
Polysulfide rejection layer from alpha-lipoic acid for high performance lithium–sulfur battery†
Jongchan
Song
a,
Hyungjun
Noh
a,
Hongkyung
Lee
a,
Je-Nam
Lee
a,
Dong Jin
Lee
a,
Yunju
Lee
b,
Chul Hwan
Kim
c,
Yong Min
Lee
*b,
Jung-Ki
Park
*a and
Hee-Tak
Kim
*a
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), 373-1, Guseong-dong, Yuseong-gu, Daejeon 305-701, Republic of Korea. E-mail: heetak.kim@kaist.ac.kr
bDepartment of Chemical and Biological Engineering, Hanbat National University, Deokmyoung-dong, Yuseong-gu, Daejeon 305-719, Republic of Korea
cOrange Power Ltd, #102, 187 Techno2-ro, Yuseong-gu, Daejeon 305-500, Republic of Korea
First published on 31st October 2014
Abstract
The polysulfide shuttle has been an impediment to the development of lithium–sulfur batteries with high capacity and cycling stability. Here, we report a new strategy to remedy the problem that uses alpha-lipoic acid (ALA) as an electrolyte additive to form a polysulfide rejection layer on the cathode surface via the electrochemical and chemical polymerization of ALA and a stable solid electrolyte interface (SEI) layer on the Li metal anode during the first discharge. The poly(ALA) layer formed in situ effectively prevents the polysulfide shuttle and consequently enhances the discharge capacity and cycling stability, owing to the Donnan potential developed between the polysulfide-concentrated cathode and the fixed negative charge-concentrated poly(ALA) layer. Also, the SEI layer additionally prevents the chemical reaction of the polysulfide and Li metal anode. The approach, based on the double effect, encompasses a new scientific strategy and provides a practical methodology for high performance lithium–sulfur batteries.
Introduction
The lithium–sulfur (Li–S) battery is considered a promising candidate to succeed lithium-ion batteries (LIBs) for portable energy storage and use. The high theoretical specific capacity of elemental sulfur (1675 mA h g−1) could result in a theoretical energy density for a cell of 2500 W h kg−1 when coupled with a Li anode, which is 3–5 times higher than those of state-of-the-art LIBs.1 Moreover, the non-toxic nature, low cost, and abundance of sulfur make Li–S batteries preferable to LIBs employing transition-metal-based cathode materials.Despite such promise, the commercialization of Li–S batteries has yet to succeed even after sustained effort spanning several decades. Significant problems encountered have included low sulfur utilization, short cycle life, low cycling efficiency, and high self-discharge rate. These are mainly attributed to a process known as the polysulfide (PS) shuttle;2 PS chains dissolved in the electrolyte diffuse to the Li anode where they directly react with the Li metal to produce lower order PS species, which diffuse back to the sulfur cathode to regenerate higher PS forms. The PS shuttle leads to incomplete charging of the sulfur electrode, corrosion of the Li electrode, and formation of electrochemically inactive lithium sulfates (LixSOy) on the sulfur electrode,3 thus resulting in poor battery performance. Prevention of the PS shuttle is therefore extremely important for the practical use of Li–S batteries.
Previous efforts to mitigate the PS shuttle were mainly focused on the following: (1) suppressing the diffusion of the dissolved PS out from the cathode by the geometrical trapping of PS in porous carbon or PS adsorption on oxides, conducting polymers,4 and by ion-selective polymers5 and (2) protecting the Li anode from reaction with PS by forming a passivation layer on the Li metal surface6 or a hybrid anode structure.7 Of the two approaches, the control of the electrolyte–electrode interface with an electrolyte additive has been demonstrated to be a simple and scalable method for LIBs.8 However, this approach has not been attempted for sulfur cathodes due to the lack of a proper electrolyte additive that could form a stable passivation layer prior to the electrochemical reduction of sulfur during the first discharge.
With an eye to identifying an electrolyte additive that could modulate the sulfur cathode interface via an in situ electrochemical reaction, similar to the additives in LIBs, we considered an antioxidant typically found in the human body. Sulfur-containing compounds have the vital function in our bodies of inhibiting oxidation in the cell.9 They capture oxygen radicals or terminate chain reactions acting as reducing agents through the redox reactions of thiol and disulfide groups. The similarity in the redox behavior between the sulfur-containing antioxidants and the sulfur in Li–S batteries motivated us to examine the possibility of controlling the sulfur cathode–electrolyte interface through their use. We considered that if a dithiol-containing antioxidant had a lower redox potential than any of the sulfur compounds in the Li–S battery, it would be oxidized to form a polymer film by disulfide bond formation during the first discharge prior to the electrochemical reduction of the sulfur cathode.
In an effort to exploit the properties of natural functional materials for technical applications, we discovered that alpha-lipoic acid (ALA), a unique antioxidant in the human body10 and commercially available in bulk quantities as a dietary supplement, forms a dense and uniform layer of poly(ALA) at the cathode–electrolyte interface under cathodic conditions prior to the electrochemical reduction of sulfur. Surprisingly, the in situ layers effectively mitigate the PS shuttle and enhance the discharge capacity and cycling stability of Li–S batteries. Owing to the Donnan potential difference between the sulfur cathode and the poly(ALA) layer imparted by the carboxylate anions attached to poly(ALA), the rejection of the PS anion at the sulfur cathode interface is realized. Moreover, the ALA also forms a stable SEI layer on the surface of the Li metal anode, which could additionally prevent the reaction between Li metal and PS. Since this approach requires only the addition of ALA to the electrolyte without any use of nanostructured carbon or coatings of additional layers, it is highly attractive in terms of performance, process simplicity, and cost. To the best of our knowledge, this is the first example of the in situ formation of a PS rejection layer on both sulfur cathode and Li metal anode surfaces with a single electrolyte additive.
Experimental section
Electrochemical measurements
All reagents used in the experiments were analytical grade and used as received. A 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 w/w Ketjen black carbon/sulfur (KB/S) composite was fabricated as the active material. The cathode comprised 80 wt% KB/S composite, 10 wt% vapor-grown carbon fiber (VGCF), and 10 wt% polyvinylpyrrolidone (PVP, Aldrich) on etched Al foil (thickness: 20 μm). The cathode was dried in a vacuum oven at 50 °C for 24 h before use. The typical mass loading of active sulfur was 1.2 mg cm−2. Li metal (Honjo Metal) was used as the anode. A solution of 1,3-1,3-dioxolane (DOL)/1,2-dimethoxyethane (DME) (50/50 v/v) containing 1 M lithium bis(trifluoromethane sulfonyl) imide (LiTFSI) (Panax Etec) was used as the reference electrolyte. To evaluate the electrochemical performance, CR2032 coin-type cells were fabricated by stacking a polypropylene (PP) separator (Celgard 2400) between the electrodes, followed by injection of the electrolyte. To measure the electrochemical impedance spectroscopy (EIS; Solartron 1255, frequency range from 10 mHz to 1 MHz) over the charge–discharge cycles, three-electrode cells were fabricated. Li metal was used as the reference and counter electrodes in the cell. All of the electrolyte preparation and cell assembly and disassembly processes were performed in a glove box filled with Ar (99.999%), with a dew point below −90 °C at room temperature. The unit cells were cycled between 1.5 and 2.8 V at a constant current density of 0.37 mA cm−2 (0.2 C rate based on the theoretical capacity of sulfur, 1675 mAh g−1) using a TOSCAT-3000U (Toyo System) at room temperature.
:80 w/w Ketjen black carbon/sulfur (KB/S) composite was fabricated as the active material. The cathode comprised 80 wt% KB/S composite, 10 wt% vapor-grown carbon fiber (VGCF), and 10 wt% polyvinylpyrrolidone (PVP, Aldrich) on etched Al foil (thickness: 20 μm). The cathode was dried in a vacuum oven at 50 °C for 24 h before use. The typical mass loading of active sulfur was 1.2 mg cm−2. Li metal (Honjo Metal) was used as the anode. A solution of 1,3-1,3-dioxolane (DOL)/1,2-dimethoxyethane (DME) (50/50 v/v) containing 1 M lithium bis(trifluoromethane sulfonyl) imide (LiTFSI) (Panax Etec) was used as the reference electrolyte. To evaluate the electrochemical performance, CR2032 coin-type cells were fabricated by stacking a polypropylene (PP) separator (Celgard 2400) between the electrodes, followed by injection of the electrolyte. To measure the electrochemical impedance spectroscopy (EIS; Solartron 1255, frequency range from 10 mHz to 1 MHz) over the charge–discharge cycles, three-electrode cells were fabricated. Li metal was used as the reference and counter electrodes in the cell. All of the electrolyte preparation and cell assembly and disassembly processes were performed in a glove box filled with Ar (99.999%), with a dew point below −90 °C at room temperature. The unit cells were cycled between 1.5 and 2.8 V at a constant current density of 0.37 mA cm−2 (0.2 C rate based on the theoretical capacity of sulfur, 1675 mAh g−1) using a TOSCAT-3000U (Toyo System) at room temperature.
Characterization
For the matrix-assisted laser desorption ionization (MALDI) mass spectroscopy and 1H nuclear magnetic resonance (NMR) analysis, Li/stainless steel (SUS) cells were fabricated with a 4 wt% ALA-containing electrolyte. After applying a galvanostatic cathodic current (0.1 mA cm−2) for 7 h into the cell from an open-circuit voltage (OCV) to 1.5 V, the SUS electrode was taken from the cell, washed with DME several times, and dried overnight. Then, the poly(ALA) layer was collected from the SUS electrode for the MALDI-mass spectroscopy and NMR analysis. MALDI-mass spectrometry (Autoflex III, Bruker) analysis was performed to clarify the structure of the poly(ALA) layer, using 2,5-dihydroxybenzoic acid (DHBA) as a matrix in the mass/charge (m/z) range of 500–6000. 1H-NMR (Agilent 400 MHz 54 mm NMR DD2) spectra were recorded in DMSO-d6 with tetramethylsilane (TMS) as an internal standard at RT. 1H NMR δ (ppm) 1.12–1.30 (m, SH–CH2–, 2H), 1.25–1.45 (m, –CH2–CH2–CH2–COOH, 2H), 1.42–1.62 (m, –CH2–CH2–CH2–CH2–COOH and –CH2–CH2–CH2–CH2–COOH, 4H), 1.65–1.81 (m, SH–CH2–CH2–, 1H), 2.05 (t, –CH2–COOH, 2H), 2.10 (t, SH–CH2–CH2–, 1H), 2.42–2.61 (m, SH–CH2–, 2H), 2.75–2.82 (m, SH–CH–, 1H). In order to measure the amount of PS ions in the cell, the electrolyte salt was replaced with LiClO4 before cycling. After cycling, cells were disassembled. The sulfur cathode, Li anode, separator, and the coin-cell casing were washed with DME (Aldrich), which was collected and diluted to 20 mL with additional DME. After oxidizing the diluted solution with concentrated HNO3 aqueous solution, the total sulfur content was measured by inductively coupled plasma-optical emission spectroscopy (ICP-OES; iCAP 6300 Duo). The resulting solutions were aspirated into aerosol mists which were conveyed in an argon gas stream through an inductively RF coupled region whereby a plasma was formed. Within the plasma, characteristic radiative emission for the analytes occurred. The resultant emitted radiation was directed through the optics of the spectrometer where it was dispersed via a grating into sulfur wavelengths of 1807 and 1820 nm. The intensity of the radiation was measured using a charge-coupled solid-state detector. The sample concentration values were calculated via the Perkin Elmer ICP WinLab software, using first order regression analysis calculations. The morphology and bonding characterization of the electrode surface was carried out via field emission-scanning electron microscopy (FE-SEM; Sirion, FEI) and X-ray photoelectron spectroscopy (XPS; Sigma Probe, Thermo VG Scientific). Before the analysis, each electrode was washed with DME several times in the glove box and dried in a vacuum chamber overnight. Then, samples were sealed in the laminated pouch bag and transferred into the SEM and XPS chambers. All XPS spectra were calibrated against the hydrocarbon peak at a binding energy of 285.0 eV and deconvoluted by a standard Shirley background function and Lorentzian–Gaussian curves using XPS-PEAK and Avantage softwares. To fabricate the poly(ALA) coated separator, PP was dipped into the ALA 10 wt% dissolved acetic acid solution for 2 h and dried in a convection oven for 30 min for solvent removal. Then, the dried separator was thermally treated at 80 °C for 3 h for polymerization of the ALA. The average thickness of the poly(ALA) layer was 6 μm.Results and discussion
A possible reaction route from ALA to poly(ALA) in a Li–S battery during the first discharge is illustrated in Fig. 1, based on previous studies on the electrochemical and chemical reaction of ALA.11 The ALA monomer contains a strained five-membered dithiolane ring and a terminal carboxylic acid. As the cathode potential decreases, the terminal carboxylic acid is first electrochemically reduced to form H2 and the carboxylate anion (reaction 1). H2 is known to readily react with ALA to form dihydroxylipoic acid (DHLA).11 Concurrently, the breakage of the passivation layer originally formed on the Li metal anode and the subsequent chemical reaction between ALA and Li metal result in the formation of Li-ALA thiolate (reaction 2). The Li-ALA thiolate diffuses across the electrolyte phase and, at the sulfur cathode, is electrochemically re-oxidized, regenerating ALA (reaction 3). The ALA dissolved in the electrolyte is chemically polymerized via ring opening due to a ring strain in the presence of thiolate which acts as an initiator for the ring opening polymerization of ALA (reaction 4).12 The resulting poly(ALA) is then precipitated on the surface of the sulfur cathode, forming a layer. The poly(ALA) precipitate was also found in the outside of the cell assembly where the excess liquid electrolyte stagnates, which may support the chemical polymerization. It also could be deposited on the Li metal surface, however, the Li anode would chemically decompose poly(ALA) to Li-ALA thiolate or form a new SEI layer.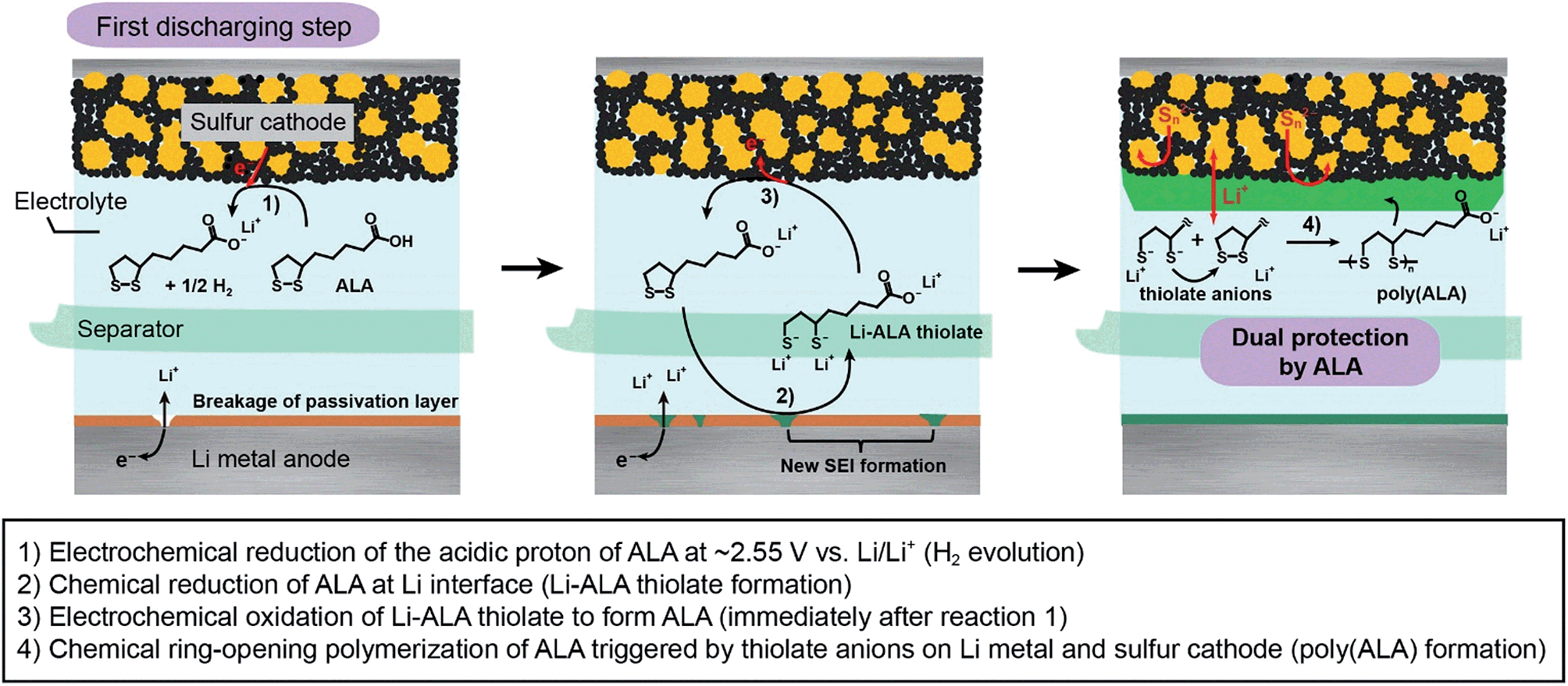 | ||
| Fig. 1 Reaction scheme for the formation of the poly(ALA) layer on the sulfur cathode and of the SEI layer on the Li metal anode during the first discharge of a Li–S battery. | ||
The electrochemical reaction of ALA in the coin-type Li–S cell was monitored by cyclic voltammetry (CV). As a reference electrolyte, 1 M LiTFSI in DOL/DME (50/50 v/v) was used. The two reduction and two oxidation peaks for the reference cell (Fig. 2a) are representative of a typical electrochemical reaction of sulfur in an aprotic electrolyte. When ALA was added to the reference electrolyte at 4 wt%, we discovered an additional cathodic peak at 2.5–2.7 V and a subsequent relatively large anodic peak during the cathodic scan (inset of Fig. 2a). The peaks originated from ALA because they did not appear in the CV curve for the reference electrolyte and the peak intensity increases as the concentration of ALA increases (Fig. S1.1†), suggesting that these peaks are associated with ALA. Taking the previously suggested mechanism11 for the ALA reaction into account, the cathodic peaks would correspond to the reduction of the terminal carboxylic acid of ALA (2H+ + 2e− → H2), which is the cathodic peak at the same potential for acetic acid (Fig. S1.2†). The sharp transition from reduction to oxidation may indicate that the passivation layer originally formed on the Li metal anode is broken and ALA starts to chemically react with the newly exposed Li. The Li-ALA thiolate generated by the chemical reaction of ALA and the Li metal anode is again electrochemically oxidized at the sulfur cathode (reaction 2), generating the large anodic peak. At high scan rates (≥0.5 mV s−1) the anodic peak became unapparent (Fig. S1.3†), supporting that the peak is associated with the chemical reaction of ALA and the Li metal anode. At the second scan, the characteristic cathodic and anodic peak disappeared (Fig. S1.4†), which indicates the complete consumption of ALA in the poly(ALA) formation.
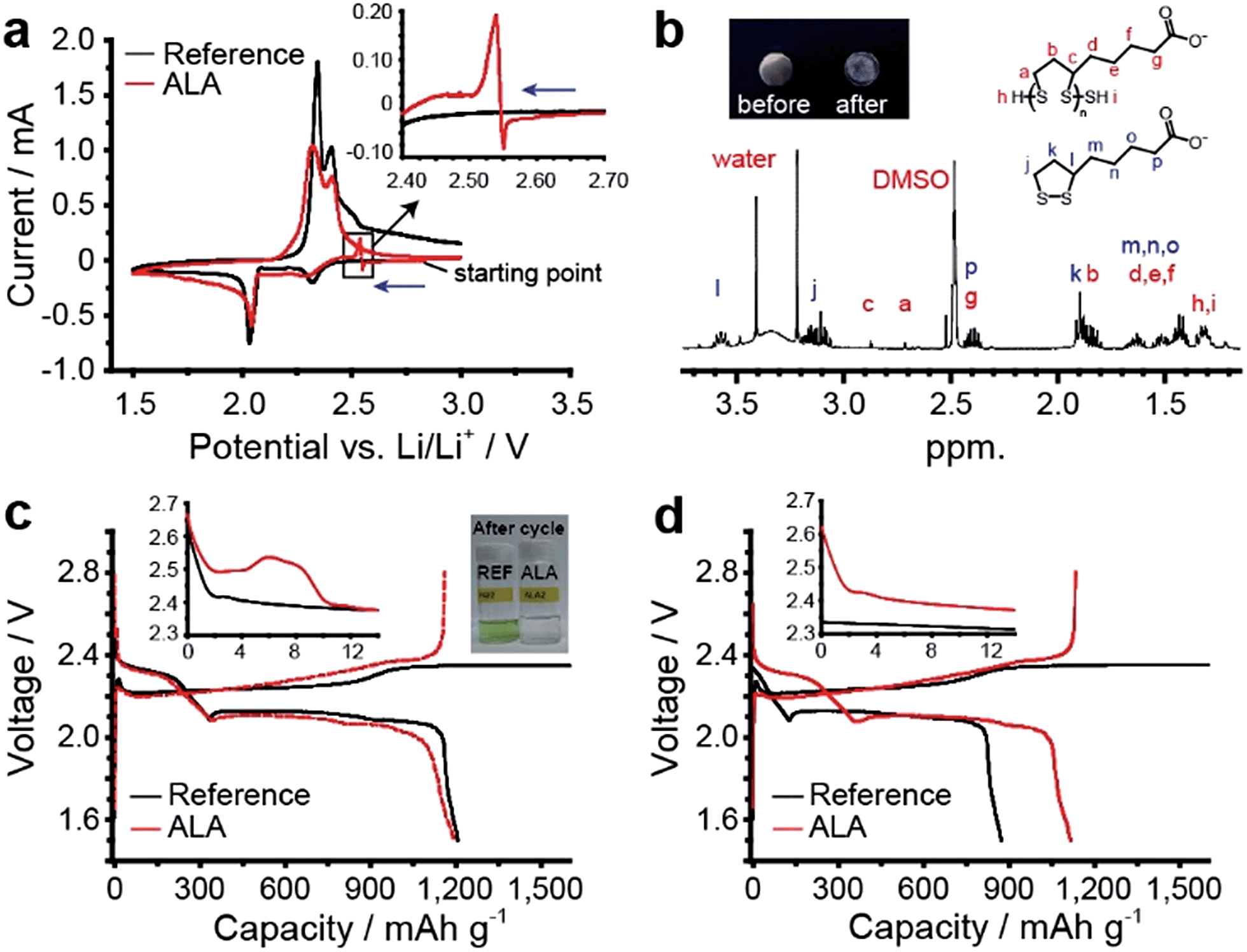 | ||
| Fig. 2 Structural and electrochemical characterization of poly(ALA) layer formation (scan rate: 0.05 mV s−1). (a) Comparison of the cyclic voltammetry curves for the Li–S cells with 4 wt% and without ALA in the electrolyte. The arrows indicate the scan direction. (b) 1H NMR spectrum of the layer formed on a SUS electrode with the 4 wt% ALA-containing electrolyte after the cathodic scan. Discharge–charge curves of Li–S cells with 4 wt% and without ALA in the electrolytes for (c) the first and (d) the second cycle at 167.5 mA g−1 (0.1 C). | ||
To confirm the formation of poly(ALA), a cathodic scan from an open-circuit voltage (ca. 2.9 V) to 1.5 V was conducted on a SUS working electrode in the 4 wt% ALA-containing electrolyte. A polymeric layer was formed on the SUS electrode, as shown in the inset of Fig. 2b. In the 1H NMR spectrum of the layer (Fig. 2b), characteristic peaks from ALA and poly(ALA) were identified. According to the MALDI mass spectroscopy results, the molecular weight (Mw) of the layer was ∼1400 and the degree of polymerization was about 7 (Fig. S2.1†). Therefore, it can be concluded that poly(ALA) was produced on the SUS electrode. In fact, a poly(ALA) layer is also formed on the sulfur cathode which is presented in the following section. For easier sampling, the SUS electrode was alternatively used for characterization of the poly(ALA) layer.
The effect of ALA on the discharge and charge processes of the Li–S battery was investigated. The discharge and subsequent charge curves of a Li–S cell between the reference and the 4 wt% ALA-containing electrolyte were compared for the first (Fig. 2c) and second cycles (Fig. 2d). The reduction/oxidation of ALA/Li-ALA thiolate was detected in the first discharge curve in the form of a slight potential increase at 2.55 V (inset of Fig. 2c), but not in the second discharge curve (inset of Fig. 2d), indicating that these reactions are nearly completed during the first discharge. The discharge curve of the reference cell was characterized by a higher voltage plateau (2.3–2.4 V) corresponding to the reduction of octet sulfur (S8) to soluble PS and a lower voltage plateau (2.0–2.1 V) corresponding to the reduction of PS to Li2S2 or Li2S.13 In addition, the reference cell exhibited a limiting voltage around 2.35 V during charge and a disappearance of the higher voltage discharge plateau in the subsequent discharge, indicating that oxidation of PS to elemental sulfur did not occur because of the PS shuttle. By contrast, the cell with the 4 wt% ALA-containing electrolyte (ALA cell) showed an abrupt rise in the charging voltage at the end of the first charge and a higher voltage plateau in the following discharge, which evidences the formation of elemental sulfur during charge and the prevention of the PS shuttle compared to the reference cell.
As direct evidence for the prevention of the PS shuttle with the introduction of ALA to the electrolyte, we collected the PS in the Li–S cell after the first discharge, the first charge, and the 30th charge by washing the disassembled components and coin-cell casing with a predetermined amount of DME as described in the Experimental section, and monitored the colors of the diluted electrolyte samples. For the reference cells after the first and 30th charges, the color of the diluted electrolyte was green as shown in the inset of Fig. 2c and S3.1;† this is representative of PS dissolution into the electrolyte from the sulfur cathode. Surprisingly, for the ALA-containing electrolyte, no apparent color changes were observed after the first or even after the 30th charge, which was indisputable evidence for the prevention of the PS shuttle. The amount of PS ions in the cell was quantitatively determined by ICP-OES. For the ALA cell, the amounts of PS ions after 30 cycles were significantly lower than those for the reference electrolytes; after the 30th charge, 10.4 wt% of the total amount of sulfur was existing in the form of PS in the cell, which was contrasted by 47.1 wt% for the reference cell. Therefore, there is no doubt that the introduction of ALA to the Li–S battery effectively confined PS ions within the cathode and prevented their passage to the bulk electrolyte, resulting in a higher conversion to solid sulfur during charging. A comparison of scanning electron microscopy (SEM) images of the sulfur cathodes in the reference and ALA cells (Fig. 3) provides convincing evidence for the formation of the poly(ALA) layer. The pristine sulfur cathode was highly porous (Fig. 3a). In the reference cell, solid discharge products were formed in the carbon matrix of the electrode (Fig. 3b). On the other hand, for the ALA cell, a smooth surface layer was observed after the first charge (Fig. 3c), which was preserved even after the 50th charge (Fig. 3d). However, little cracks were found on the ALA layer after the 50th charge, which would be probably generated by a high mechanical stress exerted on the sulfur cathode of high sulfur loading (1.2 mg cm−2), during the volume expansion (S8 → 8Li2S) and contraction (8Li2S → S8). It should be noted here that the layer was not formed inside the sulfur cathode, but was only concentrated at the interface of the sulfur cathode and the electrolyte, as seen in the cross-sectional images (Fig. S3.2†). Since the electrochemical reduction (depolymerization) of poly(ALA) can occur at around 1.7 V in the cathode as indicated by the CV analysis for an ALA-containing electrolyte with a Li/SUS cell (Fig. S3.3†), poly(ALA) mostly remains at the interface of the bulk electrolyte phase and the top surface of the cathode in which the electron conducting pathway is very limited. We speculate that the Li2S layer formed on the sulfur cathode separates the conducting surface and poly(ALA), preventing electrochemical depolymerization of poly(ALA).
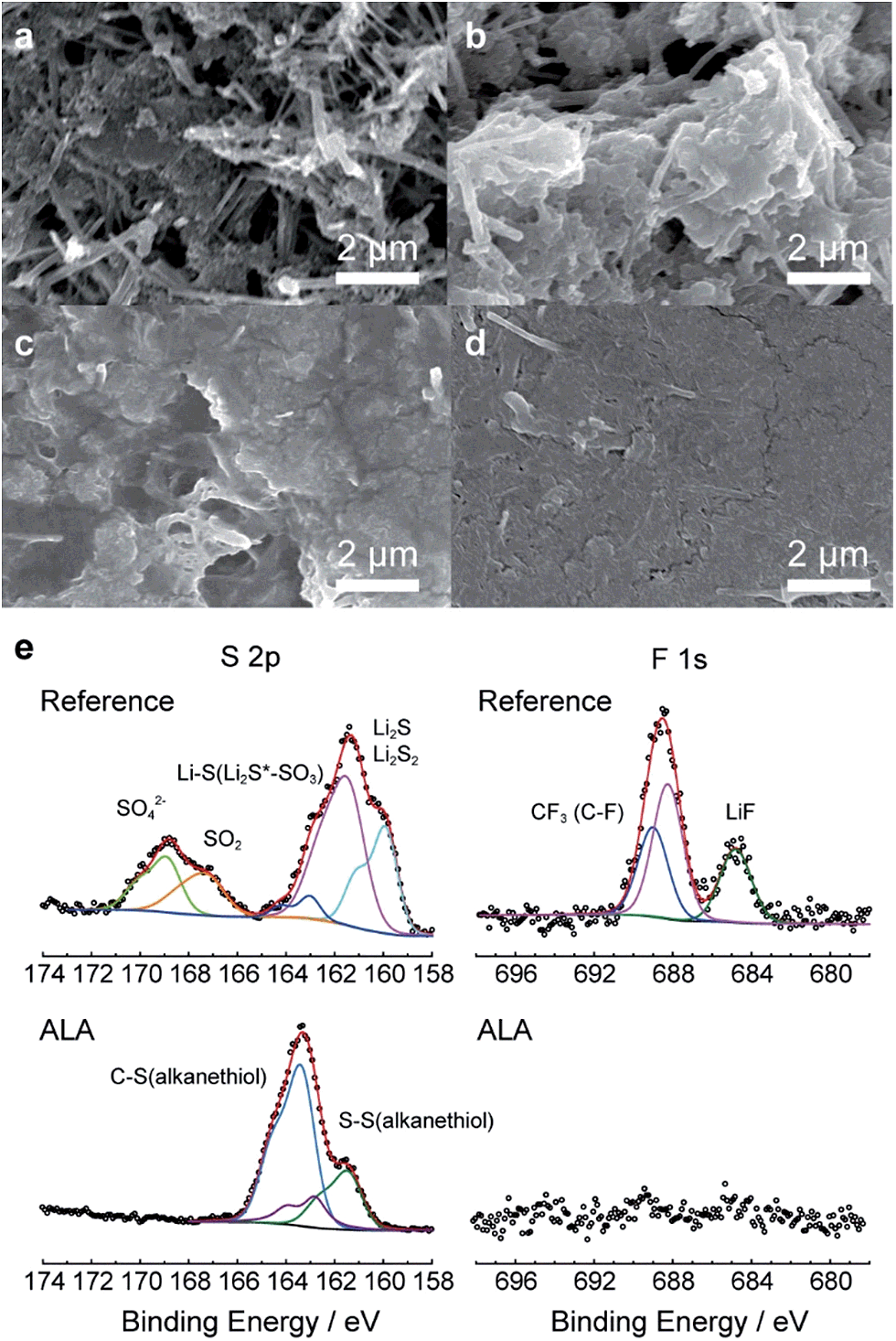 | ||
| Fig. 3 Morphology and surface characterization of the sulfur cathode. (a) SEM images of the pristine sulfur cathode before cycling. SEM images of the sulfur cathode for (b) the reference cell after the first charge and the ALA cell after the (c) first and (d) 50th charges. (e) XPS spectra of the cathode surfaces for the reference cell and the ALA cell after the 20th charge. | ||
Fig. 3e compares the S2p and F1s spectra of the cathode surfaces in both reference and ALA cells after the 20th charge, obtained from XPS. The S2p spectra exhibited a pronounced difference. The reference cell spectrum included peaks from LixSOy at 167.2–168.9 eV3 and Li2S at 159.9–161.4 eV,5,14 although they were not observed in the spectrum for the ALA cell cathode surface. The S2p spectrum of the ALA cathode surface demonstrated the presence of C–S and S–S bonds (161.415 and 163.3 eV16, respectively) which represented unreacted ALA or poly(ALA). Furthermore, peaks in the F1s spectrum that would typically result from the decomposition of LiTFSI did not appear in the spectrum for the ALA cell layer.17 Therefore, the XPS results indicate that poly(ALA) was responsible for the layer and it prevented both the dissolution of PS ions and electrolyte decomposition reactions.
One of the interesting phenomena observed with the introduction of ALA to the Li–S cell is the strong adsorption of ALA on the carbon or metal surface. ALA is known to form self-assembled monolayers and hinders ionic conduction and charge transfer reaction at the electrode interface.18 When ALA is introduced to the as-prepared Li–S cell, it adsorbs on both the sulfur cathode and the Li metal anode, generating large resistance for both electrodes (Fig. S4.1†). When the sulfur cathode was immersed in 2 wt% ALA-containing electrolyte for 12 h prior to cell assembly, the cell exhibited much larger resistance even in the absence of ALA in the bulk electrolyte phase (Fig. S4.2†), which indicates that the adsorption of ALA is responsible for the large resistance. We found that the anode SEI layer formed by ALA is highly effective in improving the anode performance. The large anode impedance prior to cycling for the ALA cell demonstrates passivation of the Li metal anode by ALA. As the cycling proceeded, the anode resistance of the ALA cell significantly decreased and became even lower than that of the reference (Fig. S4.1†). After the 50th cycle, the anode impedance of the ALA cell was invariant, suggesting the formation of a highly stable SEI layer for the ALA cell.
To further investigate the Li metal anode stabilization effect of ALA, the Li/Li symmetric cells with the reference electrolyte and the 4 wt% ALA-containing electrolyte were fabricated and cycled at 0.37 mA cm−2 (equivalent to 0.2 C of the Li–S cell) with a capacity cut of 0.74 mA h cm−2. The ALA cell showed a relatively large overvoltage during the initial few cycles; however, it continuously decreased and stabilized with cycling (Fig. 4a). After 270 h (67 cycles), the reference cell failed due to short circuiting, whereas, the ALA cell exhibited stable cycling more than 400 h (100 cycles), strongly supporting the stabilizing effect of ALA. On the other hand, the two cells operated for 20 cycles were disassembled and the surfaces of the Li metal anodes were analysed by SEM. For the reference cell, a thick and porous Li layer was observed (Fig. 4b). By contrast, the ALA cell surprisingly showed a highly uniform surface without any dendrite formation as shown in Fig. 4c. It indicates that the SEI layer formed by ALA is highly stable and uniform, effectively preventing dendritic growth and chemical reaction with the electrolyte.
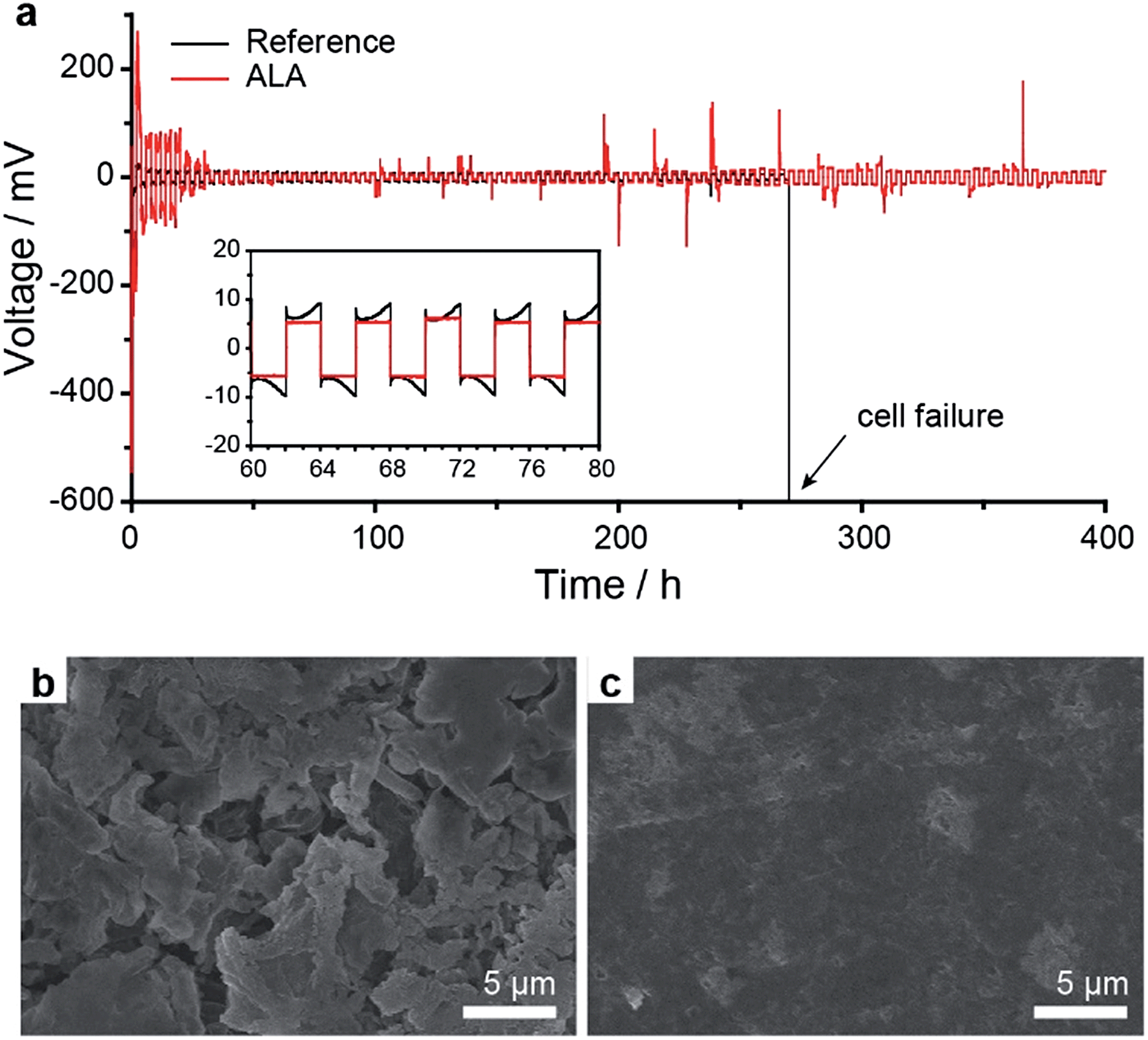 | ||
| Fig. 4 Li metal stabilization effect of ALA. (a) Galvanostatic cycling for the Li/Li symmetric cells with 4 wt% and without ALA in the electrolyte measured at a current density of 0.37 mA cm−2 with a capacity cut of 0.74 mA h cm−2. The SEM image of the surface of the Li metal anode after 20 cycles (after Li plating) for (b) the reference and (c) the 4 wt% ALA-containing electrolyte. | ||
From the evolution of the charge and discharge curves with cycling for the reference cell (Fig. 5a) and the cell employing ALA (Fig. 5b), it is notable that, with the addition of ALA, the discharge capacity from the higher voltage plateau (Qhigh) is nearly invariant with cycle, in contrast to the profound reduction in Qhigh after the 1st cycle for the reference cell. Moreover, the discharge capacity from the lower voltage plateau (Qlow) was more stably maintained for the ALA cell with cycling. As a result, the ALA cell revealed significantly enhanced discharge capacity and cycling stability compared to the reference cell as seen in Fig. 4c. In detail, the discharge capacity at the 3rd cycle was 995 mA h g−1 for the ALA cell and 829 mA h g−1 for the reference cell. At the 200th cycle, the ALA cell delivered a reversible capacity of 787 mA h g−1. In contrast, the discharge capacity of the reference cell abruptly dropped to 701 mA h g−1 within 25 cycles and then gradually decreased to 575 mA h g−1 after 200 cycles, which was 73% of the discharge capacity of the ALA cell. As seen in Fig. 5d, the capacity retention for Qhigh was higher than 80% over 200 cycles, thus indicating that the PS shuttle prevention by the poly(ALA) layer is preserved under the prolonged cycling. The higher retention of Qlow for the ALA cell (Fig. 5d) may be attributed to the confinement of a larger amount of PS within the cathode during discharge. The coulombic efficiency of the first cycle was a very high value of 97.0% for the ALA cell, as compared to 72.3% for the reference cell. During the first few cycles, the coulombic efficiency of the ALA cell decreased and reached a stable but inferior value of 90% probably due to the little crack of the poly(ALA) layer that occurred in the initial cycling as shown in Fig. 3d. Nevertheless, the majority of PS should be confined within the cathode as evidenced by the high retention of Qhigh (>80%). Next, the rate capability of the Li–S cells with 4 wt% and without ALA in the electrolyte was evaluated (Fig. 5e and f). For the reference cell, after an initial capacity of 777 mA h g−1 at 0.2 C, the reversible capacities of 616, 553, and 477 mA h g−1 were found at 0.5, 1, and 2 C, respectively. In contrast, the ALA cell showed higher capacities than the reference cell; 1005, 923, 820, and 654 mA h g−1 at 0.2, 0.5, 1, and 2 C, respectively. Moreover, after the C-rate was recovered from 2 to 0.2 C for the ALA cell again, the original capacity was mostly recovered (Fig. 5e), representing the stable confinement of PS ions within the poly(ALA) layer. However, polarization was found in the voltage–capacity curve at 2 C for the ALA cell (Fig. 5f), which may be ascribed to the highly viscous electrolyte in the cathode due to the large PS confinement in the poly(ALA) layer.
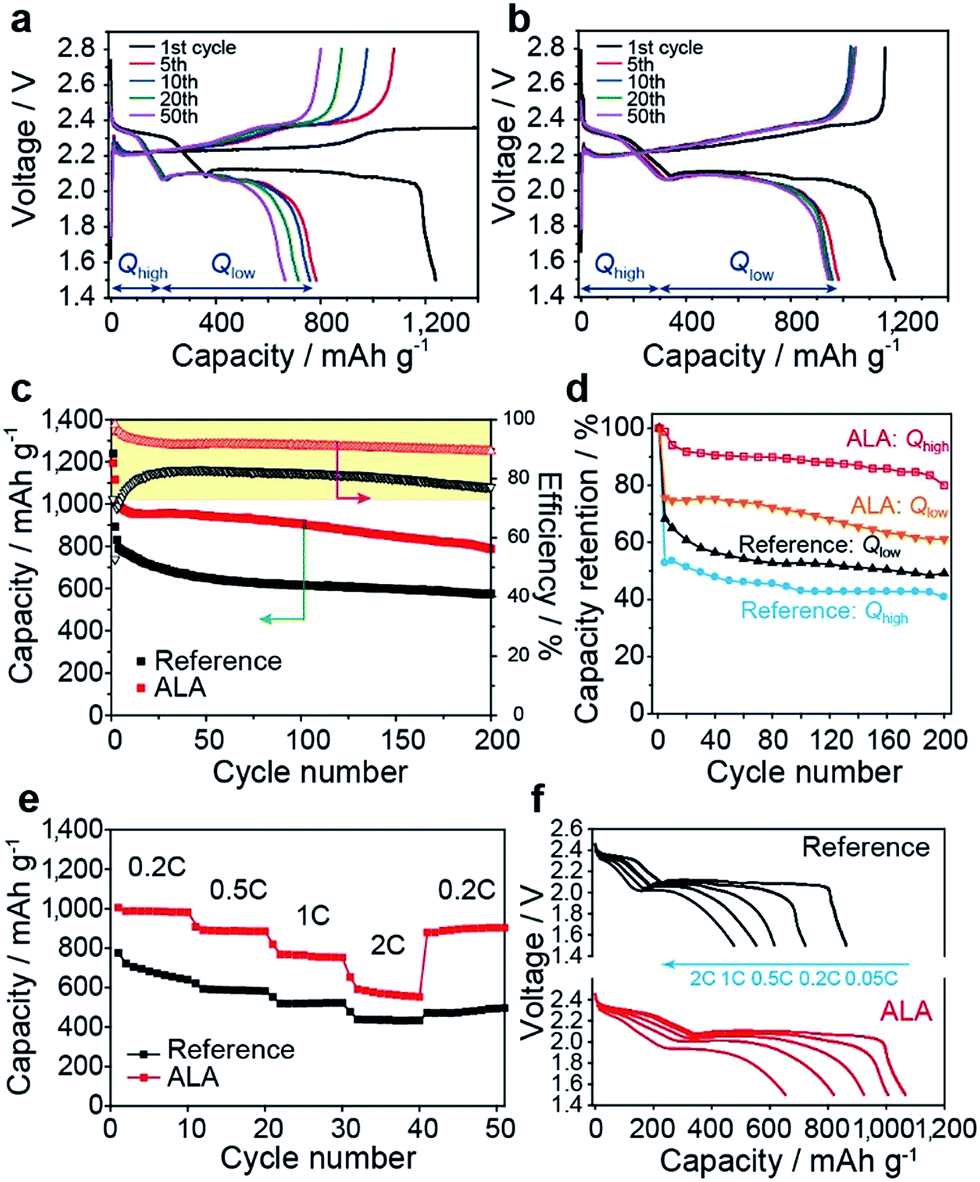 | ||
| Fig. 5 Electrochemical performance of Li–S cells with ALA. Discharge–charge curves of Li–S cells with (a) the reference electrolyte and (b) with the 4 wt% ALA-containing electrolyte at different cycle numbers when measured at a rate of 0.2 C for both charge and discharge except for the 1st cycle (0.05 C). (c) Cycling stability and cycling efficiency. (d) Comparison of the capacity retention from the high and low plateaus at 0.2 C between the reference and the 4 wt% ALA-containing cells. (e) Rate capability and (f) typical voltage–capacity profiles at various current densities. | ||
The PS rejection by the poly(ALA) layer can be explained in terms of Donnan exclusion which arises from the inability of certain ions to diffuse from one phase to another. As theoretically described in ESI S5 and Fig. S5.1,† the non-diffusible fixed carboxylate anions in poly(ALA) generate an electric potential difference, known as the Donnan potential difference, between the PS-containing cathode electrolyte phase and the poly(ALA) phase. As a result of electrochemical equilibrium between the two phases, the distributions of the PS anion, Li+ and TFSI− in two phases are determined. Owing to the lower electrical potential from the fixed carboxylate anion in the poly(ALA) phase, the concentrations of the negatively charged mobile anions (PS and TFSI−) in the poly(ALA) phase should be lower than those in the cathode electrolyte phase. Therefore, the diffusion of PS toward the bulk electrolyte from the cathode could be inhibited. Since the PS rejection is not based on geometrical trapping but rather on thermodynamic equilibrium, the low PS concentration in the poly(ALA) layer could be maintained under prolonged operation, which is demonstrated by the invariant coulombic efficiency during repeated cycles (Fig. 5c). In sharp contrast to geometric trapping by a brittle material, the flexible nature of the polymeric PS rejection layer could provide a high degree of reliability in manufacturing and operation. Here it should be noted that the PS rejection effect by the poly(ALA) layer is theoretically consistent with that by a nafion layer; according to the previous pacesetting reports,5 discharge capacity and coulombic efficiency were significantly improved with the introduction of a nafion layer on the separator or cathode surface. We believe that these behaviors can be restated in terms of Donnan exclusion; the dissociated sulfonate anion attached to the main chain of nafion generates Donnan potential and rejects PS with prevention of the PS shuttle. Compared to the nafion coating method, the in situ formation of the PS rejection layer could be advantageous, because it does not require any coatings of additional layers and expensive functional polymers such as nafion.
While rejecting PS anions, the poly(ALA) layer readily allows Li ion permeation. The ion conduction resistance through the poly(ALA) layer in the Li–S cell was quantified by the difference in ohmic resistance between the 4 wt% ALA and reference cells, determined from the impedance at the 10th and later cycles where the influence of the ALA adsorption disappeared. A small increase (7.1%) in ohmic resistance was observed with the formation of the poly(ALA) layer, implying that the layer does not significantly retard Li conduction. This is supported by the high ion conductivity of an electrolyte-swollen poly(ALA) layer coated on a porous separator measured from a symmetric cell of two blocking SUS electrodes with a poly(ALA) coated-separator (Fig. S6†). Taking the 6 μm thickness of the poly(ALA) coating on the separator into account, the ionic conductivity of the swollen poly(ALA) is 2.09 × 10−4 S cm−1, which is comparable to that of the liquid electrolyte used in this work (2.28 × 10−4 S cm−1). This result is reasonable because the poly(ALA) absorbs a large amount of electrolyte, exhibiting a weight ratio of the electrolyte to the poly(ALA) layer of 146% (Fig. S6†).
Considering the practical implications of this approach, it is worth discussing the hydrogen generation that occurs during the electrochemical reaction of ALA, which would potentially result in the swelling of the battery if all the hydrogen is not consumed by forming in the subsequent chemical reactions (H2 + ALA → DHLA). Residual hydrogen can be removed by the degassing and re-sealing processes which are currently employed in LIB manufacturing after an appropriate pre-cycling for fully consuming ALA. Considering that the hydrogen evolution as a result of SEI layer formation happens also in LIBs during pre-conditioning cycles,19 hydrogen evolution for ALA-containing Li–S batteries should not be a serious hurdle for practical use.
Conclusions
In summary, when the new electrolyte additive, ALA, was introduced to a Li–S battery, it formed a poly(ALA) layer on the interface of the cathode and electrolyte through electrochemical and chemical reactions and a SEI layer on the Li metal surface. Owing to the combined effects of the two layers, the prevention of the PS shuttle and improvements in discharge capacity and cycling stability resulted. The prevention of the PS shuttle by ALA can be regarded as an effective approach to enhancing the performance of Li–S batteries based on its impact on performance, processing simplicity, and cost-effectiveness.Acknowledgements
This research was financially supported by a National Research Foundation of Korea Grant funded by the Korean Government (MEST) (NRF-2009-0094219).Notes and references
- S. S. Zhang, J. Power Sources, 2013, 231, 153 CrossRef CAS PubMed; B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691 CrossRef; B. S. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287 Search PubMed.
- B. M. L. Rao and J. A. Shropshire, J. Electrochem. Soc., 1981, 128, 942 CrossRef CAS PubMed; Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969 CrossRef PubMed.
- Y. Diao, K. Xie, S. Xiong and X. Hong, J. Power Sources, 2013, 235, 181 CrossRef CAS PubMed; Y. Diao, K. Xie, S. Xiong and X. Hong, J. Electrochem. Soc., 2012, 159, A1816 CrossRef PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500 CrossRef CAS PubMed; C. Liang, N. J. Dudney and J. Y. Howe, Chem. Mater., 2009, 21, 4724 CrossRef; N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 50, 5904 CrossRef PubMed; G. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462 CrossRef PubMed; J. Wang, et al. , Electrochim. Acta, 2006, 51, 4634 CrossRef PubMed; J. Guo, Y. Xu and C. Wang, Nano Lett., 2011, 11, 4288 CrossRef PubMed; Y.-S. Su and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef PubMed; S. Moon, Y. H. Jung, W. K. Jung, D. S. Jung, J. W. Choi and D. K. Kim, Adv. Mater., 2013, 25, 6547 CrossRef PubMed; Z. W. She, W. Li, J. J. Cha, G. Zheng, Y. Yang, M. T. Mcdowell, P.-C. Hsu and Y. Cui, Nat. Commun., 2013, 4, 1331 CrossRef PubMed.
- J.-Q. Huang, Q. Zhang, H.-J. Peng, X.-Y. Liu, W.-Z. Qian and F. Wei, Energy Environ. Sci., 2014, 7, 347 Search PubMed; Q. Tang, Z. Shan, L. Wang, X. Qin, K. Zhu, J. Tian and X. Liu, J. Power Sources, 2014, 246, 253 CrossRef CAS PubMed; J. Song, M.-J. Choo, H. Noh, J.-K. Park and H.-T. Kim, ChemSusChem, 2014 DOI:10.1002/cssc.201402789.
- D. Aurbach, et al. , J. Electrochem. Soc., 2009, 156, A694 CrossRef CAS PubMed; S. S. Zhang, Electrochim. Acta, 2012, 70, 344 CrossRef PubMed; Z. Lin, Z. Liu, W. Fu, N. J. Dudney and C. Liang, Adv. Funct. Mater., 2013, 23, 1064 CrossRef; J.-H. Song, et al. , J. Electrochem. Soc., 2013, 160, A873 CrossRef PubMed.
- C. Huang, et al. , Nat. Commun., 2014, 5, 3015 Search PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303 CrossRef CAS; J. B. Goodenough and Y. Kim, Chem. Mater., 2009, 22, 587 CrossRef.
- S. Parcell, Altern. Med. Rev., 2002, 7, 22 Search PubMed; L. A. Komarnisky, R. J. Christopherson and T. K. Basu, Nutrition, 2003, 19, 54 CrossRef CAS.
- L. Parker, E. H. Witt and H. J. Tritschler, Free Radicals Biol. Med., 1995, 19, 227 CrossRef CAS; J. L. Evans, I. D. Goldfine, B. A. Maddux and G. M. Grodsky, Endocr. Rev., 2002, 23, 599 CrossRef PubMed; H. Moini, L. Packer and N.-E. Saris, Toxicol. Appl. Pharmacol., 2002, 182, 84 CrossRef.
- J. K. Howie, J. J. Houts and D. T. Sawyer, J. Am. Chem. Soc., 1977, 99, 6323 CrossRef CAS; S. M. Bachrach, J. T. Woody and D. C. Mulhearn, J. Org. Chem., 2002, 67, 8983 CrossRef PubMed.
- A. Kisanuki, et al. , J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5247 CrossRef CAS; R. Singh and G. M. Whitesides, J. Am. Chem. Soc., 1990, 112, 6304 CrossRef; S. M. Bachrach, J. T. Woody and D. C. Mulhearn, J. Org. Chem., 2002, 67, 8983 CrossRef PubMed.
- X. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821 RSC; H. Yamin and E. Peled, J. Power Sources, 1983, 9, 281 CrossRef CAS; K. Kumaresan, Y. Mikhaylik and R. E. White, J. Electrochem. Soc., 2008, 155, A576 CrossRef PubMed.
- H. Ota, Y. Sakata, X. Wang, J. Sasahara and E. Yasukawa, J. Electrochem. Soc., 2004, 151, A437 CrossRef CAS PubMed.
- N. S. Figoli and P. C. L'Argentiere, Catal. Lett., 1996, 38, 171 CrossRef CAS; K. V. Wollf, D. A. Cole and S. L. Bernasek, Langmuir, 2001, 17, 8254 CrossRef.
- T. M. Willey, et al. , Langmuir, 2004, 20, 4939 CrossRef CAS.
- J. G. Langan, J. A. Shorter, X. Xin, S. A. Joyce and J. I. Steinfeld, Surf. Sci., 1989, 207, 344 CrossRef CAS; M. Herstedt, M. Stjerndahl, T. Gustafsson and K. Edstrom, Electrochem. Commun., 2003, 5, 467 CrossRef; J. Mun, et al. , J. Power Sources, 2009, 194, 1068 CrossRef PubMed.
- Q. Cheng and A. B. Toth, Anal. Chem., 1992, 64, 1998 CrossRef CAS; S.-J. Ding, B.-W. Chang, C.-C. Wu, M.-F. Lai and H.-C. Chang, Anal. Chim. Acta, 2005, 554, 43 CrossRef PubMed; O. Corduneanu, A. M. C. Paquim, M. Garnett and A. M. O. Brett, Talanta, 2009, 77, 1843 CrossRef PubMed.
- M. Onuki, S. Kinoshita, Y. Sakata, M. Yanagidate, Y. Otake, M. Ue and M. Deguchi, J. Electrochem. Soc., 2008, 155, A794 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta03625e |
| This journal is © The Royal Society of Chemistry 2015 |