Switchable and reversible water adhesion on superhydrophobic titanate nanostructures fabricated on soft substrates: photopatternable wettability and thermomodulatable adhesivity†
Yasuaki
Tokudome
*,
Kenji
Okada
,
Atsushi
Nakahira
and
Masahide
Takahashi
*
Department of Materials Science, Graduate School of Engineering, Osaka Prefecture University, Sakai, Osaka 599-8531, Japan. E-mail: tokudome@photomater.com; masa@photomater.com; Fax: +81-72-254-9309; Tel: +81-72-254-9309
First published on 29th October 2013
Abstract
A titanate nanotube (TNT) film is demonstrated to provide a switchable and adhesive hydrophobic surface. The surface adhesivity is reversibly switched from adhesive to repellent by mild heating and spontaneous rehydration. A persistent superhydrophobic/superhydrophilic pattern can be fabricated on the TNT films coated on soft substrates.
Introduction
Extensive exploration dedicated to surface wettability has revealed that a superhydrophobic surface with controllable liquid-adhesion is a potential tool for versatile water manipulation.1–3 A water droplet, for example, can strongly attach onto a superhydrophobic surface with a high adhesion force, and thereby the droplet can be transferred from the surface without losing its original mass.4 Water manipulation is expected in microfluidics to open up applications, such as microreactors and oil/water separation.5–7Superhydrophobic surfaces with controlled adhesion force have been demonstrated on films made of polystyrene,8 polypropylene (PP),9 poly(dimethyl siloxane),10–12 organically modified oxides (TiO2,13 ZnO,14 ZrO2 (ref. 15 and 16)), carbon nanotubes,17 epoxy resin,18 polydopamine-coated micropatterns,19 and so on.20–23 The controlled adhesion force therein is based on tunable surface nanomorphologies in the respective systems.24,25 For further manipulation, reversible and switchable adhesion with a single surface offers considerable promise. This type of “smart” surface with reversible and switchable adhesion is one of the central issues in this field and indeed developed to date. On the other hand, the smart surface is generally dictated by functionality of organics, making it still challenging to achieve water manipulation in a system free from stimuli-responsive organic molecules and/or particles.20 A hydrophobic organic layer on nanostructured oxides, such as ZnO and TiO2, is photocatalytically decomposed, which allow wettability change without using stimuli-responsive organics. However their wetting transitions are not reversible and switchable. Elegant strategies reported on TiO2 nanorod/tube films26,27 and a Pd-coated Si nanowire film28 overcame this barrier and demonstrated reversible and switchable water adhesion on the films without any stimuli-responsive substances. Especially, the TiO2 nanorod/tube films should be highlighted. The system could afford the adhesion switching under an ambient condition (air atmosphere). The switchable adhesion therein shows cycle characteristics by taking advantage of photo-induced hydrophilization and subsequent thermally activated hydrophobization. Nevertheless, there still remains considerable demand for switchable adhesion surfaces via an alternative route; light stimulus is indispensable in the TiO2 system, which limits the possibility to use non-photo-active oxides/hydroxides with functional properties. Another technological challenge of the TiO2 system is how to use soft substrates. It is generally difficult to nanofabricate oxides (including TiO2) on plastic substrates29 because techniques, such as anodic oxidation, etching of templates, and lithography used in the nanofabrication of oxides,26,27 are hardly fused with using soft substrates. The use of soft substrates opens up attractive applications, such as mechanically tunable transparency and wettability.30
Herein, we develop a novel multifunctional oxide film, which is controllably nanofabricated on the soft substrate as well as operated by another stimulus. Nanostructured titanate is demonstrated to produce a superhydrophobic surface with switchable adhesivity. The functional surface is made up of vertically oriented titanate nanotubes (TNTs) with a high aspect ratio. After fluoroalkylsilane (FAS) modification, the surface exhibits superhydrophobicity with a high adhesion force. The FAS-modified TNT film shows a sticky superhydrophobic surface upon exposure to moisture, whereas it becomes slippy on mild heating. The adhesive force of the film is increased by water molecules adsorbed onto the TNT surface; the mild heating dehydrates the TNT surface and the exposure to moisture recovers physically adsorbed water.
Another important finding in the present work is that TNTs can grow on plastic/soft substrates because the protocol does not require severe conditions, such as anode oxidation, template removal at higher temperatures, and lithographic processing.31 The spontaneous growth of TNTs is simply induced by carrying out a mild hydrothermal reaction on amorphous TiO2 used as a precursory film. Photopatternability is provided by a photocatalytic nature of TNTs with a band gap of 3.87 eV.32,33 Because of the large band gap compared with TiO2, the pattern remains for more than a year under fluorescent tube radiation in a laboratory atmosphere. As a result, TNTs create the patternable and switchable surface through the process applicable to soft substrates. The obtained TNT film can be potentially used for water manipulation in lab-on-a-chip, microfluid devices and water/oil separation.27,34 As a proof of concept, here we show that the TNT surface can controllably catch and repel a water droplet. Deformable soft substrates are expected to induce morphological change of nanostructures on the top of the substrate. This allows anisotropic wettability35 and mechanicallytunable wettability.30
Results and discussion
TNT is a tubular nanoarchitecture formed by scrolling a titanate single layer with cations inserted between adjacent walls.36 Generally, it is impossible to grow TNTs on substrates which are less resistant to highly basic conditions, because the growth of TNTs takes place under highly basic hydrothermal conditions.37 We have recently reported that vertically aligned TNTs can be grown on amorphous TiO2 films under mild hydrothermal conditions.38 The previous protocol was further developed in the present study using an amorphous and crack-free TiO2 film on various substrates. The substrates coated with the TiO2 film was covered with a reactive NaOH liquid film, and processed at 110 °C in a closed vessel (Methods and Fig. S1, ESI†). The TiO2 film works as a protective coating for substrates as well as a Ti source for TNTs. The amorphous nature of the film allows the use of plastic substrates because no thermal treatment is required for preparing precursory TiO2 films. As a result, TNTs successfully grow on various substrates, such as rutile TiO2, PP, poly(ethylene terephthalate), and poly(tetrafluoro ethylene) (PTFE). A field emission scanning electron microscopic (FE-SEM) image of TNTs on a PTFE substrate is presented in Fig. 1a. A close look at the image reveals that the tubular nanoarchitecture is formed at the top of the film. A transmission electron microscopic (TEM) image and an X-ray diffraction pattern confirm that the obtained nanoarchitecture is composed of a layered titanate with a tubular shape, TNTs (H2Ti4O9) (Fig. S2, ESI†).39 The formation of TNT proceeds by scrolling at the top end of the nanosheet as shown in Fig. S3.† A well-grown nanosheet starts scrolling to decrease excess surface energy on the top of the sheet.40Fig. 1b shows the film appearance on a bendable PTFE substrate. The soft and bendable substrates are promising candidates to produce a surface with mechanically tunable wettability.30 TNTs formed on all of the substrates used in this study, suggesting the process versatility to substrates with less durability against basic conditions. The obtained TNTs are identical on all the employed substrates. Hereafter, detailed discussion on the film surfaces is given in the case of a rutile substrate. Fig. 1c and d show FE-SEM images of TNTs before and after FAS modification. The images confirm that FAS modification leads to no morphological change of TNTs. On the other hand, the modification forms a surface with a very low surface free energy and drastically changes surface properties. Pristine TNTs show a superhydrophilic nature, while a superhydrophobic surface with considerable adhesion force is formed by the FAS modification (the inset images of Fig. 1c and d).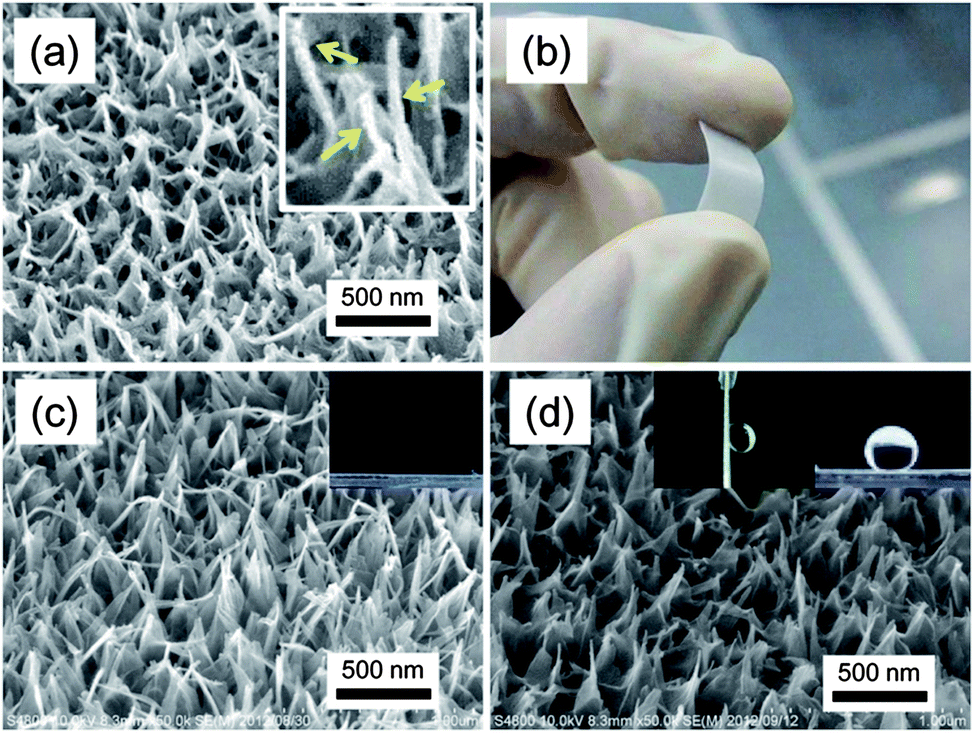 | ||
| Fig. 1 (a) FE-SEM and (b) optical images showing TNTs fabricated on a flexible PTFE substrate; the yellow arrows in the magnified image indicate TNTs. (c and d) FE-SEM images showing TNTs prepared on a rutile TiO2 substrate under a condition of 110 °C for 24 h in 1.00 M NaOH aq.: (c) before and (d) after FAS modification. The inset images in (c) and (d) represent water droplets on the corresponding films. | ||
Contact angle, θCA, and sliding angle, θSA, of a water droplet on the films are tunable by changing the NaOH concentration employed in the hydrothermal treatment (Fig. 2a). FE-SEM images of Fig. 2b and S3, ESI,† clarify that the surface properties are dictated by different nanomorphologies. Although the relatively random alignment of TNTs prevents precise theoretical evaluation, we can qualitatively state that the change of θCA with NaOH concentration is due to vertical elongation of the nanostructure, which increases a roughness factor value involved in Wenzel's mode;25 indeed, surface roughness increases with NaOH concentration (Table S1, ESI†). The values of θSA were estimated using a quite large water droplet (30 μL) in this case. The adhesion forces of the films are suggested to be >60 μN considering a previous report.8 Relatively small water droplets are strongly stuck irrespective of the nanostructures (see Fig. S4, ESI†). It is worth noting that the simple tuning of crystal growth kinetics achieves tunable surface properties without any templates. The FAS-modified TNT surface can be transformed again to a superhydrophilic surface (θCA = ∼0°) by UV illumination with a xenon lamp (Fig. 2c). The variation of θCA with increasing UV illumination time is due to photocatalytic decomposition of FAS molecules; the reference FAS-modified Si substrate is intact under the UV illumination. The decomposition of FAS molecules is confirmed by Fourier transform infrared spectroscopy (FTIR) analysis (Fig. S5, ESI†). UV illumination through a mask extends this capability of producing patterned films as shown in Fig. 2d. Superhydrophilic wetting occurs space-selectively at UV-illuminated areas. The patterned wetting was obtained by shaking off waters on the hydrophobic areas after the substrate had been immersed into a solution. It should be mentioned that the large band gap of TNTs (3.87 eV) would avoid vanishing the pattern under near UV illumination. This provides another capability of using near UV light to induce photochemical reaction only at wetting areas (Fig. S6, ESI†).
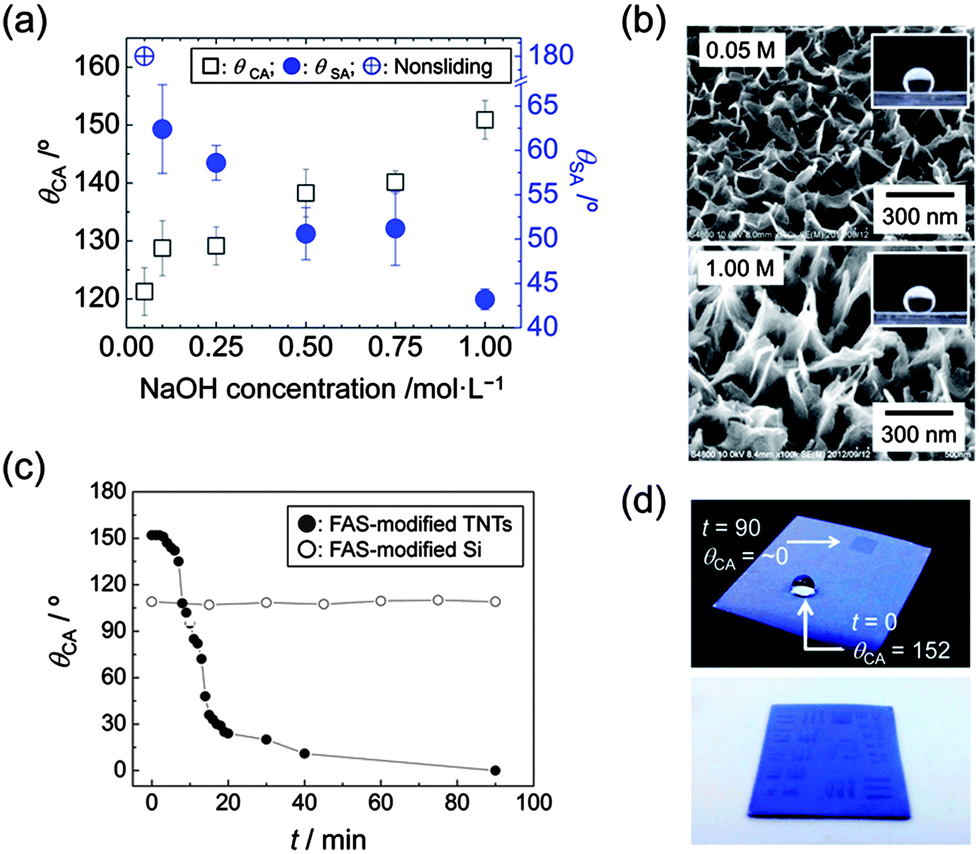 | ||
| Fig. 2 (a) Variations of contact (θCA) and sliding (θSA) angles with NaOH concentration used in the hydrothermal treatment. θSA was measured using a 30 μL water droplet. (b) FE-SEM images showing FAS-modified TNTs grown in 0.05 and 1.00 M NaOH aq. solutions. (c) θCAvs. UV illumination time, t. ●: FAS-modified TNTs; ○: reference FAS-modified Si substrate. (d) Optical images showing films UV-illuminated for 90 min through a mask. The lower patterned image represents selective wetting only on UV-illuminated areas. 1.00 M NaOH aq. was used for the TNTs growth in (c) and (d). | ||
In addition to the features which have been discussed so far: (1) structure-directed wettability tuning and (2) UV patternability, FAS-modified TNT films show reversible and switchable water adhesion on a single TNT film. Interestingly, the superhydrophobic film has a response to moisture/water and change the wettability. Reversible change of θCA is observed by alternating mild heating (80 °C, 30 min) and hydration (30 min in water) as shown in Fig. 3a; the hydrated film was dried at room temperature prior to the contact angle measurement. The film is nearly superhydrophobic (θCA > 149 ± 2°) after heating at 80 °C, while it becomes less hydrophobic (θCA = 137 ± 2°) after hydration. The change of θCA between the two states is so large that the surface with modulated wettability can catch and repel a falling water droplet (8 μL) (Fig. 3b and Video 1, ESI†). The transition also takes place even when the hydration is performed under an ambient air condition. The process is much slower compared to the case of using hydration in water. The moderate rehydration under an ambient air condition (23 °C, 85% RH, 3 h) affords to control the water repelling/catching transition in the superhydrophobic regime (θCA > 150°) as shown in Video 2, ESI.† Adhesion energy of a water droplet was estimated to be 8.94 and 8.51 mJ m−2 for the hydrated and the heated TNT surfaces, respectively. These results confirm that the hydrated TNTs form the sticker surface to catch a water droplet.
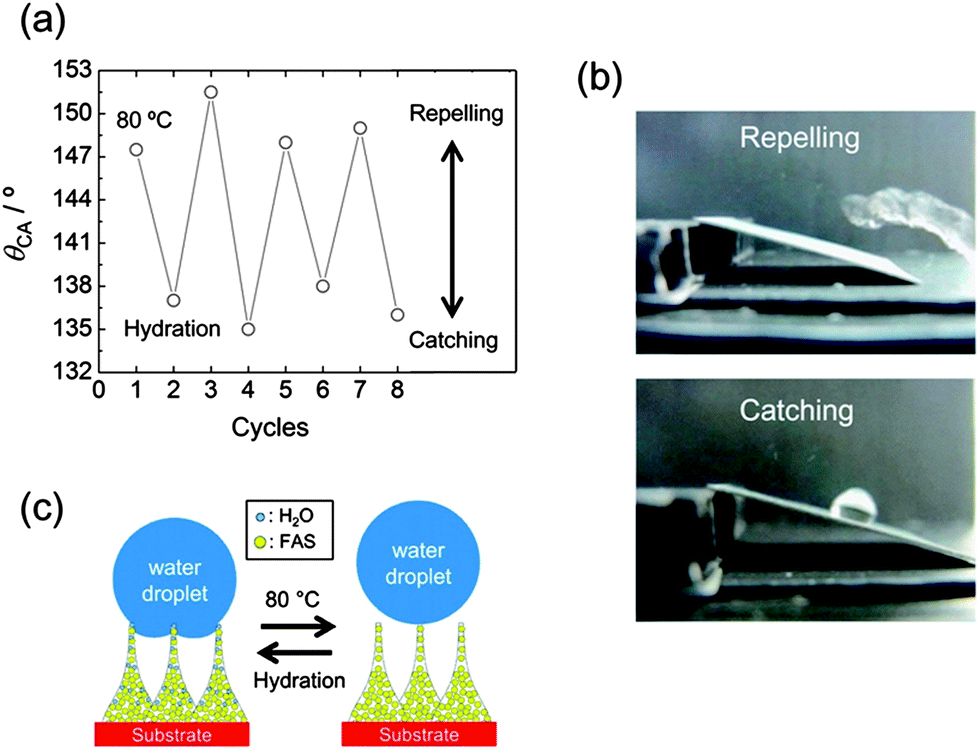 | ||
| Fig. 3 (a) Reversible water adhesion of the FAS-coated TNT film by alternating thermal-treatment (80 °C, 30 min) and hydration (30 min in water). (b) Optical images of the FAS-coated film repelling and catching a water droplet after heating (80 °C) and hydration, respectively. (c) A schematic illustration showing reversible water adhesion. | ||
A mechanism for the reversible adhesive surface is proposed in Fig. 3c. Adsorption/desorption of water molecules on the TNT film induces a slight change of surface hydrophilicity and adhesive force. Thermogravimetric (TG) analysis was performed on TNTs (Fig. S7, ESI†); powdery TNTs was used considering sample mass. The cycles 1 and 3 show weight losses measured on heated powdery TNTs, while the cycles 2 and 4 correspond to weight losses measured on hydrated powder TNTs. The clear reversible trend observed in Fig. S7† indicates that desorption of physically adsorbed water takes place at 80 °C and the hydration fully recovers water molecules onto the TNT surface. It is well-known that TNTs possess water molecules which are physically adsorbed on the surface and trapped in the interlayer; the water molecules dehydrate below 100 °C and over 150 °C, respectively (Fig. S8, ESI†).41 The dehydration of physically adsorbed water is not accompanied by structural reconstruction in contrast to that of interlayer water,42 and thus the mild heating allows the reversible cycle characteristics. Water molecules are adsorbed on the FAS-modified TNT film as well as the TNT powder; OH vibration is detected by FTIR analysis, as shown in Fig. S9, ESI.† A previous report demonstrated that θCA has reached to 120° on a FAS-coated flat substrate,43 whereas θCA is below 110° on the flat FAS-modified Si substrate as shown in Fig. 2c. The coverage of FAS is ∼85% in the present case. The partial FAS coverage on the surface ensures available adsorption sites on the TNT film.
Conclusions
We have demonstrated that the TNT film on various substrates is a novel material providing superhydrophobic surfaces with switchable adhesivity. The adhesive surface is switched by heating (80 °C) and spontaneous rehydration. This allows reversible water manipulation without using a light stimulus. Fig. 3c represents a schematic illustration summarizing the reversible water adhesion on the TNT surface. The mechanism behind is possibly applicable to other materials with high water affinity. Additionally, the procedure demonstrated here can fabricate nanostructured oxides on bendable substrates with patterned wettability. Further study is expected to open up applications taking both advantages of novel nanostructured materials and functional substrates.Acknowledgements
The present work is partially supported by JSPS KAKENHI (no. 22360276, no. 24750206, no. 2510817). M. T. is also supported by a research grant from the Murata Scientific Foundation. Special thanks go to Dr H. Uchiyama, Kansai University, Japan, for adhesion energy measurement.Notes and references
- X. Liu, Y. Liang, F. Zhou and W. Liu, Soft Matter, 2012, 8, 2070 RSC.
- X. M. Li, D. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 1350 RSC.
- Y. Y. Yan, N. Gao and W. Barthlott, Adv. Colloid Interface Sci., 2011, 169, 80 CrossRef CAS PubMed.
- X. Hong, X. Gao and L. Jiang, J. Am. Chem. Soc., 2007, 129, 1478 CrossRef CAS PubMed.
- N. Verplanck, Y. Coffinier, V. Thomy and R. Boukherroub, Nanoscale Res. Lett., 2007, 2, 577 CrossRef CAS.
- L. Zhang, Z. Zhang and P. Wang, NPG Asia Mater., 2012, 4, e8 CrossRef.
- B. L. Xue, L. C. Gao, Y. P. Hou, Z. W. Liu and L. Jiang, Adv. Mater., 2013, 25, 27 CrossRef PubMed.
- M. Jin, X. Feng, L. Feng, T. Sun, J. Zhai, T. Li and L. Jiang, Adv. Mater., 2005, 17, 1977 CrossRef CAS.
- H. Y. Erbil, A. L. Demirel, Y. Avci and O. Mert, Science, 2003, 299, 1377 CrossRef CAS PubMed.
- X. J. Huang, D. H. Kim, M. Im, J. H. Lee, J. B. Yoon and Y. K. Choi, Small, 2009, 5, 90 CrossRef CAS PubMed.
- C. Greiner, A. del Campo and E. Arzt, Langmuir, 2007, 23, 3495 CrossRef CAS PubMed.
- W. K. Cho and I. S. Choi, Adv. Funct. Mater., 2008, 18, 1089 CrossRef CAS.
- Y. Lai, X. Gao, H. Zhuang, J. Huang, C. Lin and L. Jiang, Adv. Mater., 2009, 21, 3799 CrossRef CAS.
- Y. Li, M. Zheng, L. Ma, M. Zhong and W. Shen, Inorg. Chem., 2008, 47, 3140 CrossRef CAS PubMed.
- S. A. Kulinich and M. Farzaneh, Langmuir, 2009, 25, 8854 CrossRef CAS PubMed.
- S. A. Kulinich and M. Farzaneh, Appl. Surf. Sci., 2009, 255, 4056 CrossRef CAS PubMed.
- Z. Wang, N. Koratkar, L. Ci and P. M. Ajayan, Appl. Phys. Lett., 2007, 90, 143117 CrossRef.
- B. Bhushan and E. K. Her, Langmuir, 2010, 26, 8207 CrossRef CAS PubMed.
- S. M. Kang, I. You, W. K. Cho, H. K. Shon, T. G. Lee, I. S. Choi, J. M. Karp and H. Lee, Angew. Chem., Int. Ed., 2010, 49, 9401 CrossRef CAS PubMed.
- M. Liu and L. Jiang, Adv. Funct. Mater., 2010, 20, 3753 CrossRef CAS.
- D. Ishii, H. Yabu and M. Shimomura, Chem. Mater., 2009, 21, 1799 CrossRef CAS.
- Z. G. Guo and W. M. Liu, Appl. Phys. Lett., 2007, 90, 223111 CrossRef.
- N. D. Boscher, D. Duday, S. Verdier and P. Choquet, ACS Appl. Mater. Interfaces, 2013, 5, 1053 CAS.
- M. Miwa, A. Nakajima, A. Fujishima, K. Hashimoto and T. Watanabe, Langmuir, 2000, 16, 5754 CrossRef CAS.
- Z. Yoshimitsu, A. Nakajima, T. Watanabe and K. Hashimoto, Langmuir, 2002, 18, 5818 CrossRef CAS.
- G. Caputo, B. Cortese, C. Nobile, M. Salerno, R. Cingolani, G. Gigli, P. D. Cozzoli and A. Athanassiou, Adv. Funct. Mater., 2009, 19, 1149 CrossRef CAS.
- D. A. Wang, Y. Liu, X. J. Liu, F. Zhou, W. M. Liu and Q. J. Xue, Chem. Commun., 2009, 7018 RSC.
- J. Seo, S. Lee, H. Han, H. B. Jung, J. Hong, G. Song, S. M. Cho, C. Park, W. Lee and T. Lee, Adv. Mater., 2013, 25, 4139 CrossRef CAS PubMed.
- H. Kozuka, J. Mater. Res., 2013, 28, 673 CrossRef CAS.
- X. Yao, Y. Hu, A. Grinthal, T.-S. Wong, L. Mahadevan and J. Aizenberg, Nat. Mater., 2013, 12, 529 CrossRef CAS PubMed.
- X. Feng and L. Jiang, Adv. Mater., 2006, 18, 3063 CrossRef CAS.
- D. V. Bavykin, S. N. Gordeev, A. V. Moskalenko, A. A. Lapkin and F. C. Walsh, J. Phys. Chem. B, 2005, 109, 8565 CrossRef CAS PubMed.
- S. Uchida, Y. Yamamoto, Y. Fujishiro, A. Watanabe, O. Ito and T. Sato, J. Chem. Soc., Faraday Trans., 1997, 93, 3229 RSC.
- K. Tsougeni, D. Papageorgiou, A. Tserepi and E. Gogolides, Lab Chip, 2010, 10, 462 RSC.
- H. Yabu, Y. Nakamichi, Y. Hirai and M. Shimomura, Phys. Chem. Chem. Phys., 2011, 13, 4877 RSC.
- T. P. Feist and P. K. Davies, J. Solid State Chem., 1992, 101, 275 CrossRef CAS.
- A. Nakahira, W. Kato, M. Tamai, T. Isshiki, K. Nishio and H. Aritani, J. Mater. Sci., 2004, 39, 4239 CrossRef CAS.
- K. Okada, Y. Takamatsu, Y. Tokudome, A. Nakahira and M. Takahashi, J. Sol-Gel Sci. Technol., 2012, 65, 36 CrossRef.
- K. Okada, Y. Tokudome, P. Falcaro, Y. Takamatsu, A. Nakahira and M. Takahashi, Chem. Commun., 2012, 48, 6130 RSC.
- D. V. Bavykin, V. N. Parmon, A. A. Lapkin and F. C. Walsh, J. Mater. Chem., 2004, 14, 3370 RSC.
- Y. J. Xie, C. Liu, H. B. He and X. H. Lu, J. Therm. Anal. Calorim., 2012, 110, 671 CrossRef CAS.
- H. G. Yang and H. C. Zeng, J. Am. Chem. Soc., 2005, 127, 270 CrossRef CAS PubMed.
- K. Tadanaga, N. Katata and T. Minami, J. Am. Ceram. Soc., 1997, 80, 3213 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods and additional data. See DOI: 10.1039/c3ta13536e |
| This journal is © The Royal Society of Chemistry 2014 |