Heating and mechanical force-induced luminescence on–off switching of arylamine derivatives with highly distorted structures†
Received
22nd July 2013
, Accepted 16th October 2013
First published on 17th October 2013
Abstract
A triphenylamine-based organic luminophor (TPA-CO) with a highly distorted structure has been designed and effortlessly obtained by an Ullmann reaction. The luminophor exhibits a stimuli-induced emission enhancement effect and intramolecular charge transfer properties. The fluorescence efficiency of its crystals is dramatically increased from 0.4% to 12.3% upon grinding. The emission enhancement is also realized by a heating process. The “bright” state can recover its original state and turn “dark”. The luminescence “on–off” behaviour is repeatedly transformed by a grinding–vapour process or by a heating process. The XRD patterns of the “bright” and “dark” states show that the change of emission intensity is related to the reversible transition between the crystalline state and the metastable amorphous state. At the molecular level, the emission enhancement upon external stimuli may be attributed to conformational planarization and weak intermolecular interactions.
Introduction
Stimuli-responsive materials are attracting considerable attention, especially organic dyes whose luminescence can be repeatedly switched by external stimuli.1 In principle, it could be readily realized that luminescent efficiency and colour are tuned simultaneously, which accompanies the altering of the molecular stacking pattern upon external stimuli.2 However, the overwhelming majority of materials present transformation of colour3 rather than a change of emission efficiency, which makes it difficult to satisfy the needs of high-contrast luminescence recording. Using a dramatic switching in emission intensity has recently been presented by Park's group to settle this intractable issue.4 However, examples of utilizing external stimuli such as vapour,5 heating6 and mechanical force4,7 to reversibly tune the emission intensity are rather scarce at this stage. Among the mechanical force-responsive luminescent materials, one typical example based on a binary complex is a donor (D)–acceptor (A) system, which produces high-contrast fluorescence.7a This D–A binary system, with rather ill-defined composition, shows various emission colours by a change in donor molecule. Encouragingly, luminophors with well-defined structures obtained by Park's group4 and Jia's group7b also exhibit the intriguing fluorescence on–off switching property, and it is proposed that the distinct fluorescent properties can be attributed to varying excited states (Jia's group) and different rates of electron transfer (Park's group). Regrettably, their single-crystal structures are not obtained due to poor solubility or ill-defined composition, which limits the deeper understanding of the stimuli-responsive behaviour at the molecular level. Therefore, exploring new design strategies and innovative materials with high-contrast luminescence switching remains a challenge.
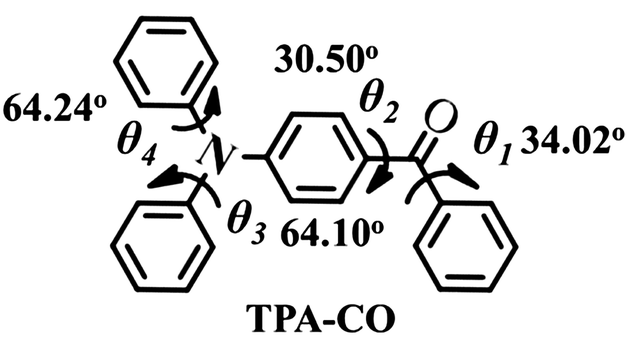
In this paper, we have prepared a stimuli-responsive luminophor (TPA-CO, Chart 1) consisting of benzophenone and arylamine that exhibits a weak luminescence with a quantum yield (ΦPL) of 0.4% due to its highly distorted structure. After grinding, the emission efficiency is increased to 12.3%. Its luminescence on–off switching can be reversibly transformed by a grinding–vapour process or by a pure thermal treatment, which is rarely observed in traditional mechanochromic materials. Moreover, based on the single-crystal structures and optical properties analysis, a new design strategy to explore stimuli-responsive materials with high contrast is proposed.
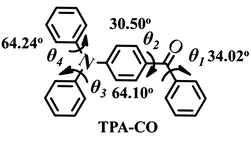 |
| | Chart 1 Molecular structure and dihedral angle of TPA-CO single-crystals. | |
Results and discussion
Intramolecular charge transfer
The desired luminophor TPA-CO was prepared conveniently by an Ullmann reaction with yields over 74%. As depicted in Fig. S1,† as the solvent polarity increased from nonpolar toluene to highly polar acetonitrile, the emission peaks strongly depend on the solvent polarity, revealing an evident bathochromic effect. The fluorescence intensity was also obviously reduced in highly polar media. Additionally, Fig. S2† shows that electron clouds of the highest occupied molecular orbital (HOMO) levels were mainly concentrated on the electron-donating arylamine units; however, those of lowest unoccupied molecular orbital (LUMO) levels were mostly spread to the electron-accepting benzophenone. These results showed that luminophor TPA-CO was a D–π–A dipole molecule with obvious intramolecular charge transfer (ICT) properties.8
Multi-stimuli-responsive fluorescence switching
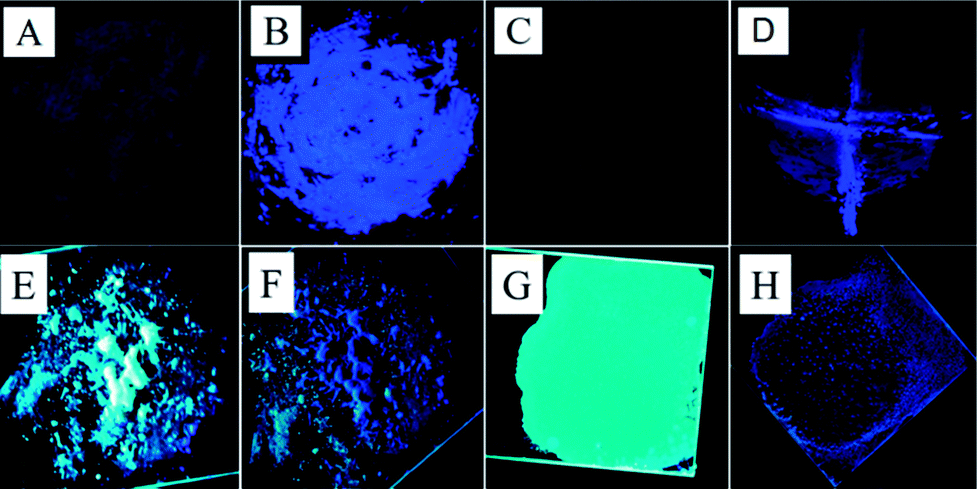
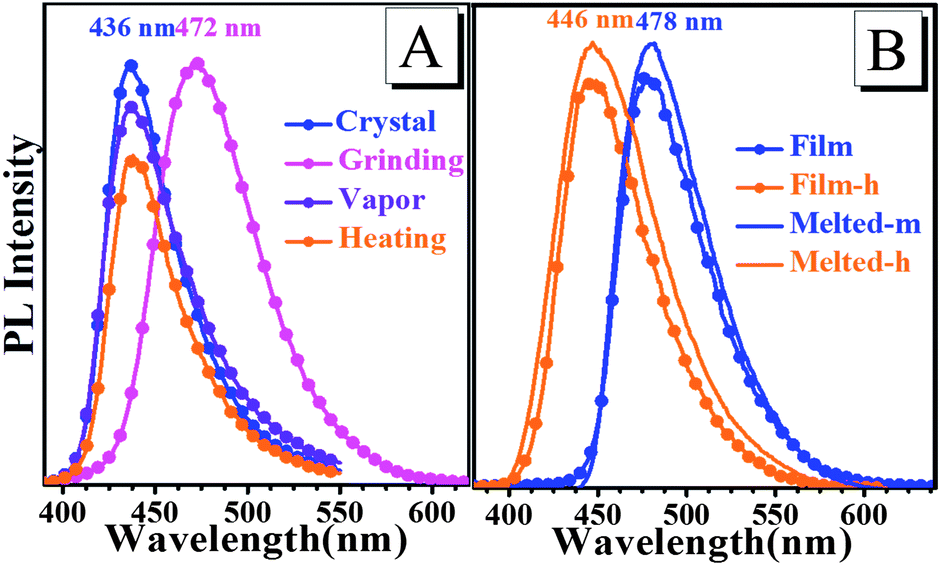
Upon crystallization from ethanol solution, the TPA-CO molecules produced a white powder, which exhibited a faint luminescence at 436 nm (Fig. 1a). Interestingly, as shown in Fig. 1b, its emission intensity was dramatically enhanced with a ΦPL as high as 12.3% after grinding with a pestle. Such “dark”–“bright” switching was sufficiently obvious as to be easily distinguished by the naked eye, and this change occurred only at the grinding area. The photographic image of TPA-CO in Fig. 1d, shaped in a “┼” on a mortar after pressing with a spatula, further indicated the high-contrast luminescence switching. Upon wetting-processing with ethanol, the luminescence recovered its original state and turned “dark” (Fig. 1c). The fluorescence quenching was also achieved by exposing the ground sample to a heating process (80 °C for 2 min). Importantly, this reversible luminescence on–off switching can be repeated many times by grinding–vapour or grinding–heating treatment (Fig. S3 and S4†) with almost no fatigue. When the non-luminescent crystals of TPA-CO were heated to the melted state and rapid cooling at room temperature (R.T.) in air, the obtained sample, termed as Melted-m, exhibited intense sky-blue emission at 478 nm with a ΦPL of 41.6% (Fig. 1e). However, it was relatively less stable at R.T., and a tiny variation in fluorescence was observed over a period of time. The rate of change could be accelerated by increasing the temperature. For instance, after heating above 80 °C for 4 h, the bright emission of Melted-m was obviously weakened and blue-shifted (Melted-h, Fig. 1f and 2b). Moreover, if the pristine powder was thermally melted and then slowly cooled down, its emission remained “dark”. As shown in Fig. 1g, the drop-casting film also indicated strong sky-blue luminescence (ΦPL = 44.2%), which resembles the Melted-m sample with poor stability. Upon heating above 80 °C for 4 h, its luminescence was completely turned off (Film-h, Fig. 1h) and the emission spectrum of the film was blue-shifted and overlapped with that of Melted-h (Fig. 2b). The fluorescence of the Melted-h and Film-h samples can be turned “bright” again after melting and then quickly cooling at R.T. The results showed that the emission on–off switching could also be realized by only heating treatment. Fig. S5† shows the NMR spectroscopic experiments of the above-mentioned samples. These spectra showed no differences, thus revealing that the external stimuli did not change the chemical structures of the samples.
 |
| | Fig. 1 Photograph of the TPA-CO powder under 365 nm UV light: (A) pristine powder, (B) pristine powder after grinding, (C) sample B upon exposure to alcohol vapour, (D) pristine powder after being pressed with a spatula into a “┼” shape, (E) Melted-m, powder heated until melted and solidified at R.T., (F) Melted-h, the melted powder annealed above 80 °C for 4 h, (G) the film prepared by drop-casting, (H) Film-h, the as-prepared film heated above 80 °C for 4 h. | |
 |
| | Fig. 2 Luminescence spectra of TPA-CO in different states. | |
Photophysical properties and XRD patterns
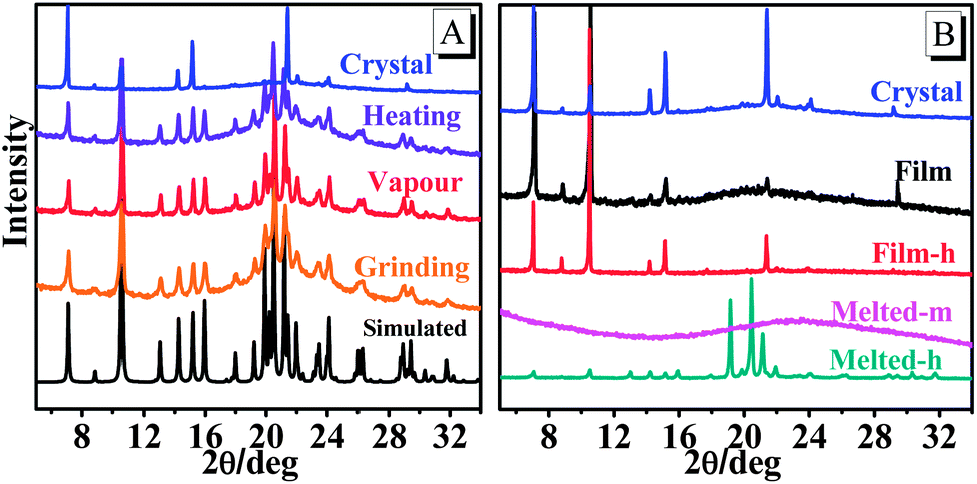
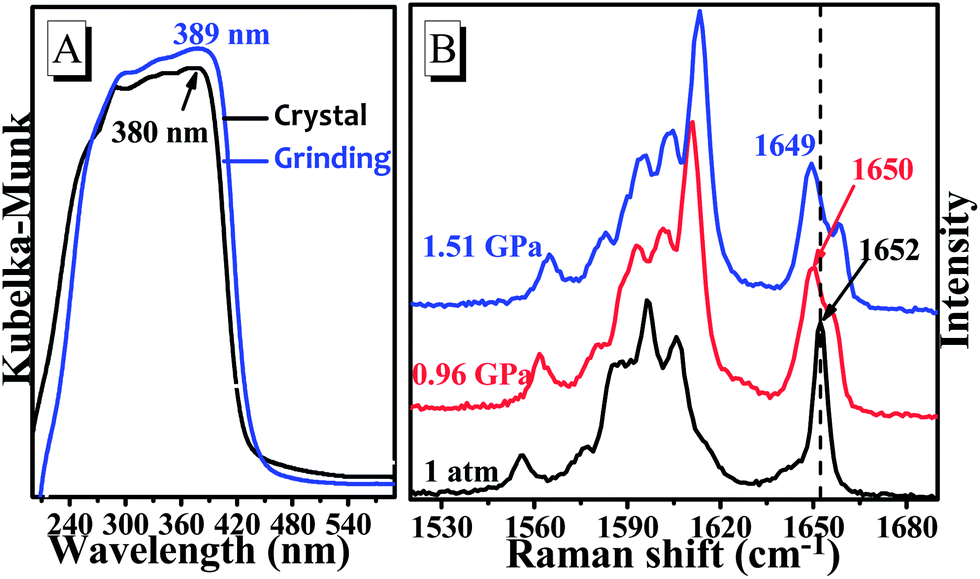
The reversible emission color switching was also easily realized by the grinding–vapor treatment or the only heating treatment. As depicted in Fig. 2a, the emission peak of pristine crystals after the grinding treatment indicated an obvious red-shift with a change of more than 36 nm, and the emission wavelength was red-shifted to 478 nm via melting and cooling at room temperature (Fig. 2b). To understand these stimuli-responsive fluorescent behaviours, powder X-ray diffraction (PXRD) patterns of TPA-CO in various states were performed. The pristine crystals exhibited intense and sharp reflection peaks indicative of microcrystalline order (Fig. 3). As depicted in Fig. 3a, the PXRD patterns of the ground powders were similar to the simulated XRD from single crystals, except for a diffuse, broad halo ranging from 17° to 25°. After vapour treatment, the broad peak disappeared. The results showed that the partially amorphous phase and crystalline phase co-existed in the ground powder. Thus, the stimuli-responsive behaviour may be attributed to the transformation of the crystalline phase to the partial amorphous phase.9 For the drop-casting film and Melted-m with high ΦPL, the signals of PXRD were rather weak, suggesting disordered molecular packing (Fig. 3b). Obvious reflection peaks were observed upon heating (Film-h and Melted-h). The samples became crystalline with rather low ΦPL. Clearly, the metastable amorphous state was crucial to the fluorescence intensity of the luminogens.
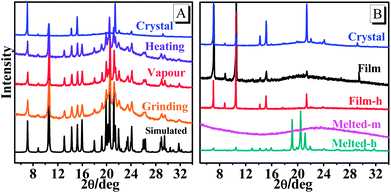 |
| | Fig. 3 Powder XRD patterns of TPA-CO, and the simulated powder XRD pattern of single crystals (A, black line). | |
At the molecular level, the emission intensity of luminophors is usually related to intramolecular effects and intermolecular packing.3b,10 Intramolecular effects on fluorescence enhancement could be attributed to the various molecular conformations of the luminophors. For example, the weak fluorescence of the special luminophors in solution may be a consequence of their twisted conformations, which led to a large non-radiative rate constant. However, the molecular conformation became planar after aggregation, which blocked the non-radiation process, and the fluorescence intensity was enhanced.10a,b,11 In this case, the pristine powders yielded a fluorescence lifetime of τ = 0.65 ns (ΦPL = 0.4%) with a relatively high non-radiative rate constant of knr = (1 − ΦPL)/τF = 1.5 × 109 s−1 as shown in Fig. S6.† Indeed, the single-crystal structures revealed that the TPA-CO molecules, with a dihedral angle (θ) ranging from 30° to 64°, were highly twisted (Chart 1), which was in accordance with the experimental results above. Upon grinding, the non-radiative deactivation pathways were obviously blocked, with a non-radiative rate constant of knr = 2.9 × 108 s−1 (Fig. S6,†τ = 3.0 ns, ΦPL = 12.3%), which was obviously smaller that of the original crystals. Thus, it was suggested that the molecular conformation of the TPA-CO crystals became planar upon grinding. This proposal was further verified by a slight red-shift of approximately 9 nm in the diffuse reflectance absorption spectrum (Fig. 4a).12 The molecular conformational change of the TPA-CO crystals in response to hydrostatic pressure was further studied by Raman spectroscopy. The high-pressure experiments were performed in a diamond anvil cell (DAC) with 0.5 mm diamond culets. A small piece of crystal was loaded into the DAC with a ruby chip to determine in situ pressure calibration. A 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of methanol–ethanol was used as the pressure transmitting medium. As depicted in Fig. 4b, the Raman absorption band at 1652 cm−1 was assigned to the C
:1 mixture of methanol–ethanol was used as the pressure transmitting medium. As depicted in Fig. 4b, the Raman absorption band at 1652 cm−1 was assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching mode at normal pressure. Interestingly, this band was shifted to the lower frequency of 1649 cm−1, whereas other bands were shifted to higher frequencies as the pressure was increased up to 1.51 GPa. The change corresponded to extension of the electron delocalization, which resulted in conformational planarization of benzo-phenone.13 On the basis of the structure data of single-crystal diffraction, the packing geometries of TPA-CO at 0.0 GPa, 1.0 GPa and 2.0 GPa along the c axis were optimized by DFT calculations. The optimized structure at 0.0 GPa was in agreement with that of single crystals (see Table S1 and S2†). Notably, the dihedral angle (θ1, θ2) between the carbonyl group and the neighbouring benzene ring was smaller at 2.0 GPa than at 0 GPa, as shown in Table S2.† Clearly, pressurization reduced the void volumes and densified the molecular packing, which induced the structural planarization.
O stretching mode at normal pressure. Interestingly, this band was shifted to the lower frequency of 1649 cm−1, whereas other bands were shifted to higher frequencies as the pressure was increased up to 1.51 GPa. The change corresponded to extension of the electron delocalization, which resulted in conformational planarization of benzo-phenone.13 On the basis of the structure data of single-crystal diffraction, the packing geometries of TPA-CO at 0.0 GPa, 1.0 GPa and 2.0 GPa along the c axis were optimized by DFT calculations. The optimized structure at 0.0 GPa was in agreement with that of single crystals (see Table S1 and S2†). Notably, the dihedral angle (θ1, θ2) between the carbonyl group and the neighbouring benzene ring was smaller at 2.0 GPa than at 0 GPa, as shown in Table S2.† Clearly, pressurization reduced the void volumes and densified the molecular packing, which induced the structural planarization.
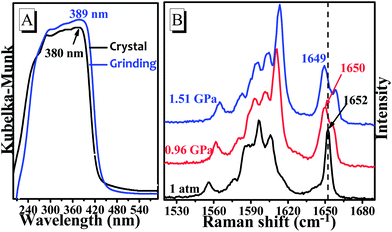 |
| | Fig. 4 (A) The absorption spectra with a diffuse reflectance method of TPA-CO before and after grinding; (B) Raman spectra in the range of 1520–1690 cm−1 of crystals at different pressures. | |
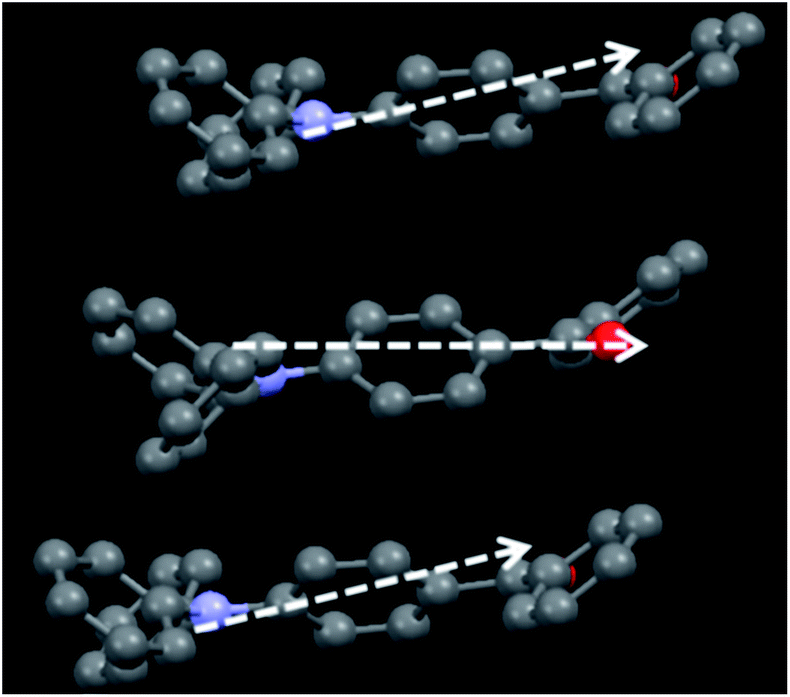
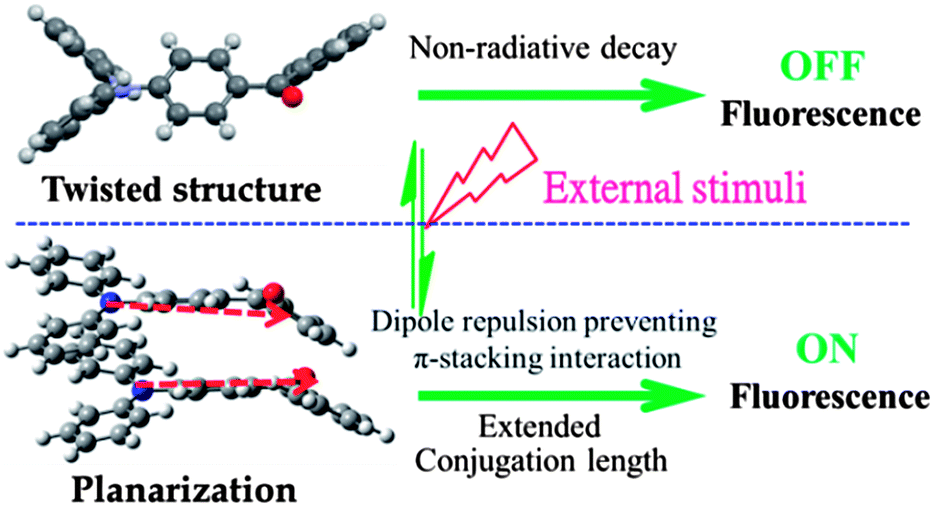
Single crystals of TPA-CO were obtained by slow evaporation of n-hexane–CH2Cl2. As depicted in Fig. S8B and S9C,† the aromatic C–H⋯π and multiple C–H⋯O interactions made the adjacent twisted molecules connect together and form “molecular sheets”. Another C–H⋯O interaction with a distance of 2.8616 Å was also observed between the neighbouring sheets (Fig. S8B†). Usually, for D–π–A dipole molecules, the dipole interaction places the “A” group of one molecule above the “D” moiety of its neighbouring molecule, which contributes to the stability of the crystalline state.14 In this case, the A (D) moiety of one molecule was placed above the A (D) moiety of another adjacent molecule, as shown in Fig. 5. As a result, the D–π–A dipole–dipole interactions were mutually repulsive, which implied that these neighbouring molecules could not be clustered closely together even under external stimuli. Thus, the repulsive force was sufficient to block intermolecular excitonic coupling,15 which caused fluorescence quenching. Fusing all the results together enabled us to draw a clear overall picture for stimuli-responsive behaviours, as depicted in Fig. 6. In the crystalline state, the luminogens may adopt a more twisted conformation in order to fit into the crystalline lattice. Thus, the pristine crystals exhibit a faint luminescence. Upon amorphization by mechanical force, the molecular structure slightly turned planar and the effective intramolecular conjugation was increased, which significantly blocked the non-radiative deactivation pathways. Also, the neighbouring molecules did not get close to one another due to mutually repulsive dipole interactions, which weakened the intermolecular interactions. As a result, the luminescence of ground powders was turned “bright”. The luminescence on–off switching was mainly attributed to the synergy between the planarization of the twisted conformation and weak intermolecular interactions.
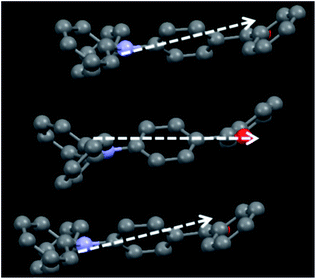 |
| | Fig. 5 An illustration of the repulsive dipole–dipole interactions, the white arrows represent the dipole moments. | |
 |
| | Fig. 6 Proposed mechanism for external stimuli induced luminescence on–off switching in TPA-CO, the red arrows represent the dipole moments. | |
For the melted powders, due to the rapid cooling, the molecules may have quickly aggregated to become amorphous, which tends towards a more planar structure.9a This proposal was further supported by the poor reflection peaks of the PXRD pattern in Fig. 3B. During the long annealing process (Film-h), the molecules in the powder might restructure themselves and adopt a more distorted conformation to fit into the crystalline lattice, which enables the powder to emit a weak fluorescence.16 Uniquely, the quantum yields of samples Melted-m and the drop-casting film were evidently higher than that of the ground powder, which could be attributed to the uneven grinding force applied to the pristine powder17 and the spontaneous recovery ability of the ground powder. As depicted in Fig. S7,† the emission intensity of the freshly ground powder was spontaneously reduced with a slight blue-shift as time passed. Thus, the obtained quantum yields become smaller. In a word, the solid-state TPA-CO with larger twist angles exhibited a tunable molecular conformation, which led to a change in intramolecular conjugation and intermolecular interactions under external stimuli.
In summary, we have designed a novel luminophor, TPA-CO, with a highly distorted structure. Its luminescence on–off switching with high contrast (ΦPL, 0.4% → 12.3%) was repeatedly achieved by simple grinding–vapour cycles. Both the “bright” and “dark” states were separately obtained by tuning the solidification speed of the melted powders. Moreover, the “bright” state, including the films and the ground powders with poor stability, could turn “dark” upon annealing. The photophysical, optical and structural properties were systematically investigated. The fluorescence “bright” and “dark” states of luminophor TPA-CO corresponded to the partially amorphous state and the crystalline state, respectively. In the crystalline state, the luminogens adopted a more twisted conformation and exhibited weak emission. Upon amorphization by external stimuli, the dye molecules might relax to a more planar conformation and then emit a strong fluorescence. Thus, developing highly distorted molecules with tunable conformations may be a design strategy for developing new stimuli-responsive materials with high contrast.
Experimental section
Diphenylamine (3.4 g, 20 mmol), potassium tert-butoxide (0.43 g, 4.5 mmol), Pd(dppf)Cl2 (0.15 mmol), dppf (1 mmol) and dried toluene (60 mL) were mixed in a three-necked flask. The mixture was heated at 70 °C for 10 min with the addition of 4-bromobenzophenone (5.2 g, 20 mmol). The resulting mixture was vigorously stirred for 24 h at 120 °C. After the reaction finished, a certain amount of hydrochloric acid was slowly added. Subsequently, the residue was extracted with dichloromethane. The extraction solution was washed with brine, and then the organic layer was dried over MgSO4 and filtered. The crude product was purified by column chromatography using a dichloromethane–hexane (1/50) mixture as eluent to obtain the desired compound in 44.2% yield (3.1 g); 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J = 7.0 Hz, 2H), 7.72 (dd, J = 7.0 Hz, J = 1.5 Hz, 2H), 7.56 (t, J = 7.5 Hz, 1H), 7.48 (t, J = 7.5 Hz, 2H), 7.35 (t, J = 9.0 Hz, 4H), 7.20 (d, J = 7.5 Hz, 4H), 7.16 (t, J = 7.5 Hz, 2H), 7.03 (dd, J = 7.0 Hz, J = 2.0 Hz, 2H). 13C NMR (500 MHz, CDCl3); δ 195.2, 151.9, 146.5, 138.5, 131.9, 131.7, 129.6, 128.1, 126.0, 124.6, 119.6; crystallographic data for TPA-CO: C25H19NO, M = 349.41, orthorhombic, a = 16.740(5) Å, b = 12.432(5) Å, c = 9.236(3) Å; V = 1922.2(11) Å3, T = 296(2) K, space group Pca2(1), DC = 1.207 g cm−3, Z = 4, 10430 reflections collected, 3385 unique reflection (Rint = 0.0308), the final R indices were R1 = 0.0384, wR2 = 0.0799 [I > 2σ(I)], CCDC = 872836.
Acknowledgements
The authors gratefully thank the supporting of National Natural Science Foundation of China (51203138, 51273179), International S&T Cooperation Program, China (2012DFA51210) and National Basic Research Program of China (2011CBA00700) and scientific research project of Huzhou teachers college (2013002).
Notes and references
-
(a) H. Tian and S. Wang, Chem. Commun., 2007, 781–792 RSC;
(b) Y. Sagara and T. Kato, Nat. Mater., 2009, 1, 605–610 CAS;
(c) O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef CAS PubMed;
(d) Y. S. Zhao, J. S. Wu and J. X. Huang, J. Am. Chem. Soc., 2009, 131, 3158–3159 CrossRef CAS PubMed;
(e) G. G. Shan, H. B. Li, H. Tao Cao, D. X. Zhu, P. Li, Z. M. Su and Y. Liao, Chem. Commun., 2012, 48, 2000–2002 RSC;
(f) L. Y. Bu, M. X. Sun, D. T. Zhang, W. Liu, Y. L. Wang, M. Zheng, S. F. Xue and W. J. Yang, J. Mater. Chem. C, 2013, 1, 2028–2035 RSC;
(g) Y. J. Zhang, G. L. Zhuang, M. Ouyang, B. Hu, Q. B. Song, J. W. Sun, C. Zhang, C. Gu, Y. X. Xu and Y. G. Ma, Dyes Pigm., 2013, 98, 486–492 CrossRef CAS PubMed;
(h) R. M. Rao, C. W. Liao and S. S. Sun, J. Mater. Chem. C, 2013, 1, 6386–6394 RSC.
-
(a) S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS;
(b) Z. Q. Xie, B. Yang, F. Li, G. Cheng, L. L. Liu, H. Xu, L. Ye, M. Hanif, S. Liu, D. G. Ma and Y. G. Ma, J. Am. Chem. Soc., 2005, 127, 14152–14153 CrossRef CAS PubMed;
(c) Z. Q. Xie, W. J. Xie, F. Li, L. L. Liu, H. Wang and Y. G. Ma, J. Phys. Chem. C, 2008, 112, 9066–9071 CrossRef CAS;
(d) W. Liu, Y. L. Wang, M. X. Sun, D. T. Zhang, M. Zheng and W. J. Yang, Chem. Commun., 2013, 49, 6042–6044 RSC.
-
(a) Z. G. Chi, X. Q. Zhang, B. J. Xu, X. Zhou, C. P. Ma, Y. Zhang, S. Liu and J. R. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC;
(b) S. P. Anthony, ChemPlusChem, 2012, 77, 518–531 CrossRef CAS;
(c) Y. F. Zhao, H. Z. Gao, Y. Fan, T. L. Zhou, Z. M. Su, Y. Liu and Y. Wang, Adv. Mater., 2009, 21, 3165–3169 CrossRef CAS.
- M. S. Kwon, J. Gierschner, S. J. Yoon and S. Y. Park, Adv. Mater., 2012, 24, 5487–5492 CrossRef CAS PubMed.
- M. Kato, A. Omura, A. Toshikawa, S. Kishi and Y. Sugimoto, Angew. Chem., Int. Ed., 2002, 41, 3183–3185 CrossRef CAS.
-
(a) S. Hirata and T. Watanabe, Adv. Mater., 2006, 18, 2725–2729 CrossRef CAS;
(b) T. Mutai, H. Satou and K. Araki, Nat. Mater., 2005, 4, 685–687 CrossRef CAS PubMed;
(c) Y. F. Zhao, H. Z. Gao, Y. Fan, T. L. Zhou, Z. M. Su, Y. Liu and Y. Wang, Adv. Mater., 2009, 21, 3165–3169 CrossRef CAS;
(d) R. M. Rao, C. W. Liao, W. L. Su and S. S. Sun, J. Mater. Chem. C, 2013, 1, 5491–5501 RSC.
-
(a) J. Luo, L. Y. Li, Y. L. Song and J. Pei, Chem.–Eur. J., 2011, 17, 10515–10519 CrossRef CAS PubMed;
(b) M. J. Teng, X. R. Jia, S. Yang, X. F. Chen and Y. Wei, Adv. Mater., 2012, 24, 1255–1261 CrossRef CAS PubMed;
(c) Y. J. Zhang, J. W. Sun, X. J. Lv, M. Ouyang, F. Cao, G. X. Pan, L. P. Pan, G. B. Fan, W. W. Yu, C. He, S. S. Zheng, F. Zhang, W. Wang and C. Zhang, CrystEngComm, 2013, 15, 8998–9002 RSC.
-
(a) W. Rettig, Angew. Chem., Int. Ed., 1986, 25, 971–988 CrossRef;
(b) G. Qian, B. Dai, M. Luo, D. B. Yu, J. Zhan, Z. Q. Zhang, D. G. Ma and Z. Y. Wang, Chem. Mater., 2008, 20, 6208–6216 CrossRef CAS;
(c) Y. J. Zhang, J. W. Sun, G. F. Bian, Y. Y. Chen, M. Ouyang, B. Hua and C. Zhang, Photochem. Photobiol. Sci., 2012, 11, 1414–1421 RSC;
(d) P. Y. Gu, C. J. Lu, Z. J. Hu, N. J. Li, T. T. Zhao, Q. F. Xu, Q. H. Xu, J. D. Zhang and J. M. Lu, J. Mater. Chem. C, 2013, 1, 2599–2606 RSC.
-
(a) W. Z. Yuan, Y. Q. Tan, Y. Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. F. Feng, H. H. Y. Sung, Y. W. Lu, I. D. Williams, J. Z. Sun, Y. M. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837–2843 CrossRef CAS PubMed;
(b) Y. Y. Gong, Y. Q. Tan, J. Liu, P. Lu, C. F. Feng, W. Z. Yuan, Y. W. Lu, J. Z. Sun, G. F. He and Y. M. Zhang, Chem. Commun., 2013, 49, 4009–4011 RSC.
-
(a) D. Oelkrug, A. Tompert, J. Gierschner, H. J. Egelhaaf, M. Hanack, M. Hohloch and E. Steinhuber, J. Phys. Chem. B, 1998, 102, 1902 CrossRef CAS;
(b) B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed;
(c) M. Levitus, K. Schmieder, H. Ricks, K. D. Shimizu, U. H. F. Bunz and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2001, 123, 4259–4265 CrossRef CAS PubMed;
(d) R. Deans, J. Kim, M. R. Machacek and T. M. Swager, J. Am. Chem. Soc., 2000, 122, 8565–8566 CrossRef CAS.
-
(a) D. Oelkrug, A. Tompert, H. Egelhaaf, M. Hanack, E. Steinhuber, M. Hohloch, H. Meier and U. Stalmach, Synth. Met., 1996, 83, 231–237 CrossRef CAS;
(b) M. M. Souza, G. Rumbles, I. R. Gould, H. Amer, I. D. W. Samuel, S. C. Moratti and A. B. Holmes, Synth. Met., 2000, 111–112 Search PubMed;
(c) S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kin, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed.
-
(a) X. Q. Zhang, Z. G. Chi, H. Y. Li, B. J. Xu, X. F. Li, W. Zhou, S. W. Liu, Y. Zhang and J. R. Xu, Chem.–Asian J., 2011, 6, 808–811 CrossRef CAS PubMed;
(b) X. Q. Zhang, Z. G. Chi, J. Y. Zhang, H. Y. Li, B. J. Xu, X. F. Li, S. W. Liu, Y. Zhang and J. R. Xu, J. Phys. Chem. B, 2011, 115, 7606–7611 CrossRef CAS PubMed;
(c) F. K. Chen, J. Zhang and X. H. Wan, Chem.–Eur. J., 2012, 18, 4558–4667 CrossRef CAS PubMed;
(d) B. J. Xu, Z. G. Chi, J. Y. Zhang, X. Q. Zhang, H. Y. Li, X. F. Li, S. W. Liu, Y. Zhang and J. R. Xu, Chem.–Asian J., 2011, 6, 1470–1478 CrossRef CAS PubMed.
-
(a) M. Piacenza, D. Comoretto, M. Burger, V. Morandi, F. Marabelli, C. Martinelli, G. M. Farinola, A. Cardone, G. Gigli and F. D. Sala, ChemPhysChem, 2009, 10, 1284–1290 CrossRef CAS PubMed;
(b) G. Heimel, D. Somitsch, P. Knoll, J. Brédas and E. Zojer, J. Chem. Phys., 2005, 122, 114511–114519 CrossRef PubMed;
(c) K. Honda, Y. Furukawa and H. Nishide, Vib. Spectrosc., 2006, 40, 149 CrossRef CAS PubMed.
-
(a) A. Patra, S. P. Anthony and T. P. Radhakrishnan, Adv. Funct. Mater., 2007, 17, 2077–2084 CrossRef CAS;
(b) Q. Dai, W. M. Liu, L. T. Zeng, C. S. Lee, J. S. Wu and P. F. Wan, CrystEngComm, 2011, 13, 4617–4624 RSC.
-
(a) Z. J. Ning, Z. Chen, Q. Zhang, Y. L. Yan, S. X. Qian, Y. Cao and H. Tian, Adv. Funct. Mater., 2007, 17, 3799–3807 CrossRef CAS;
(b) A. Dreuw, J. Plöner, L. Lorenz, J. Wachtveit, J. E. Djanhan, J. Brüning, T. Metz, M. Bolte and M. U. Schmidt, Angew. Chem., Int. Ed., 2005, 44, 7783–7786 CrossRef CAS PubMed;
(c) N. S. S. Kumar, S. Varghese, N. P. Rath and S. Das, J. Phys. Chem. C, 2008, 112, 8429–8437 CrossRef CAS.
- Y. Q. Dong, J. W. Y. Lam, A. J. Qin, J. X. Sun, J. Z. Liu, Z. Li, J. Z. Sun, H. H. Y. Sung, I. D. Williams, H. S. Kwokc and B. Z. Tang, Chem. Commun., 2007, 3255–3257 RSC.
- X. T. Chen, R. R. Wei, Y. Xiang, Z. J. Zhou, K. Li, P. S. Song and A. J. Tong, J. Phys. Chem. C, 2011, 115, 14353–14359 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 872836. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3tc31416b |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.