layer which is the ternary complex of [CuI(BCS)(PEI-TA)], the yellow middle layer which has lost the blue colour due to the diffusion of sodium ascorbate, and finally the blue bottom layer which has not yet been influenced by the diffusion layer. For PEI-GA and PEI-GA-BT, however, the dark colour of the original polymers makes such an observation more difficult.
 | ||
| Fig. 6 (a) Cu(II) absorption from a 1 mM CuSO4 aqueous solution under stagnant condition for 12 hours; (b) reduction by sodium ascorbate for 12 hours by a BCS:EDTA:sodium ascorbate (2 mM:10 mM:1 mM). | ||
To exclude the diffusion effect and to make some comparisons between these crosslinked structures, the same experiment as in Fig. 6 was repeated with shaking. In this case, 50 mg of each polymer was loaded with Cu(II) ions. After washing with deionized water, the polymer was shaken for 12 hours in BCS:EDTA:sodium ascorbate (2 mM:10 mM:1 mM) and the UV-vis absorption spectra of the solution phase was measured. As shown in Fig. 7, even though this experiment is also qualitative in nature, while keeping all other parameters constant, the red colour evolution in the solution together with analysis of the sediment is an indication of the ability of the crosslinked structure to retain Cu(I) upon reduction of Cu(II) ions with sodium ascorbate in competition with a water soluble ligands such as BCS and EDTA. The UV-Vis spectra of the titrating solutions in Fig. 7d clearly shows that PEI-GA has a much lower capacity to retain Cu(I) in comparison to PEI-TA which in turn retains almost all the Cu(I) within the structure. PEI-GA-BT stands somewhere in between PEI-GA and PEI-TA. This also suggests that the partitioning of Cu(I) between the crosslinked polymer phase and the solution that contains a strong Cu(I)-coordinating compound can be tuned by introducing a third functionality into the crosslinked PEI-GA system.
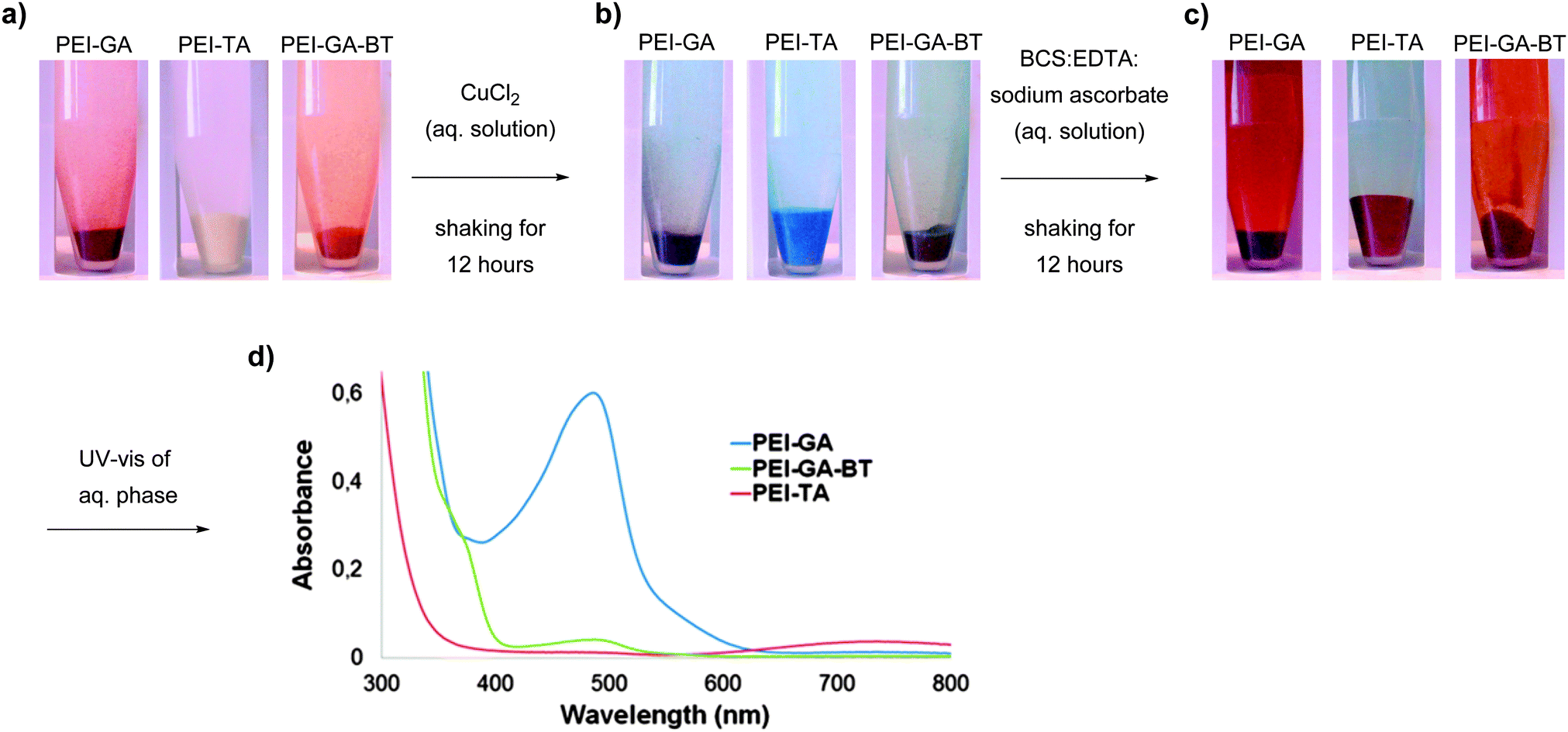 | ||
| Fig. 7 (a) PEI-GA, PEI-TA, and PEI-GA-BT; (b) Cu(II) loaded PEI-GA, PEI-TA, and PEI-GA-BT; (c) titration of Cu-loaded PEI-GA, PEI-TA, and PEI-GA-BT crosslinked polymers with BCS:EDTA:sodium ascorbate solution; (d) UV-Vis spectroscopy of the aq. phase from the titration step. | ||
The Cu content of the sediment in Fig. 7c has been also measured using EDS. The results are presented in Table 4. As can be seen, the Cu(I) content in the case of PEI-TA is the highest (3.6 wt%) while it was found to be lower for both PEI-GA and PEI-GA-BT (2.2 wt% and 1.4 wt% respectively).
The weight-percent of Cu(I) in all of the polymer networks are lower after the titration. One reason for this, as stated earlier, is that the copper ions leave the polymer structure upon formation of water-soluble [Cu(BCS)2]+ and/or [Cu(EDTA)]2+ complexes, which is mainly the case for PEI-GA and PEI-GA-BT. However, for PEI-TA, the coordination of BCS to the copper ions in the polymer structure is the main reason that the total Cu(I) weight percent in the final sediment is lower. This happens to a much lesser extent for PEI-GA and PEI-GA-BT. Measuring the total weight increase of the polymer sediments after titration also demonstrates that although the total weight increase is 4% for PEI-GA and PEI-GA-BT, the weight of PEI-TA increases by 20% after titration, mainly due to the coordination of BCS to the Cu ions in the polymer network.
4 Conclusions and outlook
We have shown that chemical modification of polyethyleneimine with a triazole-based crosslinker results in a material with a very strong affinity to Cu(I) and Cu(II) which are the most common and biologically most important oxidation states of copper. In particular, the crosslinker increased the affinity to Cu(I). Furthermore, we have prepared a polyethyleneimine-glutaraldehyde-(benzyl)triazole polymer that allows effective release of Cu(I). Finally, we have shown that a combination of the polymers can be used to fine tune the uptake and release of copper across an interface, which opens up for a new approach to antibiofouling. Our proposed antibiofouling mechanism has the potential to lead to biocide-free antibiofouling coatings for a wide range of freshwater applications as well as static marine constructions and vessels, with the added benefit of remediating copper from copper-contaminated harbours once a vessel is exposed to the open sea. Yet there are also three significant environmental dimensions of this science. First, the advance should enable us to supplant the use of existing coatings, which release highly toxic substances into the ocean, causing untold environmental damage. Second, the new generation of coatings will minimise the spread of invasive organisms on the hulls of ocean-going vessels. The third environmental benefit is related to the deleterious effects of copper on marine and aquatic biodiversity: in the longer term, the technology will significantly decrease copper concentrations in polluted water, resulting in increased biodiversity.Acknowledgements
This work is funded by the Swedish Research Council FORMAS. We would like to thank Mr Anders Mårtensson for SEM images and Dr Alexander Idström for assistance with photography of the samples. We are also grateful to Prof Lars Öhrström and Prof Krister Holmberg for fruitful discussions.References
- M. R. Bleackley and R. T. A. MacGillivray, BioMetals, 2011, 24, 785–809 CrossRef CAS PubMed.
- K. Balamurugan and W. Schaffner, Biochim. Biophys. Acta, Mol. Cell Res., 2006, 1763, 737–746 CrossRef CAS PubMed.
- M. M. O. Pena, J. Lee and D. J. Thiele, J. Nutr., 1999, 129, 1251–1260 CAS.
- E. Almeida, T. C. Diamantino and O. de Sousa, Prog. Org. Coat., 2007, 59, 2–20 CrossRef CAS PubMed.
- L. D. Chambers, K. R. Stokes, F. C. Walsh and R. J. K. Wood, Surf. Coat. Technol., 2006, 201, 3642–3652 CrossRef CAS PubMed.
- N. Voulvoulis, M. D. Scrimshaw and J. N. Lester, Appl. Organomet. Chem., 1999, 13, 135–143 CrossRef CAS.
- E. Ytreberg, J. Karlsson and B. Eklund, Sci. Total Environ., 2010, 408, 2459–2466 CrossRef CAS PubMed.
- W. G. Sunda and R. R. L. Guillard, J. Mar. Res., 1976, 34, 511–529 CAS.
- V. F. Vetere, M. C. Perez, R. Romagnoli, M. E. Stupak and B. del Amo, J. Coat. Technol., 1997, 69, 39–45 CrossRef CAS.
- L. Ciavatta, D. Ferri and R. Palombari, J. Inorg. Nucl. Chem., 1980, 42, 593–598 CrossRef CAS.
- J. W. Moffett and R. G. Zika, Mar. Chem., 1983, 13, 239–251 CrossRef CAS.
- J. F. Jiang, I. A. Nadas, M. A. Kim and K. J. Franz, Inorg. Chem., 2005, 44, 9787–9794 CrossRef CAS PubMed.
- Y. Nose, E. M. Rees and D. J. Thiele, Trends Biochem. Sci., 2006, 31, 604–607 CrossRef CAS PubMed.
- K. D. Karlin, J. C. Hayes, S. Juen, J. P. Hutchinson and J. Zubieta, Inorg. Chem., 1982, 21, 4106–4108 CrossRef CAS.
- M. A. Trojer, A. Movahedi, H. Blanck and M. Nyden, J. Chem., 2013, 946739 Search PubMed.
- J. T. Rubino and K. J. Franz, J. Inorg. Biochem., 2012, 107, 129–143 CrossRef CAS PubMed.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS PubMed.
- P. S. Donnelly, S. D. Zanatta, S. C. Zammit, J. M. White and S. J. Williams, Chem. Commun., 2008, 2459–2461 RSC.
- A. Movahedi, K. Moth-Poulsen, J. Eklöf, M. Nyden and N. Kann, React. Funct. Polym., 2014, 82, 1–8 CrossRef CAS PubMed.
- J. Jia, A. H. Wu and S. J. Luan, Colloids Surf., A, 2014, 449, 1–7 CrossRef CAS PubMed.
- B. L. Rivas, E. D. Pereira and I. Moreno-Villoslada, Prog. Polym. Sci., 2003, 28, 173–208 CrossRef CAS.
- F. Sabermahani and M. A. Taher, Anal. Chim. Acta, 2006, 565, 152–156 CrossRef CAS PubMed.
- Y. L. Chen, B. C. Pan, S. J. Zhang, H. Y. Li, L. Lv and W. M. Zhang, J. Hazard. Mater., 2011, 190, 1037–1044 CrossRef CAS PubMed.
- W. J. Tong, C. Y. Gao and H. Möhwald, Polym. Adv. Technol., 2008, 19, 817–823 CrossRef CAS PubMed.
- J. Jia, A. H. Wu and S. J. Luan, Phys. Chem. Chem. Phys., 2014, 16, 16158–16165 RSC.
- D. M. Saad, E. M. Cukrowska and H. Tutu, Toxicol. Environ. Chem., 2011, 93, 914–924 CrossRef CAS PubMed.
- D. M. G. Saad, E. M. Cukrowska and H. Tutu, Toxicol. Environ. Chem., 2012, 94, 1916–1929 CrossRef CAS PubMed.
- D. M. G. Saad, E. M. Cukrowska and H. Tutu, Toxicol. Environ. Chem., 2013, 95, 409–421 CrossRef CAS PubMed.
- J. B. Linden, M. Larsson, B. R. Coad, W. M. Skinner and M. Nyden, RSC Adv., 2014, 4, 25063–25066 RSC.
- S. J. Miklavcic, M. Nyden, J. B. Linden and J. Schulz, RSC Adv., 2014, 4, 60349–60362 RSC.
- A. F. AbdelMagid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS PubMed.
- T. R. Chan and V. V. Fokin, QSAR Comb. Sci., 2007, 26, 1274–1279 CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS PubMed.
- B. Delley, J. Phys. Chem. A, 2006, 110, 13632–13639 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS PubMed.
- D. Kim, D. Y. Lee, K. Lee and S. Choe, Macromol. Res., 2009, 17, 250–258 CrossRef CAS.
- K. Kim, Y. Kim, N. R. Ko and S. Choe, Polymer, 2011, 52, 5439–5444 CrossRef CAS PubMed.
- B. Peng and A. Imhof, Soft Matter, 2015, 11, 3589–3598 RSC.
- J. W. Moffett, R. G. Zika and R. G. Petasne, Anal. Chim. Acta, 1985, 175, 171–179 CrossRef CAS.
| This journal is © the Owner Societies 2015 |