Formation of hexagonal and cubic fluorescent periodic mesoporous organosilicas in the channels of anodic alumina membranes
Yan
Li
a,
Andreas
Keilbach
b,
Norihiro
Mizoshita
cd,
Shinji
Inagaki
cd and
Thomas
Bein
*a
aDepartment of Chemistry and Center for NanoScience (CeNS), University of Munich (LMU), Butenandtstrasse 5–13 (E), Munich 81377, Germany. E-mail: tbein@lmu.de; Fax: +49-89-2180-77622
bAnton Paar GmbH, Anton-Paar-Str. 20, A-8054 Graz, Austria
cToyota Central R&D Laboratories Inc., Nagakute, Aichi 480-1192, Japan. E-mail: inagaki@mosk.tytlabs.co.jp; Fax: +81-561-63-6507
dCore Research for Evolutional Science and Technology (CREST), Japan Science and Technology Agency (JST), Kawasaki, Saitama 332-0012, Japan
First published on 14th August 2013
Abstract
The synthesis of periodic mesoporous organosilicas (PMOs) in the confinement of porous anodic alumina membranes (AAMs) was successfully achieved through a modified evaporation-induced self-assembly (EISA) process. 1,3,5-Tris(4-triethoxysilylstyryl)benzene, (a three-armed oligo(phenylenevinylene) organosilane compound, abbr. 3a-OPV), the precursor of the first reported charge-conducting PMO, was used as an organosilica source. Triblock-copolymers Pluronic F127 (EO106 PO70 EO106) or F108 (EO132 PO50 EO132) were used as structure directing agents. The block-copolymer F127 led to a 2D-hexagonal circular mesostructure within the AAM channels and the block copolymer Pluronic F108 resulted in a mesophase with a body-centered cubic (Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) structure. Compared to the previously reported 3a-OPV-PMO film, the resulting hierarchical PMO/AAM systems have improved features, that is, the synthesized PMOs have a pore size of around 10 nm and the compounds are found to be stable against thermal treatment at temperatures of up to 200 °C and they are also stable in the electron beam of the electron microscope. Additionally, both of the resulting hierarchical mesoporous composites show fluorescence in the visible region due to the strongly interacting phenylenevinylene chromophores in the PMO frameworks.
m) structure. Compared to the previously reported 3a-OPV-PMO film, the resulting hierarchical PMO/AAM systems have improved features, that is, the synthesized PMOs have a pore size of around 10 nm and the compounds are found to be stable against thermal treatment at temperatures of up to 200 °C and they are also stable in the electron beam of the electron microscope. Additionally, both of the resulting hierarchical mesoporous composites show fluorescence in the visible region due to the strongly interacting phenylenevinylene chromophores in the PMO frameworks.
Introduction
Mesoporous organic–inorganic materials are attractive because they can combine desirable features of organic and inorganic building blocks in a single system. Thus, many hybrid porous materials feature the versatile functionalities of organic molecules as well as the thermal stability and mechanical robustness of the inorganic parts.1,2 Periodic mesoporous organosilicas (PMOs) are such a class of hybrid materials with organic units directly incorporated into the mesoporous silica pore walls, and have attracted much interest since their initial discovery in 1999.3–5 The method to obtain such mesostructured hybrid materials is polycondensation of alkoxysilane precursor molecules in the presence of structure directing agents (e.g., surfactants such as tetraalkylammonium halides or nonionic copolymers). The noteworthy point regarding the PMO materials is the successful integration of versatile organic groups into their pore walls, such as alkyl, unsaturated and aromatic systems.6–9 The resulting functional porous materials can interact in different ways with guest molecules that reside in the pores, leading to potential applications of PMOs as catalysts,10,11 adsorbents,12,13 and optical14–17 systems. In recent years, great efforts have been made towards developing novel PMOs with photoactive or electroactive functions within their frameworks.18 Recently, a novel PMO material was reported by some of us with oligo(phenylenevinylene) molecules being incorporated into the pore walls and it was found to be charge-conducting (its charge carrier mobility was on the order of 10−5 cm2 V−1 s−1).19 This PMO was synthesized as a film material on flat substrates. The synthesized film had mesopores with a fairly small diameter of 2.4 nm and possessed a hexagonal packing of the mesochannels judged by indexing the diffraction peaks in the X-ray diffraction pattern. However, direct imaging of such a PMO film by electron microscopy was difficult due to the collapse of the mesostructure under electron-beam irradiation. Therefore, improving the stability of this PMO material, tuning the mesopore size and controlling the mesochannel orientation remain as challenges to further develop its potential.Recently, great efforts have been made to realize control over the domain size and orientation of mesoporous systems, including application of external electric20,21 or magnetic fields,22,23 or use of chemically24,25 or lithographically26 treated substrates. Moreover, an alternative strategy uses confinement in anisotropic hosts such as porous anodic alumina membranes (AAMs).27 The resulting mesoporous composites were reported to be able to offer high mechanical and thermal stability due to the support of the alumina host. Furthermore, the confinement effects can lead to enhanced control over the resulting mesophase morphologies, with the possibility to realize large single-phase domains and high aspect ratios of mesoporous systems.28–34 Given the favorable interaction of silica species with AAM channel walls found in inorganic silica/AAM systems, we reasoned that combining PMO systems with porous AAM hosts might lead to a different phase behavior and phase stability, thus improving the quality of PMO materials and meanwhile opening the way to design novel hierarchical nanosystems. Based on our previous work on PMO/AAM composite systems,35,36 the present study focuses on the synthesis of the reported charge-conducting PMO within the confinement of AAM channels to achieve a new hierarchical PMO/AAM nanosystem that might move us closer to the application of PMO systems in optoelectronic devices.
In this study, 1,3,5-tris(4-triethoxysilylstyryl)benzene (a three-armed oligo(phenylenevinylene) organosilane compound, denoted as 3a-OPV) was used as an organosilica source to form PMO materials with Pluronic F127 or F108 as structure directing agents. Through a modified evaporation-induced self-assembly (EISA) process,37 highly ordered and stable 3a-OPV-PMO materials were successfully synthesized within AAM host systems. The resulting hierarchical 3a-OPV-PMO/AAM systems were found to be stable against thermal treatment at temperatures of up to 200 °C and also stable in the electron beam of an electron microscope. The pore diameters of the new PMO materials are around 10 nm due to the large sizes of the structure directing agents. With such large pore sizes, the resulting PMO materials offer compatibility with many other guest molecules, i.e., the mesochannels of PMOs can accommodate large molecules such as photo-reactive precursors or charge-transporting molecules, so that these PMO materials could offer potential for applications in photocatalytic and photovoltaic fields. Furthermore, all the 3a-OPV-PMO/AAM hierarchical materials showed fluorescence in the visible region due to the phenylenevinylene chromophores in the condensed organosilica frameworks.
Experimental section
Preparation of 1,3,5-tris(styryl)benzene-bridged PMO in AAM channels
The synthesis of PMO confined in AAM channels was achieved by the evaporation-induced self-assembly (EISA) approach. Whatman Anodiscs (47 mm diameter, 60 μm thickness, 150–250 nm pore diameter) were used as porous alumina substrates. 1,3,5-Tris(4-triethoxysilylstyryl)benzene (3a-OPV) (Scheme 1) served as an organosilica precursor (provided by Toyota Central R&D Laboratories Inc., Japan).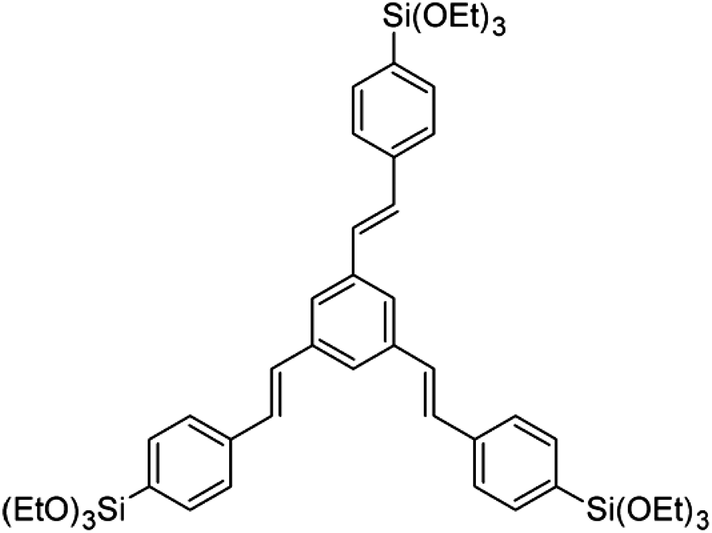 | ||
| Scheme 1 Chemical structure of 1,3,5-tris(4-triethoxysilylstyryl)benzene, a three-armed oligo(phenylenevinylene) precursor (denoted as 3a-OPV). | ||
The preparation method for 1,3,5 tris(4-triethoxysilyl-styryl)benzene was reported in the ESI of the previous paper (ref. 18). Triblock co-polymer F127 (poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide), EO106PO70EO106, Sigma-Aldrich) and F108 (EO132PO50EO132, Sigma-Aldrich) were used as structure directing agents (SDA). All chemicals were used without further purification. In the following, the 3a-OPV-PMO materials are named with the used SDA followed by the description of the formed mesostructure, i.e., the samples synthesized by F127 and F108 are denoted as F127-circular and F108-cubic, respectively.
For the preparation of the sample F127-circular, 3a-OPV (0.06 g, 0.067 mmol) and F127 (0.06 g, 0.005 mmol) were first dissolved in 2.316 g absolute EtOH. Then, distilled H2O (0.030 g, 1.7 mmol) was added and the solution was stirred for 10 min. Next, 1 M HCl (4.2 μL, 4.2 × 10−3 mmol) was added and the resulting solution was stirred for 2 h. Finally, the AAM was placed on a Teflon plate and soaked with the above prepared precursor solution by drop-casting 0.75 mL of the solution over the whole membrane surface (47 mm diameter, with an area of 17 cm2). During the EISA-process, the ambient conditions were maintained at 45–55% relative humidity and 25 °C.
For the preparation of the sample F108-cubic, the synthesis procedures followed the same protocol as above with the exception that 0.075 g F108 (0.005 mmol) was added to the precursor solution instead of F127.
In order to remove the structure directing agents to get accessible mesopores, the PMO/AAM composites were calcined up to 200 °C with a heating ramp of 0.5 °C min−1. Before reaching 200 °C, the samples were kept at 120 °C for 5 h and finally kept for 10 h at 200 °C.
Characterization
The samples were characterized by 2D small-angle X-ray scattering (2D-SAXS) using the SAXSess system by Anton Paar with a CCD detector (PI-SCX:4300, Roper Scientific). The wavelength of the incident beam is 0.154 nm (Cu Kα), and the sample-detector distance was set to 308 mm. Samples were measured with a tilt angle of 10° with respect to the primary beam. Transmission electron microscopy (TEM) was performed using a JEOL 2011 with an acceleration voltage of 200 kV. Nitrogen sorption measurements were carried out at −196 °C using an Autosorb-1 by Quantachrome instruments. Prior to the measurement, the PMO/AAM composites were broken into small pieces (without dissolving the AAM host matrix) to fit the measurement cell. All samples were degassed at 150 °C for 12 h in a vacuum. The Brunauer–Emmett–Teller (BET) surface area was calculated using experimental points at a relative pressure range of p/p0 = 0.05–0.20. The total pore volume was calculated by the N2 amount adsorbed at the point of p/p0 = 0.95. NLDFT pore size distributions were calculated using an SiO2 kernel assuming a cylindrical pore geometry for the F127-circular sample and a cylindrical/sphere pore geometry for the F108-cubic sample. Solid-state 13C and 29Si NMR experiments were carried out with the broken and ground PMO/AAM composites (without dissolution of the AAM matrix) and performed on a Bruker Avance-III 500 spectrometer (11.7 Tesla) operating at frequencies of 125.8 MHz for 13C and 99.4 MHz for 29Si. 13C(1H) CP-MAS spectra were acquired using a 90° pulse length of 2.5 μs (3.7 dB) with a cross-polarization contact time of 5 ms and a recycle delay of 2 s. 29Si(1H) CP-MAS experiments were conducted using a 90° pulse length of 2.5 μs (2.7 dB) with a cross-polarization contact time of 5 ms and a recycle delay of 4 s. The fluorescence spectra were measured with a fluorescence spectroscopy system (PTI 814 from Photon Technology international) with a xenon arc lamp.Results and discussion
F127-circular 3a-OPV-PMO
Using Pluronic F127 as a structure directing agent, a circular hexagonal phase was observed within the AAM channels. In order to obtain an open pore system, the composite membranes were mildly calcined up to 200 °C. The highly ordered hexagonal circular structure was identified as the major phase by 2D small angle X-ray scattering (2D-SAXS) measurements. The 2D-SAXS pattern of the calcined sample (Fig. 1A) shows both in-plane (01) and out-of-plane (10) peaks that are characteristics of a circular hexagonal structure confined in AAM channels.35,38,39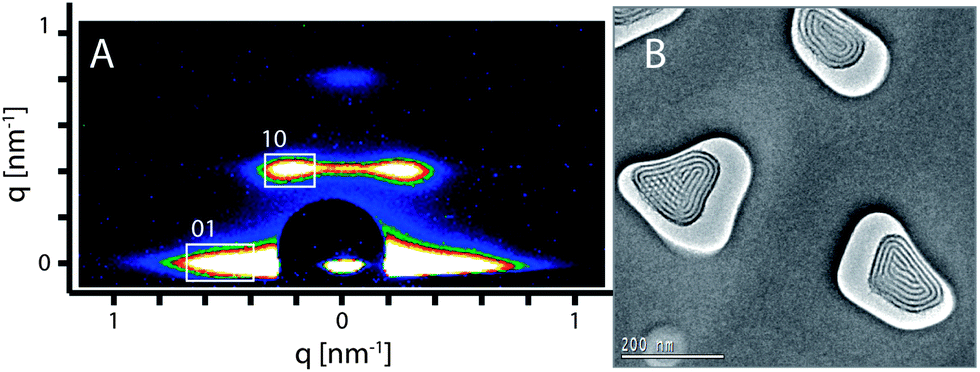 | ||
| Fig. 1 (A) 2D-SAXS pattern and (B) plan-view TEM micrograph of the calcined PMO/AAM composite showing the highly ordered hexagonal circular structure of the 3a-OPV-PMO formed with F127 as a structure directing agent. | ||
The d-values calculated from the diffraction pattern are 14.5 nm and 15.6 nm, corresponding to d01 and d10 respectively. The TEM image (Fig. 1B) of the calcined composite displays an electron-beam stable PMO with a highly ordered hexagonal circular structure as the predominant phase that is formed in the AAM channels. The measured d-value is 14 nm, which is slightly smaller than the values resulting from the X-ray diffraction. This small difference can be attributed to the shrinkage of the porous system during the ion milling procedure, which is a crucial step in the TEM specimen preparation, as well as the effect of exposure to the electron beam under high vacuum in the TEM. We note that the orientation of the hexagonal mesostructures could be influenced by the size and shape of AAM channels.39 In a small number of the AAM channels, a hexagonal columnar structure was observed as a side phase (e.g. the channel on the left side in Fig. 1B).
The nitrogen sorption isotherm (Fig. 2A) of the calcined F127-circular PMO shows the typical type IV isotherm shape commonly observed with mesoporous materials. The Brunauer–Emmett–Teller (BET) surface area calculated from the isotherm is 57.3 m2 g−1 and the pore volume is 0.073 cm3 g−1 (the mass for the calculation includes the AAM host). The isotherm has a large hysteresis loop suggesting the existence of ink-bottle shaped pores in the PMO material. This can be confirmed by comparing the DFT pore-size distributions from the adsorption branch (av. pore diameter ∼ 9.4 nm) and the desorption branch (av. pore diameter ∼ 5.3 nm), both of which show sharp pore size distributions (Fig. 2B).
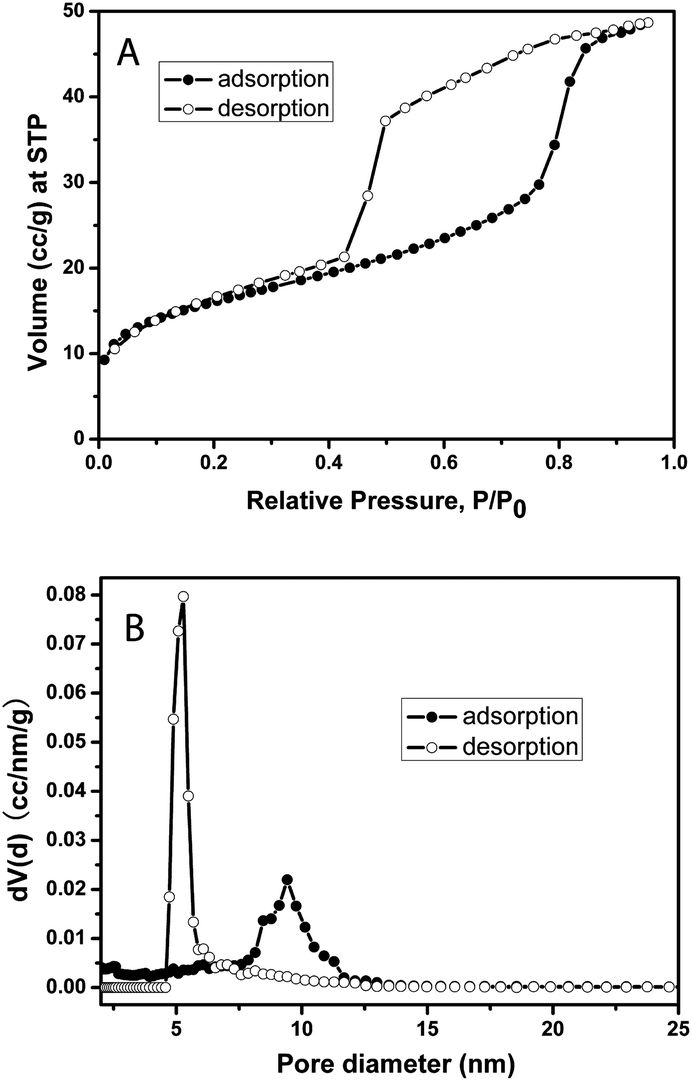 | ||
| Fig. 2 (A) Nitrogen adsorption (●) and desorption (○) isotherms of the calcined F127-circular sample and (B) pore size distributions calculated by a NLDFT method from both the adsorption branch (●) and desorption branch (○) by using an SiO2 kernel assuming a cylindrical pore geometry. | ||
In the 13C-MAS NMR spectrum (Fig. 3A), the peaks at around 130 ppm and their spinning side bands (indicated by asterisk) correspond to the 1,3,5-tris(styryl)benzene group, showing the preservation of the organic units in the mildly calcined F127-circular PMO material. In addition, weak signals (at 17.6 and 75.3 ppm) can be attributed to small amounts of residual F127 molecules, indicating the incomplete removal of the structure directing agent. This incomplete removal of F127 possibly caused partial blocking of the large mesopores and thus led to the ink-bottle shaped pores, which is in agreement with the above nitrogen sorption result. The 29Si NMR spectrum (Fig. 3B) shows resonances attributed to T2-silicon species (RSi(OSi)2(OH)) at −71.3 ppm and T3 species (RSi(OSi)3) at −80.2 ppm, suggesting a high degree of polycondensation of the organosilane precursors in the formed PMO pore walls. It is also noteworthy that no resonance appears in the Q-site region (around 100 ppm), indicating that the Si–C bonds were quite stable in the framework of the 3a-OPV-PMO material under all the conditions of preparation and thermal treatment.
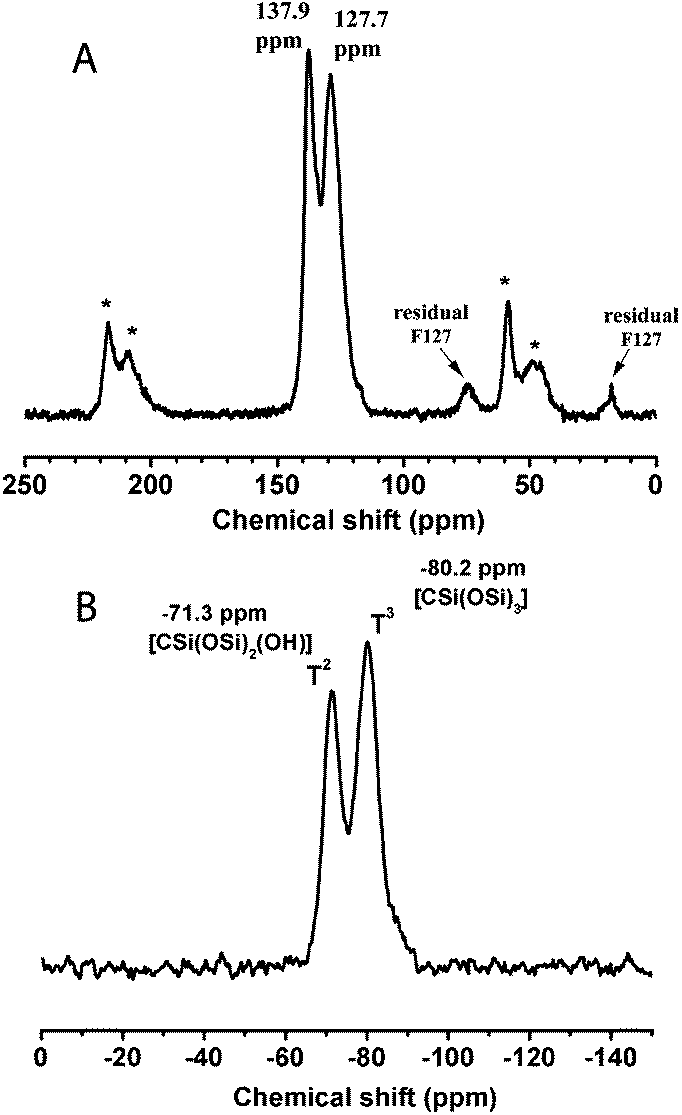 | ||
| Fig. 3 (A) 13C-MAS NMR and (B) 29Si NMR spectra of the calcined F127-circular sample, showing the almost complete removal of the surfactant F127, the highly condensed Si–O–Si network and the preserved Si–C bonds in the obtained stable PMO material. (Signals marked with asterisk denote the spinning side bands of the tris(styryl)benzene groups.) | ||
F108-cubic 3a-OPV-PMO
A second mesophase was created by using the triblock co-polymer F108 as a structure directing agent. Compared to Pluronic F127 (EO106PO70EO106), template F108 (EO132PO50EO132) is a relatively hydrophilic surfactant with longer hydrophilic EO chains and shorter hydrophobic PO parts, which leads to micelles with higher curvature in the solution.40 Thus, we reasoned that after replacing F127 with F108 into a solution that originally could result in a 2D hexagonal (circular) structure, a new structure would be formed. 2D-SAXS measurements were carried out to identify the mesostructure of the PMO templated by F108. The SAXS pattern recorded with the calcined sample shows the typical peaks assigned to a body centered cubic Imm phase (Fig. 4A), similar to recent observations with other cubic phases in AAM hosts,36,39 indicating that a new 3a-OPV-PMO with a cubic structure was formed. The formation of the cubic mesostructure was also confirmed by the TEM micrographs. The average d-value resulting from the SAXS pattern is 17.4 nm, which is in good agreement with the value (d110 = 17 nm) obtained from the TEM image. A plan-view TEM micrograph of the calcined F108-cubic sample is displayed in Fig. 4B. It is remarkable that the F108-cubic PMO material within the AAM channels shows minor shrinkage; we attribute this to the three-dimensional connection existing in the cubic structure combined with the spatial confinement in the AAM host.
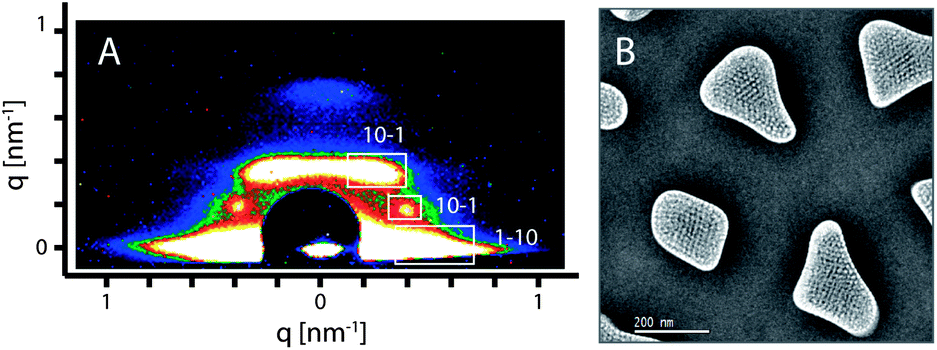 | ||
| Fig. 4 (A) 2D-SAXS pattern and (B) plan-view TEM micrograph of the calcined F108-cubic PMO, representing a highly ordered body centered cubic structure with only minor shrinkage after thermal treatment. | ||
The porosity properties were determined by nitrogen sorption. Fig. 5 depicts the isotherms of the calcined F108-cubic sample, showing the type IV shape with a very wide adsorption–desorption hysteresis that is attributed to large mesopores connected by smaller open windows in the cubic system. The BET surface area of the calcined F108-cubic composite is 37 m2 g−1 and the pore volume is 0.071 cm3 g−1 (the mass includes the AAM membrane). The pore-size distribution (calculated from the adsorption branch) shows a pore diameter of 14.0 nm.
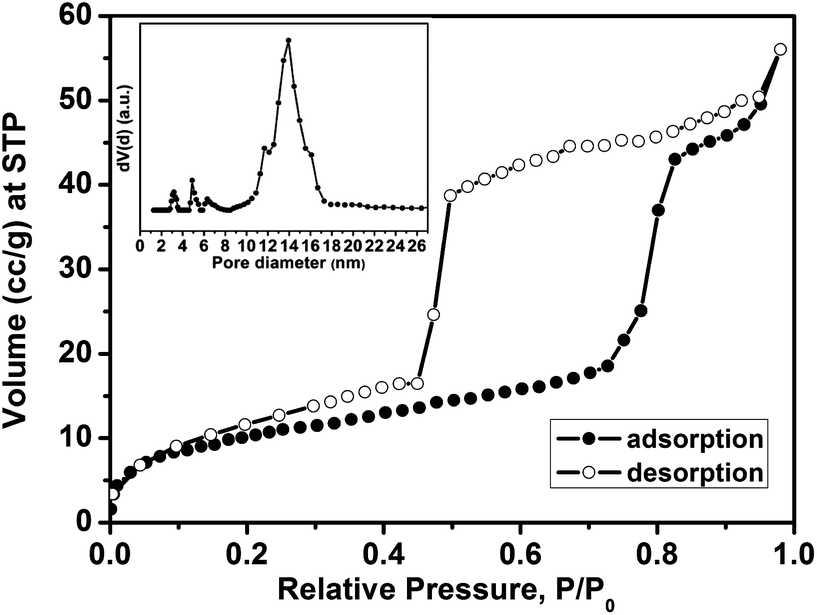 | ||
| Fig. 5 Nitrogen sorption isotherms of the calcined F108-cubic sample with an inset showing the corresponding pore size distribution calculated with a NLDFT method using the adsorption branch with a SiO2 kernel assuming a cylindrical/spherical pore geometry. | ||
The 13C and 29Si NMR data (Fig. 6) of the calcined F108-cubic sample are consistent with those obtained from the F127-circular system. All the characterization results show that a body centered cubic 3a-OPV-PMO is successfully formed with electron beam stability as well as thermal stability up to 200 °C.
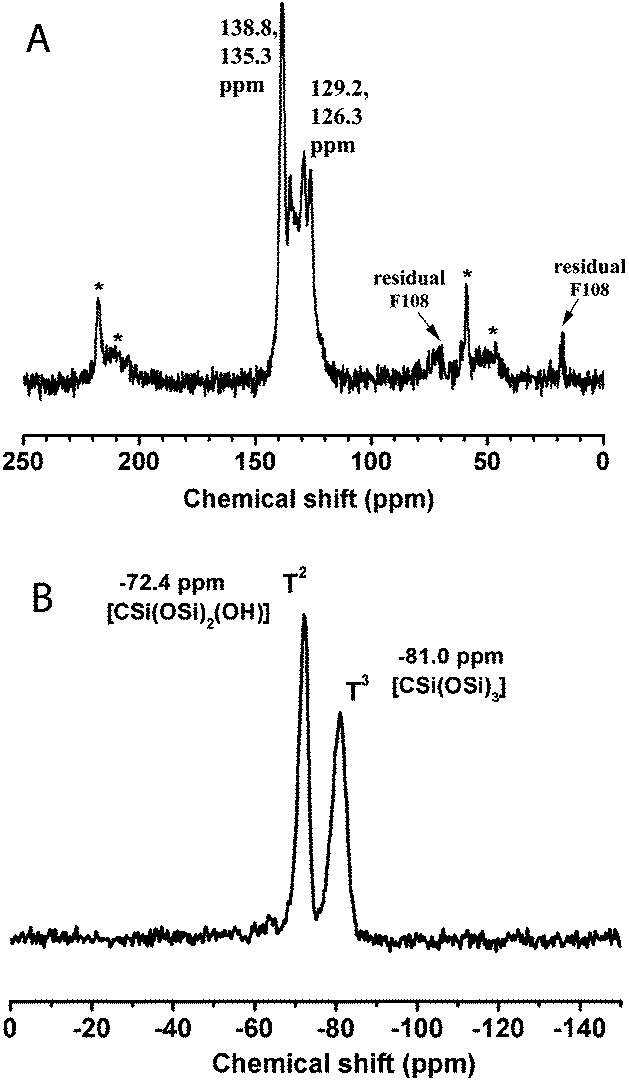 | ||
| Fig. 6 (A) 13C-MAS NMR and (B) 29Si NMR spectra of the calcined F108-cubic sample, showing the almost complete removal of the template Pluronic F108 and the high degree of condensation of the Si–O–Si network. (Signals marked with asterisk denote the spinning side bands.) | ||
Fluorescence emission of 3a-OPV-PMO/AAM composites
Fluorescence emission spectra were recorded from both 3a-OPV-PMO/AAM composites and from a diluted isopropanol solution of 3a-OPV precursor (Fig. 7).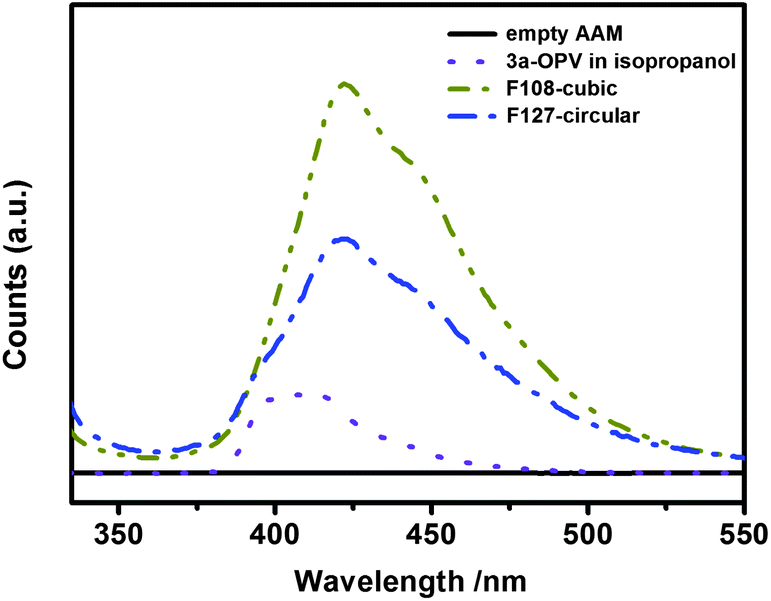 | ||
| Fig. 7 Fluorescence spectra (excited at 320 nm) of the circular and cubic-structured 3a-OPV-PMO confined in AAM channels and a diluted 3a-OPV precursor solution in isopropanol (10−5 M) (the intensities are not to scale). | ||
The resulting hierarchically structured 3a-OPV-PMO/AAM composites were observed to be fluorescent in the visible light region. The fluorescence emission bands of both circular and cubic PMO/AAM composites peak at a wavelength of 422 nm, which indicates a slight shift to longer wavelength with respect to the fluorescence emission of the diluted precursor solution (10−5 M in isopropanol) peaking at 410 nm. The broadening and energy shift of the emission bands of the obtained PMOs indicate that the phenylenevinylene chromophores are still active and that they experience intermolecular electronic interactions in the confined organosilica networks.
Conclusions
In summary, phenylenevinylene-bridged periodic mesoporous organosilica materials with tunable mesostructures were successfully synthesized within the tubular channels of anodic alumina membranes through an evaporation-induced self-assembly (EISA) process. Depending on the block copolymer structure directing agent employed, different mesophases could be observed. 2D-SAXS experiments and TEM measurements carried out on the calcined sample templated by Pluronic F127 showed that a 2D-hexagonal circular phase was predominantly formed within AAM channels, and that a hexagonal columnar structure was created as a minor phase. When using the template F108, which can form micelles with a higher curvature than Pluronic F127, a body centered cubic (Imm) mesophase was achieved in the confined environment of the AAM channels.
It is noteworthy that the resulting circular and cubic 3a-OPV-PMO/AAM composites were both structurally stable against thermal treatments at temperatures of up to 200 °C and also stable in the electron beam of the electron microscope, which is a remarkable enhancement over the previously reported flat-substrate supported 3a-OPV-PMO films. Additionally, both the hierarchically synthesized nanostructured 3a-OPV-PMO/AAM composites were observed to be fluorescent in the visible light region. The fluorescence emission of the PMO/AAM composites showed a red-shift compared to that of the molecular precursor solution, indicating electronic interactions between the phenylenevinylene chromophores in the condensed organosilica networks. The success of achieving such stable, fluorescent and highly oriented mesoporous 3a-OPV-PMO materials suggests that these PMO/AAM composites and similar chromophore-containing hierarchical nanostructures might offer potential for application in optoelectronic systems.
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft through the SFB 486, the LMUexcellent program, the Nanosystems Initiative Munich (NIM cluster), and the China Scholarship Council (CSC) for supporting this work.References
- F. Hoffmann and M. Fröba, Chem. Soc. Rev., 2011, 40, 608–620 RSC.
- P. Van Der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867–871 CAS.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611–9614 CrossRef CAS.
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302–3308 CrossRef CAS.
- J. Morell, M. Gungerich, G. Wolter, J. Jiao, M. Hunger, P. J. Klar and M. Froba, J. Mater. Chem., 2006, 16, 2809–2818 RSC.
- A. Sayari and W. H. Wang, J. Am. Chem. Soc., 2005, 127, 12194–12195 CrossRef CAS PubMed.
- T. Tani, N. Mizoshita and S. Inagaki, J. Mater. Chem., 2009, 19, 4451–4456 RSC.
- C. Vercaemst, P. E. de Jongh, J. D. Meeldijk, B. Goderis, F. Verpoort and P. Van Der Voort, Chem. Commun., 2009, 4052–4054 RSC.
- A. Kuschel and S. Polarz, J. Am. Chem. Soc., 2010, 132, 6558–6565 CrossRef CAS PubMed.
- Q. H. Yang, J. Liu, L. Zhang and C. Li, J. Mater. Chem., 2009, 19, 1945–1955 RSC.
- C. Li, J. Liu, X. Shi, J. Yang and Q. Yang, J. Phys. Chem. C, 2007, 111, 10948–10954 CAS.
- M. Park, S. S. Park, M. Selvaraj, D. Y. Zhao and C. S. Ha, Microporous Mesoporous Mater., 2009, 124, 76–83 CrossRef CAS PubMed.
- R. Hernandez, A. C. Franville, P. Minoofar, B. Dunn and J. I. Zink, J. Am. Chem. Soc., 2001, 123, 1248–1249 CrossRef CAS.
- S. Inagaki, O. Ohtani, Y. Goto, K. Okamoto, M. Ikai, K. Yamanaka, T. Tani and T. Okada, Angew. Chem., Int. Ed., 2009, 48, 4042–4046 CrossRef CAS PubMed.
- P. N. Minoofar, R. Hernandez, S. Chia, B. Dunn, J. I. Zink and A. C. Franville, J. Am. Chem. Soc., 2002, 124, 14388–14396 CrossRef CAS PubMed.
- N. Mizoshita, Y. Goto, T. Tani and S. Inagaki, Adv. Funct. Mater., 2008, 18, 3699–3705 CrossRef CAS.
- W. Wang, J. E. Lofgreen and G. A. Ozin, Small, 2010, 6, 2621 CrossRef.
- N. Mizoshita, M. Ikai, T. Tani and S. Inagaki, J. Am. Chem. Soc., 2009, 131, 14225–14227 CrossRef CAS PubMed.
- K. Kuraoka, Y. Tanaka, M. Yamashita and T. Yazawa, Chem. Commun., 2004, 1198–1199 RSC.
- M. Trau, N. Yao, E. Kim, Y. Xia, G. M. Whitesides and I. A. Aksay, Nature, 1997, 390, 674–676 CAS.
- A. Firouzi, D. J. Schaefer, S. H. Tolbert, G. D. Stucky and B. F. Chmelka, J. Am. Chem. Soc., 1997, 119, 9466–9477 CrossRef CAS.
- Y. Yamauchi, M. Sawada, T. Noma, H. Ito, S. Furumi, Y. Sakka and K. Kuroda, J. Mater. Chem., 2005, 15, 1137–1140 RSC.
- H. Miyata, T. Noma, M. Watanabe and K. Kuroda, Chem. Mater., 2002, 14, 766–772 CrossRef CAS.
- H. Miyata, T. Suzuki, A. Fukuoka, T. Sawada, M. Watanabe, T. Noma, K. Takada, T. Mukaide and K. Kuroda, Nat. Mater., 2004, 3, 651–656 CrossRef CAS PubMed.
- R. L. Rice, D. C. Arnold, M. T. Shaw, D. Iacopina, A. J. Quinn, H. Amenitsch, J. D. Holmes and M. A. Morris, Adv. Funct. Mater., 2007, 17, 133–141 CrossRef CAS.
- B. Platschek, A. Keilbach and T. Bein, Adv. Mater., 2011, 23, 2395–2412 CrossRef CAS PubMed.
- A. Y. Ku, S. T. Taylor and S. M. Loureiro, J. Am. Chem. Soc., 2005, 127, 6934–6935 CrossRef CAS PubMed.
- K. Maeda, K. Ichinose, T. Yamazaki and T. Suzuki, Microporous Mesoporous Mater., 2008, 112, 603–611 CrossRef CAS PubMed.
- B. Platschek, N. Petkov and T. Bein, Angew. Chem., Int. Ed., 2006, 45, 1134–1138 CrossRef CAS PubMed.
- B. Platschek, N. Petkov, D. Himsl, S. Zimdars, Z. Li, R. Köhn and T. Bein, J. Am. Chem. Soc., 2008, 130, 17362–17371 CrossRef CAS PubMed.
- Y. Y. Wu, G. S. Cheng, K. Katsov, S. W. Sides, J. F. Wang, J. Tang, G. H. Fredrickson, M. Moskovits and G. D. Stucky, Nat. Mater., 2004, 3, 816–822 CrossRef CAS PubMed.
- A. Yamaguchi, H. Kaneda, W. S. Fu and N. Teramae, Adv. Mater., 2008, 20, 1034–1037 CrossRef CAS.
- B. Yao, D. Fleming, M. A. Morris and S. E. Lawrence, Chem. Mater., 2004, 16, 4851–4855 CrossRef CAS.
- A. Keilbach, M. Döblinger, R. Köhn, H. Amenitsch and T. Bein, Chem.–Eur. J., 2009, 15, 6645–6650 CrossRef CAS PubMed.
- Y. Li, A. Keilbach, M. Kienle, Y. Goto, S. Inagaki, P. Knochel and T. Bein, J. Mater. Chem., 2011, 21, 17338–17344 RSC.
- C. J. Brinker, Y. F. Lu, A. Sellinger and H. Y. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
- B. Platschek, R. Köhn, M. Döblinger and T. Bein, ChemPhysChem, 2008, 9, 2059–2067 CrossRef CAS PubMed.
- J. Schuster, A. Keilbach, R. Köhn, M. Döblinger, T. Dörfler, T. Dennenwaldt and T. Bein, Chem.–Eur. J., 2011, 17, 9463–9470 CrossRef CAS PubMed.
- Y. Q. Wang, Y. J. Wang, C. M. Yang, G. Z. Lu and F. Schuth, Langmuir, 2006, 22, 5491–5496 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |