A novel CoN electrocatalyst with high activity and stability toward oxygen reduction reaction†
Li
An
a,
Weifeng
Huang
b,
Nanlin
Zhang
a,
Xin
Chen
c and
Dingguo
Xia
*a
aCollege of Engineering, Peking University, Beijing 100871, P. R. China. E-mail: dgxia@pku.edu.cn
bNational Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei Anhui, 230029, P. R. China
cCollege of Environmental and Energy Engineering, Beijing University of Technology, Beijing 100124, P. R. China
First published on 25th October 2013
Abstract
Carbon-supported CoN (CoN/C) nanoparticles have been synthesized by heating at reflux in the solution of o-xylene and subsequent thermal annealing under a NH3 reducing atmosphere. The as-prepared CoN/C composite exhibited high oxygen reduction reaction (ORR) activity and excellent stability as a new efficient non-precious metal electrocatalyst.
Highly efficient electrocatalysts of oxygen reduction reaction (ORR) play a crucial role in fuel cells (FCs) and metal–air batteries owing to their sluggish kinetics.1 Although platinum and its alloys exhibit good electrochemical properties as ORR catalysts, their high cost, scarce supply, and finite durability severely hinder their applicability to broad commercialization.2 Hence, it is highly desirable to explore alternative non-precious metal catalysts with comparable ORR activity and durability to Pt-based catalysts.
In previous studies, metal chalcogenides (Co1−xS, CoSe2, CoTe, etc.), transition metal oxides (MnO, Fe3O4, CoxMn3−xO4, etc.), transition metal nitrides and non-platinum alloys have been explored as non-precious metal electrocatalysts toward ORR.3–5 Considerable improvements in catalytic performance have been achieved. However, the high activity and long-term stability of catalysts, due to the metal dissolution, particle growth and corrosion of carbon at high potential, remain a major challenge.6
In the present work, for the first time, we reported a new type of electrocatalyst for ORR, carbon-supported CoN (CoN/C) nanoparticles, showing high electrocatalytic activity, absolute tolerance to methanol and superior long-term stability. The CoN/C catalyst was prepared by a facile two-step method. Firstly, the o-xylene solution of cobalt acetylacetonate (Co(acac)2) was heated by refluxing at 140 °C for 4.5 hours. After being separated from the solution and dried at 80 °C for 10 h in a vacuum oven, the resultant precursor powders were heat treated at 450–650 °C for 3 hours under a flowing NH3 atmosphere.
The X-ray diffraction (XRD) patterns in Fig. 1a confirm that the precursor can be assigned to pure metal Co (JCPDS, no. 65-9722), while the annealed products at 550 °C can be assigned to the crystalline of CoN species (JCPDS, no. 16-0116) (Fig. 1b). The diffraction peaks centered at about 25° are all attributed to the (002) planes of graphitic carbon. When the annealing temperature is up to 650 °C, a pure metal Co phase occurred, which is correlated with reduction of CoN (Fig. S1, ESI†).
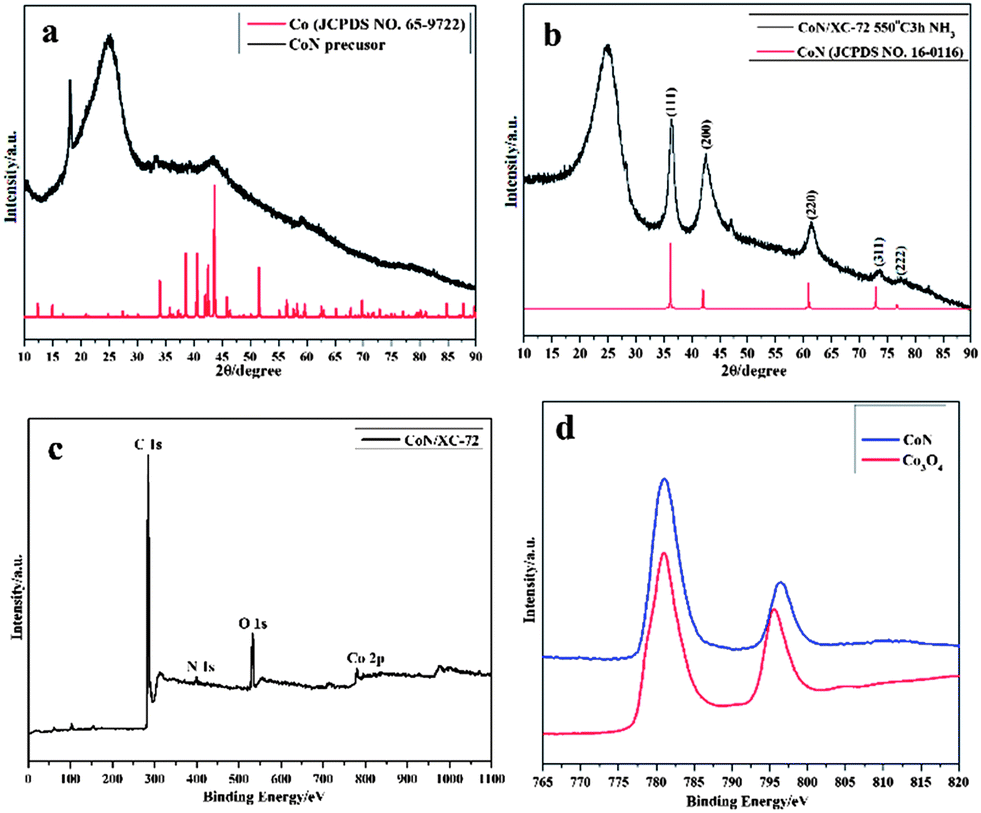 | ||
| Fig. 1 Powder XRD patterns of (a) as-prepared supported CoN precursor, (b) CoN compound (JCPDS no. 16-0116) annealed at 550 °C under a NH3 atmosphere, (c) XPS spectrum of the CoN/C composite and (d) the deconvolution of the Co XPS spectra of the CoN/C composite and Co3O4. | ||
To determine the chemical composition of the CoN/C composite, X-ray photoelectron spectroscopy (XPS) was performed to analyze the composition of materials. As displayed in Fig. 1c, the wide survey spectra reveal the presence of C, Co, N, and O in the CoN composite. The weight of CoN in the composite by using thermogravimetry/differentiate thermogravimetry (TG/DTG) analyses (Fig. S2, ESI†) was estimated to be 13 wt%, which is consistent with the value determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES). Moreover, by the deconvolution of the Co XPS spectra of the CoN/C composite and Co3O4 (Fig.1d), the presence of Co3+ in the CoN/C composite, which is characterized by two peaks at 781.1 and 796.2 eV, corresponding to the characteristic doublet of core-level Co 2p1/2 and Co 2p3/2, can be inferred.7 The XANES of Co K-edge for CoN further confirms the dominant existence of Co3+ by the comparison of the edge energy position of CoO and Co3O4 (Fig. S3, ESI†). The surface Co3+ species on the CoN might promote the ORR process owing to the higher d electron vacancy over Co, which is similar to the catalytic mechanism of Pt-based alloys.8
Fig. 2a and b show typical TEM images of the CoN/C, which clearly indicated that CoN nanoparticles are uniformly dispersed on the carbon with a narrow size distribution of 7.5 nm (Fig. 2d). No obvious particle aggregation is observed although the annealing temperature is up to 550 °C under the NH3 atmosphere. Detailed information of the CoN nanoparticles was further examined using a scanning transmission electron microscope (STEM) equipped with an aberration-correction system, as shown in Fig. 2c, demonstrating that the well-defined crystalline lattice spacing of 0.257 nm can be observed, which matches well with the d spacing values for the (111) plane of CoN. The high angle annular dark-field (HAADF) images (Fig. 2e and f) further reveal the characterization of the porous structure of the nanoparticles, which might dramatically increase the specific surface area. In addition, the BET measurements showed that the surface area of CoN/C is 454 m2 g−1 and is higher than that of XC-72 (232 m2 g−1) (Fig. S6, ESI†). These two factors are highly advantageous for the ORR process.
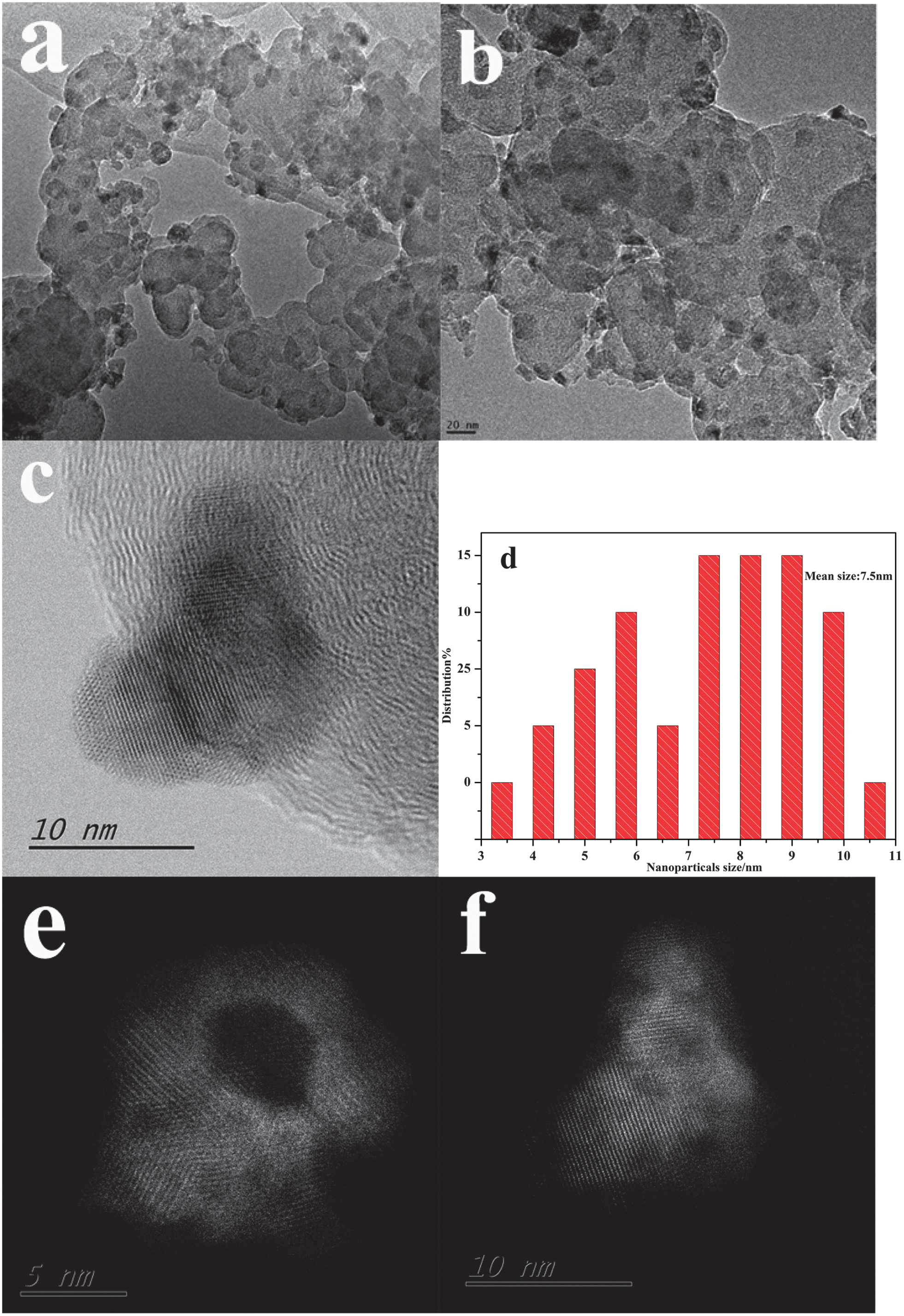 | ||
| Fig. 2 TEM images (a and b) of the CoN/C nanoparticles at different magnifications. HRTEM images and size distribution (c and d) of CoN/C nanoparticles. HAADF images (e and f) of representative CoN/C nanoparticles. | ||
The electrocatalytic activity of the CoN/C composite for the ORR was evaluated with respect to the commercial Pt/C catalyst using cyclic voltammograms (CVs). Fig. 3a shows CVs of the CoN/C electrode in N2- and O2-saturated 0.1 M KOH at a scan rate of 50 mV s−1, respectively. The CoN/C catalyst showed one well-defined redox peak at around 0.90 V in N2- and O2-saturated 0.1 M KOH solution that can be ascribed to redox of Co species.9,10 While the cathodic peak of 0.77 V in 0.1 M KOH saturated with O2 can be attributed to electrocatalytic reduction of oxygen on the CoN/C catalyst, comparable to that of commercial Pt/C (Fig. S4a, ESI†), suggesting high electrocatalytic activity for ORR.
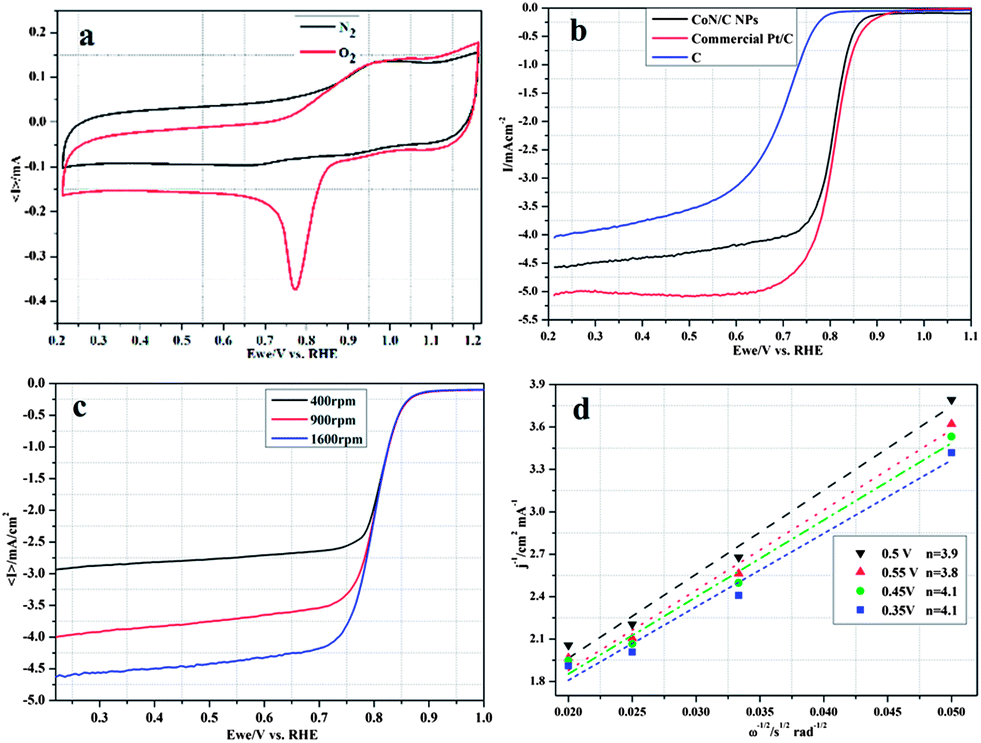 | ||
| Fig. 3 (a) CV curves of the CoN/C catalyst at a scan rate of 50 mV s−1 in N2-(black line) or O2 (red line)-saturated 0.1 M KOH solutions. (b) ORR polarization curves for CoN/C (black line), commercial Pt/C (red line) and C (blue line) catalysts in O2-saturated 0.1 M KOH at a scan rate of 10 mV s−1 at 1600 rpm for comparison. (c) RDE voltammetric response for the ORR in O2-saturated 0.1 M KOH at a scan rate of 10 mV s−1 at different rotation rates; (d) K–L plots for the CoN/C catalyst at different potentials. | ||
The linear scan polarization curves of ORR for CoN/C, commercial Pt/C and C were recorded in O2-saturated 0.1 M KOH solution with a scan rate of 10 mV s−1 at 1600 rpm, as shown in Fig. 3b. It can be found that the CoN/C catalyst displays the onset potential of 0.85 V for ORR (vs. RHE), much higher than that of C and comparable with that of commercial Pt/C. This value is also equal to or more positive than that of other non-precious metal electrocatalysts reported previously in 0.1 M KOH solution.3a,12Fig. 3c displays the polarization curves of the CoN/C catalyst at different rotating rates. The electron transfer number (n) from the slopes of Koutckey–Levich (K–L) plots was calculated to be 4.0, demonstrating that the CoN/C catalyst exhibits a dominant four-electron oxygen reduction process, similar to ORR catalyzed by the commercial Pt/C catalyst (Fig. S4b, ESI†). The kinetic currents (ik) are about 0.18 and 0.12 mA cm−2 for CoN/C and Pt/C evaluated from the disk current at 0.9 V vs. RHE, respectively. In addition, the K–L plot of J−1vs. ω−1/2 calculated at the potential from 0.30 V to 0.60 V on the CoN/C catalyst exhibits good linearity as shown in Fig. 3d, suggesting first-order reaction kinetics for ORR.11,12 The more positive onset potential, higher kinetic current and electron transfer number indicate that the CoN/C catalyst is an excellent potential electrocatalyst for ORR.
The high catalytic activity of the CoN/C catalyst toward ORR could be explained by the following aspects: (1) the increase of the d electron vacancy in Co3+ would result in an increased O2 adsorption and a weakening of the O–O bond, enhancing the ORR activity. (2) The surface Co3+ cations can produce surface electronic states by the Jahn–Teller effect to form excited cationic states [Co3+ + e−] in alkaline media, which favours the ORR in alkaline solution.11 (3) The highly porous structure of CoN nanoparticles dramatically increases the surface-to-volume ratio and abundance of catalytic sites, and promotes the electrocatalytic process toward ORR.13,14
Furthermore, long-time stability of CoN/C and commercial Pt/C catalysts toward ORR was evaluated using chronoamperometric responses (i–t) at 0.75 V in O2-saturated 0.1 M KOH. As shown in Fig. 4a, the CoN/C catalyst caused a slight activity loss, retaining more than 90% of the initial current even after 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s; whereas the commercial Pt/C catalyst lost nearly 40% of its initial activity. These results indicate superior stability of the CoN/C catalyst compared to the commercial Pt/C catalyst in alkaline solution, which is favorable for the development of alkaline fuel cells. Furthermore, after 10000 cycles from 0.6 to 1.0 V, the ORR measurements show only 10 mV and 20 mV loss in its onset potential and half wave potential, respectively (Fig. S5a, ESI†). Combining the results of XPS of stability test samples, the binding energy of Co varies a little and further confirms that the CoN/C catalyst exhibits better stability for the ORR (Fig. S7, ESI†). In addition, the ORR polarization curves of the CoN/C catalyst exhibit no obvious change after the addition of 1 M methanol as shown in Fig. 4b, while the commercial Pt/C catalyst instantaneously jumps to positive values due to the occurrence of methanol oxidation reaction on the Pt/C catalyst (Fig. S5b, ESI†), suggesting better catalytic selectivity to ORR of the CoN/C catalyst.
000 s; whereas the commercial Pt/C catalyst lost nearly 40% of its initial activity. These results indicate superior stability of the CoN/C catalyst compared to the commercial Pt/C catalyst in alkaline solution, which is favorable for the development of alkaline fuel cells. Furthermore, after 10000 cycles from 0.6 to 1.0 V, the ORR measurements show only 10 mV and 20 mV loss in its onset potential and half wave potential, respectively (Fig. S5a, ESI†). Combining the results of XPS of stability test samples, the binding energy of Co varies a little and further confirms that the CoN/C catalyst exhibits better stability for the ORR (Fig. S7, ESI†). In addition, the ORR polarization curves of the CoN/C catalyst exhibit no obvious change after the addition of 1 M methanol as shown in Fig. 4b, while the commercial Pt/C catalyst instantaneously jumps to positive values due to the occurrence of methanol oxidation reaction on the Pt/C catalyst (Fig. S5b, ESI†), suggesting better catalytic selectivity to ORR of the CoN/C catalyst.
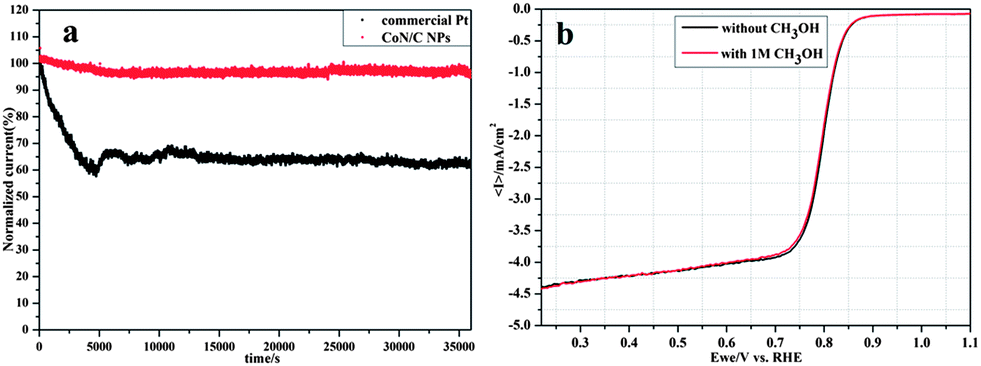 | ||
| Fig. 4 (a) Chronoamperometric responses of CoN/C and commercial Pt/C catalysts at 0.75 V in O2-saturated 0.1 M KOH, which are normalized to the initial current responses. (b) RDE voltammetric responses to injection of 1 M methanol into O2-saturated 0.1 M KOH solution at a scan rate of 10 mV s−1 for CoN/C catalyst modified electrodes. | ||
In summary, we reported a novel non-precious ORR catalyst CoN/C composite synthesized via heat refluxing and a subsequent heat treatment. The surface Co3+ species and the highly porous structure in the CoN/C composite are advantageous for oxygen adsorption during the ORR process. The as-prepared catalyst exhibited superior catalytic activity, high selectivity and excellent long-time durability toward ORR in alkaline solution, which may provide a promising cathode catalyst candidate for alkaline methanol fuel cell applications.
Acknowledgements
This work was supported by the major program of Beijing Municipal Natural Science Foundation (no. 2110001), National Natural Science Foundation of China (no. 11179001) and National High Technology Research and Development Program (863, no. 2012AA052201). We gratefully acknowledge Prof. Lin Gu (Institute of Physics Chinese Academy of Sciences) for assistance in the HAADF-STEM imagines. Dr Yu Wang at SSRF and Dr Caihao Hong at BSRF for the assistance in XAS experiment, Dr Wei Xia for measurement of BET are also acknowledged.Notes and references
- (a) B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345 CrossRef CAS PubMed; (b) M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed.
- (a) K. A. Kuttiyiel, K. Sasaki, Y. M. Choi, D. Su, P. Liu and R. R. Adzic, Nano Lett., 2012, 12, 6266 CrossRef CAS PubMed; (b) J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552 CrossRef CAS PubMed; (c) C. H. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765 CrossRef CAS PubMed; (d) S. J. Guo, D. G. Li, H. Y. Zhu, S. Zhang, N. M. Markovic, V. R. Stamenkovic and S. H. Sun, Angew. Chem., Int. Ed., 2013, 52, 3465 CrossRef CAS PubMed.
- (a) Y. J. Feng and N. A. Vante, Eletrochim. Acta, 2012, 72, 129 CrossRef CAS PubMed; (b) H. L. Wang, Y. Y. Liang, Y. G. Li and H. J. Dai, Angew. Chem., Int. Ed., 2011, 50, 10969 CrossRef CAS PubMed; (c) G. Wu, G. F. Cui, D. Y. Li, P. K. Shen and N. Li, J. Mater. Chem., 2009, 19, 6581 RSC.
- (a) Y. M. Tan, C. F. Xu, G. X. Chen, X. L. Fang, N. F. Zheng and Q. J. Xie, Adv. Funct. Mater., 2012, 22, 4584 CrossRef CAS; (b) Z. S. Wu, S. B. Yang, Y. Sun, K. Parvez, X. L. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082 CrossRef CAS PubMed; (c) F. Y. Cheng, J. Shen, B. Peng, Y. D. Pan, Z. L. Tao and J. Chen, Nat. Chem., 2011, 3, 79 CrossRef CAS PubMed.
- (a) D. A. Slanac, W. G. Hardin, K. P. Johnston and K. J. Stevenson, J. Am. Chem. Soc., 2012, 134, 9812 CrossRef CAS PubMed; (b) Y. J. Lu, N. L. Zhang, L. An, X. Li and D. G. Xia, J. Power Sources, 2013, 240, 606 CrossRef CAS PubMed; (c) S. Dong, X. Chen, X. Zhang and G. Cui, Coord. Chem. Rev., 2013, 257, 1946 CrossRef CAS PubMed; (d) D. Xia, S. Liu, Z. Wang, G. Chen, L. Zhang, L. Zhang, S. Hui and J. Zhang, J. Power Sources, 2008, 177, 296 CrossRef CAS PubMed; (e) Y. Lai, D. B. Zhou, J. W. Hu and L. L. Cui, Acta Chim. Sin., 2008, 66, 1015 CAS.
- (a) Z. W. Chen, M. Waje, W. Z. Li and Y. S. Yan, Angew. Chem., 2007, 119, 4138 CrossRef; (b) S. Y. Huang, P. Ganesan, S. Park and B. N. Popov, J. Am. Chem. Soc., 2009, 131, 13898 CrossRef CAS PubMed.
- Y. X. Ye, L. Kuai and B. Y. Geng, J. Mater. Chem., 2012, 22, 19132 RSC.
- (a) T. Toda, H. Igarashi, H. Uchida and M. Watanabe, J. Electrochem. Soc., 1999, 146, 3750 CrossRef CAS PubMed; (b) S. J. Yoo, S. K. Kim, T. Y. Jeon, S. J. Hwang, J. G. Lee, S. C. Lee, K. S. Lee, Y. H. Cho, Y. E. Sung and T. H. Lim, Chem. Commun., 2011, 47, 11414 RSC; (c) P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. F. Yu, Z. C. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454 CrossRef CAS PubMed.
- J. B. Xu, P. Gao and T. S. Zhao, Energy Environ. Sci., 2012, 5, 5333 CAS.
- Y. Y. Liang, H. L. Wang, P. Diao, W. Chang, G. S. Hong, Y. G. Li, M. Gong, L. M. Xie, J. G. Zhou, J. Wang, T. Regier, F. Wei and H. J. Dai, J. Am. Chem. Soc., 2012, 134, 15849 CrossRef CAS PubMed.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS PubMed.
- L. Wang, J. Yin, L. Zhao, C. G. Tian, P. Yu, J. Q. Wang and H. G. Fu, Chem. Commun., 2013, 49, 3022 RSC.
- D. L. Wang, Y. C. Yu, H. L. Xin, R. Hovden, P. Ercius, J. A. Mundy, H. Chen, J. H. Richard, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nano Lett., 2012, 12, 5230 CrossRef CAS PubMed.
- Y. B. Cho, J. E. Kim, J. H. Shim, C. Lee and Y. Lee, Phys. Chem. Chem. Phys., 2013, 15, 11461 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c3ta13877a |
| This journal is © The Royal Society of Chemistry 2014 |