Organic spin transporting materials: present and future
Yonghao
Zheng
ab and
Fred
Wudl
*ab
aCalifornia NanoSystems Institute, University of California, Santa Barbara, California 93106, USA. E-mail: wudl@chem.ucsb.edu
bTohoku University WPI-AIMR and CNSI Joint Center, University of California, Santa Barbara, California 93106, USA
First published on 17th September 2013
Abstract
Spintronics involves the study of spin directions manipulation in the solid-state and has been a fascinating and attractive field for scientists. In the past decade, organic materials, unlike inorganic solids, have emerged as one of the promising candidates for spintronics due to their low spin–orbit coupling and weak hyperfine interaction. Organic semiconductors have shown great potential for spin transport in recent publications. In this article, we address the issues of influence of organic materials in spin transport and introduce a variety of possible candidate organic materials for further development of the field.
 Yonghao Zheng | Yonghao Zheng is currently a joint scientist at Tohoku University WPI-AIMR Joint Center with the California NanoSystems Institute (CNSI), University of California, Santa Barbara under the supervision of Prof. Fred Wudl (UCSB) and Prof. Katsumi Tanigaki (Tohoku University, Japan). He received his MChem (2008) and Ph.D. (2011) degrees from Durham University, where his dissertation work was done under the supervision of Prof. Martin R. Bryce. He was appointed as a postdoctoral researcher in the group on the project for developing novel conjugated materials for organic electronics. In February 2012, he moved to Prof. Fred Wudl's group as a postdoctoral researcher. |
 Fred Wudl | Fred Wudl, Research Professor of Materials at the University of California, Santa Barbara (UCSB), received his B.S. (1964) and Ph.D. (1967) degree from the University of California, Los Angeles (UCLA) where his dissertation work was done with Professor Donald J. Cram. After postdoctoral research with R. B. Woodward at Harvard, he joined the faculty of the State University of New York at Buffalo. He then moved, first in 1972 to AT&T Bell Laboratories, and subsequently to UCSB in 1982. He was a Chair Professor at UCLA from 1997 to 2006, moving back to UCSB that year. He is widely known for his work on organic conductors and superconductors with the discovery of the electronic conductivity of the precursor to the first organic metal and superconductor. His interest in electronically conducting polymers resulted in the discovery of the first transparent organic conductor and the first self-doped polymers. Currently he is interested in the optical and electro-optical properties of processable conjugated polymers as well as in the organic chemistry of fullerenes and the design and preparation of self-mending and self-healing materials. He has received numerous awards and honors and has published over 500 papers. |
Introduction
Spintronics is the study of the manipulation of spin directions in the solid state and has been a frontier research field for scientists.1 Organic materials for spintronics have emerged as a new fascinating opportunity due to their low spin–orbit coupling and weak hyperfine interaction in contrast to inorganic materials.2 Organic semiconductors (OSCs) have clearly been the key ingredient in organic electronics for the past two decades. The performance of organic devices has improved remarkably due to device fabrication but also due to a better understanding of the properties of organic materials.3–6 Over the past decade, physicists and engineers have been working closely to understand the impact of OSCs in spintronics. The study of spin transport in organic spintronics has quickly become one of the most popular topics in the field in both fundamental and applied sciences.7,8 However, spin transport in organic spintronics is now facing the same early stage development of organic electronics, exciting results were obtained and analyzed only based on a handful of organic materials.8–15 Many OSCs still remain unexplored. Therefore, collaboration between chemists and physicists is expected to provide an accelerating effect in driving the field forward. The challenges in organic spintronics are to understand the role of different OSCs in the devices, to obtain high-quality and repeatable devices and, significantly, to improve the low hole and electron mobilities in comparison to inorganic materials. In this article, we address the issues that influence spin transport in organic materials used in spintronics from a chemist's viewpoint to introduce a variety of interesting organic materials that are potentially applicable to the field.Organic spin valves
A basic spin valve device consists of a non-magnetic spacer sandwiched by soft and hard magnetic layers (Scheme 1), and the resistance of the device is switchable between two different values by rotating the magnetization vector of the ferromagnetic (FM) layers.16 Efficient spin injection for inorganic spin valves has been considered as one of the major challenges,17,18 mainly due to the mismatching conductivity between the electrodes and the spacer.19 On the other hand, OSCs as a spacer have provided a solution for improving the spin injection from electrodes in spin valves, efficient spin injection at room temperature was observed due to their relatively long spin relaxation time.13,15 The spin valve effect was measured in thin layers of OSCs as tunneling magnetoresistance (TMR).16,20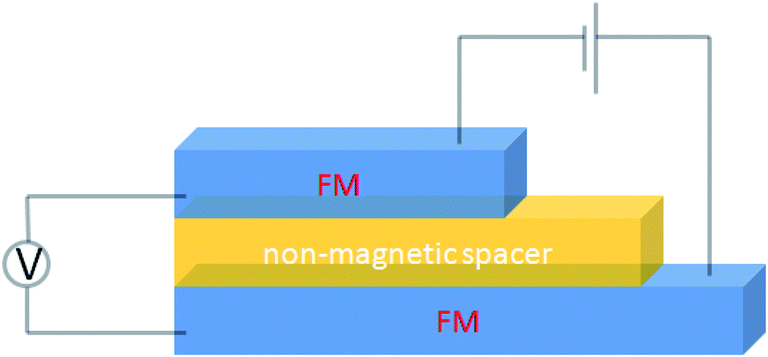 | ||
| Scheme 1 A basic device structure of organic spin valve. | ||
The only limited number of organic semiconductors that were studied in spin valves as the non-magnetic spacer in the literature, covered a wide range of materials from small molecules to polymers, and carbon based materials.8 The information of the OSCs and their properties in spin valve are listed in Table 1. In 2004, Xiong et al. have applied tris(8-hydroxyquinolinato) aluminum (Alq3) as the spacer layer between ferromagnetic La0.67Sr0.33MnO3 (LSMO) and Co electrodes and measured a giant inverse magnetoresistance (GMR) of 40% at 11 K that turned to zero when the temperature was raised above 200 K. A spin diffusion length of 45 nm was estimated at helium temperature.11 Since then, scientists have drawn attention to popular organic electronic materials such as poly[2,5-dioctyloxy-1,4-phenylenevinylene] (DOO-PPV),21 usually used in organic light emitting diodes (OLEDs), rubrene22 and 6,13-bis(triisopropylsilylethynyl)pentacene (TIPS-pentacene),23 usually used in organic field-effect transistors (OFETs) and the benchmark donor poly[3-hexylthiophene] (P3HT),24 normally used in organic photovoltaics (OPVs). The temperature at which magnetoresistance (MR) ratio and spin diffusion length can be observed has significantly improved. At room temperature, a MR ratio of 6% was obtained in a device structure of rubrene as the spacer layer between ferromagnetic Co and Fe electrodes with an estimated spin diffusion length of 13.3 nm was estimated.22 Very recently, Zhang et al. have observed a MR ratio of 5% at room temperature in a device structure with C60 as the spacer between ferromagnetic Fe3O4 and Co electrodes, where a long spin diffusion length of 110 nm was estimated.15 Interestingly, improving the spin diffusion length was achieved by chemical modification of the DOO-PPV polymer, with the deuterated DOO-PPV polymer possessing a spin diffusion length of 49 nm, 3 times higher than the hydrogenated DOO-PPV polymer and with a longer spin diffusion time.21
| Organic semiconductors | Spin-polarized electrodes, spin-diffusion length/time | MR ratio and temperature | Organic electronics | HOMO/LUMO energy level | Carrier mobility (cm2 V−1 s−1) |
|---|---|---|---|---|---|
| Alq3 | |||||
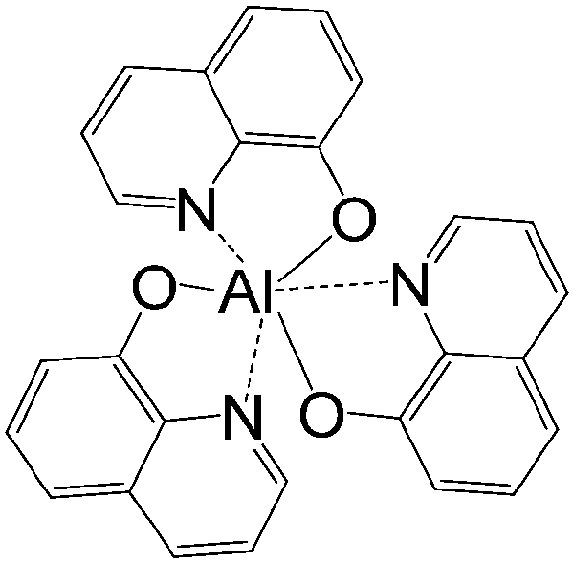
|
LSMO/Co11 | 40% at 11 K11 | Electron transporting (OLEDs) | HOMO = −5.7 eV | 10−5 |
| L s ≈ 45 nm | 0% above 200 K11 | LUMO = −2.7 eV | n-Type27 | ||
| Fe/Co25 | 5% at 11 K25 | ||||
| Co/Ni26 | 0% above 90 K25 | ||||
| τ s over 1 s | |||||
| C 60 | |||||
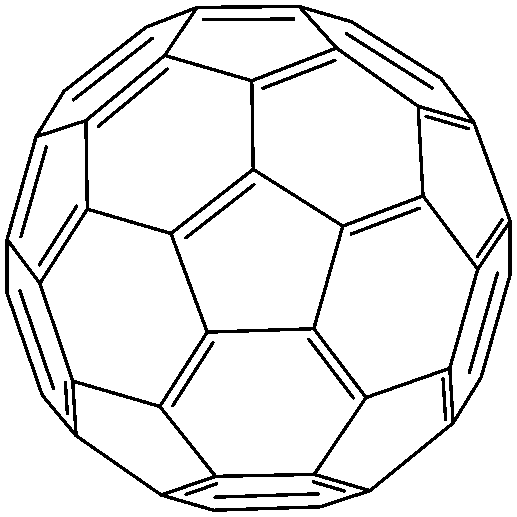
|
Fe3O4/Co15 | 5% at RT15 | Electron acceptor (OPVs) | HOMO = −6.2 eV | 0.25–1.0 |
| L s ≈ 110 nm | >5% at RT13 | LUMO = −4.5 eV28 | n-Type29 | ||
| Co/Py13 | |||||
| Carbon nanotube | |||||
|
LSMO/LSMO7 | 61% at 5 K7 | Electron and hole transporting | ||
| L s ≈ 50 μm | |||||
| τ s ≈ 30 ns | |||||
| Graphene | |||||
|
Trilayer-graphene/MgO/Py30 | Electron and hole transporting | |||
| L s ≈ 1.5 μm | |||||
| T 6 | |||||
|
LSMO/LSMO31 | Up to 30% at RT31 | Electron donor (OPVs) | HOMO = −4.9 eV | 10−1 |
| L s ≈ 70 nm | LUMO = −2.3 eV | p-Type | |||
| τ s ≈ 10−6 μs | |||||
| P3HT | |||||
|
LSMO/Co24 | 25% at 5 K24 | Electron donor (OPVs) | HOMO = −4.9 eV | 10−1 |
| L s ≈ 80 nm | 0.5% at RT24 | LUMO = −3.0 eV32 | p-Type | ||
| Rubrene | |||||
|
Co/Fe22 | 16% at 4.2 K22 | OFETs | HOMO = −5.4 eV | 40 |
| L s ≈ 13.3 nm | 6% at RT22 | LUMO = −3.2 eV33 | p-Type34 | ||
| 0.8 | |||||
| n-Type35 | |||||
| Pentacene | |||||
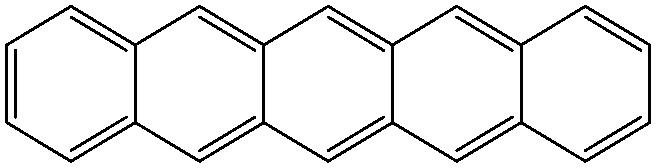
|
Co/Fe36 | OFETs | HOMO = −4.9 eV | 5.5 | |
| Electron donor (OPVs) | LUMO = −3.0 eV37 | p-Type38 | |||
| TIPS-pentacene | |||||
|
CoPt/TIPS-pentacene/Co23 | 0.32% at 200 K23 | OFETs | HOMO = −5.1 eV | 4.6 |
| L s ≈ 24 nm | LUMO = −3.4 eV39 | p-Type40 | |||
| τ s ≈ 3.5 μs | |||||
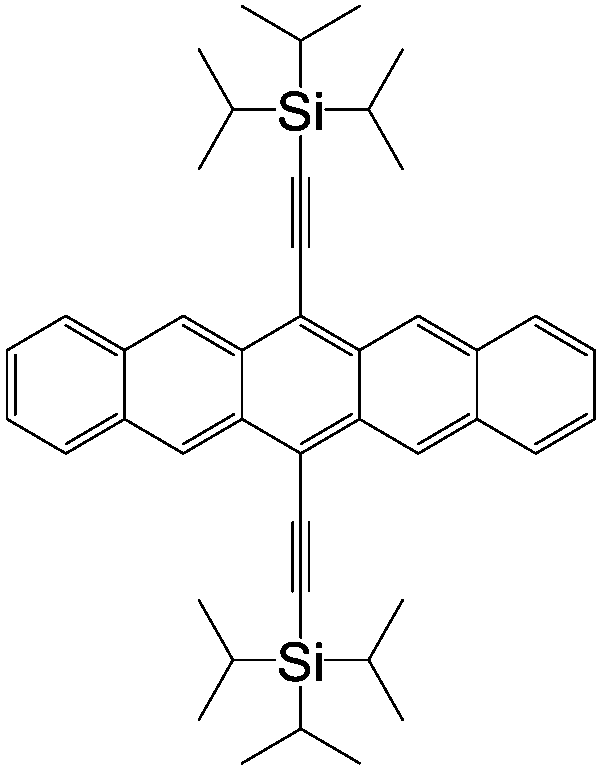
| CVB | |||||
|
LSMO/Co12 | 18 ± 3% at 14 K12 | Blue dopant (OLEDs) | HOMO = −5.5 eV | 10−3 |
| LUMO = −2.5 eV8 | p-Type8 | ||||
| αNPD | |||||
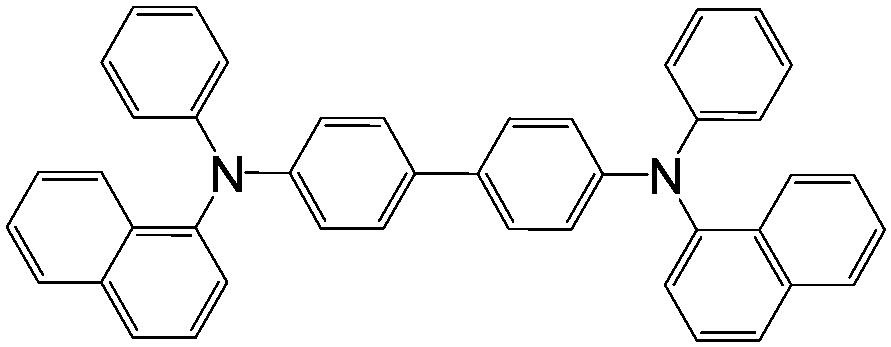
|
LSMO/Co12 | 14 ± 4% at 14 K12 | Hole transporting (OLEDs) | HOMO = −5.4 eV | 10−5 |
| LUMO = −2.3 eV8 | p-Type8 | ||||
| Tetraphenylporphyrin (TPP) | |||||
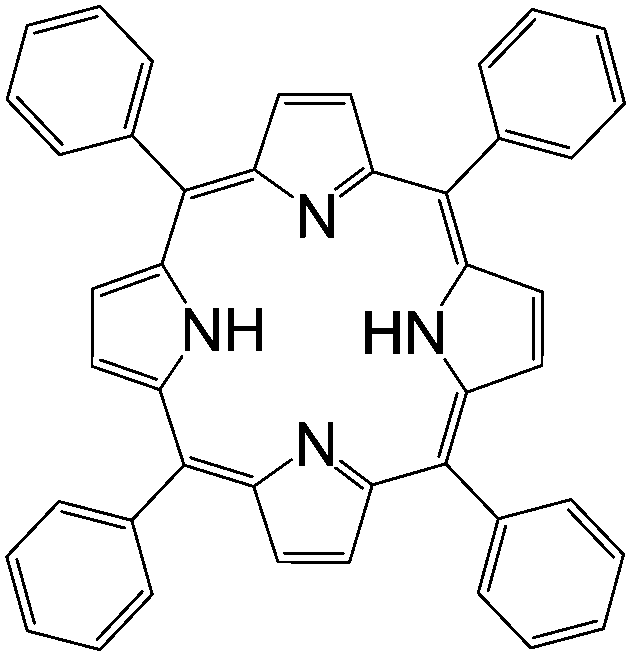
|
LSMO/Co41 | Red emitter (OLEDs) | HOMO = −4.9 eV | 10−5 | |
| LUMO = −3.1 eV42 | n-Type8 | ||||
| 1 H DOO-PPV | |||||
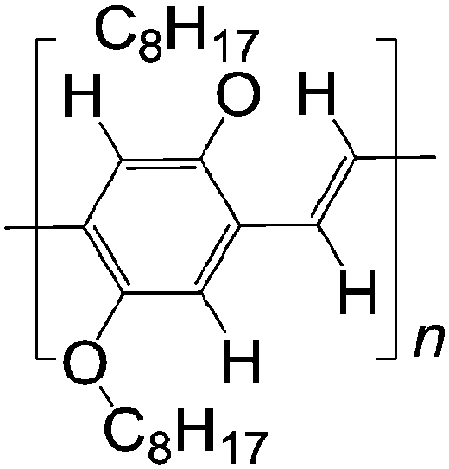
|
LSMO/Co21 | ≈2.5% at 10 K21 | Yellow emitter (OLEDs) | HOMO = −5.3 eV | 10−7 for MEH-PPV |
| L s ≈ 16 nm | LUMO = −3.0 eV43 | p-Type44 | |||
| 2 D DOO-PPV | |||||
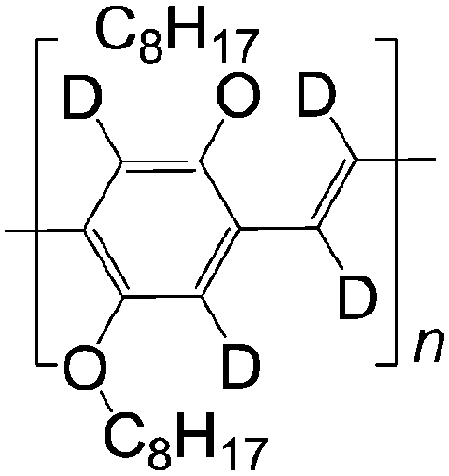
|
LSMO/Co21 | ≈18% at 10 K21 | Yellow emitter (OLEDs) | HOMO = −5.3 eV | |
| L s ≈ 49 nm | LUMO = −3.0 eV | ||||
| τ s is much longer than 1H DOO-PPV | |||||
| 13 C DOO-PPV | |||||
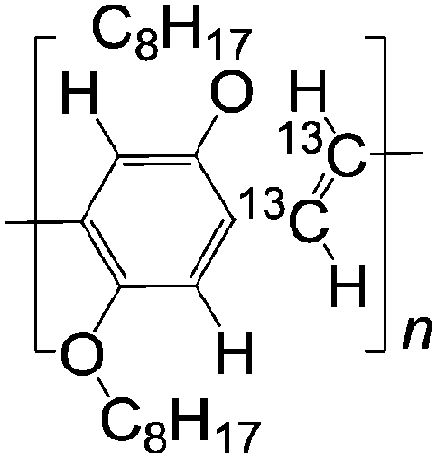
|
LSMO/Co21 | ≈2.5% at 10 K21 | Yellow emitter (OLEDs) | HOMO = −5.3 eV | |
| LUMO = −3.0 eV | |||||
A not unexpected result due the fact that the deuterium atom does not have a spin 1/2 nucleus, giving rise to a much smaller hyperfine interaction than hydrogen. Therefore, the strength of MR ratio and spin diffusion length at room temperature can be improved by appropriate selection of organic materials. The Fermi-level of the usual ferromagnetic electrodes ranges from 4.8 to 5.2 eV.15,24 A careful selection of organic semiconductors that are well-matched in their highest occupied molecular orbital (HOMO) or the lowest unoccupied molecular orbital (LUMO) to the electrodes is necessary for the fabrication of spin valves with large MR ratio and long spin-diffusion length. Fullerene C60 with a LUMO at −4.5 eV and P3HT with a HOMO at −5.1 eV, have shown relative long spin-diffusion lengths in spin valves.15,24 So far, there is no clear evidence whether the spin transport is more favorable in the HOMO or LUMO energy level. In this article, from the point of view of chemists, we suggest a range of interesting materials based on their structures, electrical properties and energy levels that should have a great impact on the performance and a better understanding of the role of organic materials in spin valves.
Charge transfer complexes
Charge transfer complexes are well known for their high electrical conductivity.45–47 The most famous complex is tetrathiafulvalene (TTF):tetracyanoquinodimethane (TCNQ)48 that crystallizes in segregated stacks. An electron from the HOMO of TTF is partially transferred to the LUMO of TCNQ, the HOMO of the complex, corresponding to TTF is at −4.8 eV49 and the LUMO, corresponding to that of TCNQ is at −4.6 eV.50 The TTF and TCNQ chains behave as one dimensional electronic systems, and at room temperature the material is highly conducting, showing metallic electrical conductance.51 Also, metallic conduction was observed at the interface of the crystals of TTF and TCNQ.52 Recently, TTF–TCNQ and other complexes have been used as source and drain in OFETs. In comparison to metal electrodes, these organics have shown improvement in terms of carrier mobility.53–55 The segregated stacked bis(ethylenedithio)tetrathiafulvalene (BEDT-TTF):TCNQ (formed by solution casting) exhibited balanced ambipolar field-effect mobility of 10−2 cm2 V−1 s−1.56 Recently, the mixed stacked charge-transfer complex of 1,4-bis(3,5-dimethylstyryl)-benzene (4M-DSB):(2Z,2′Z)-3,3′-(1,4-phenylene)bis(2-(3,5-bis-(trifluoromethyl)phenyl)acrylonitrile) (CN-TFPA) was fabricated into OFETs with ambipolar field-effect mobility of 6.7 × 10−3 and 6.7 × 10−2 cm2 V−1 s−1 for holes and electrons, respectively.57 All in all, charge-transfer complexes have shown their utility in OFETs such as relative high hole and electron mobilities. Also, the energy levels of the complex can be simply tuned to match with the FM electrodes in spin valves by a selection of different donor and acceptor. The conductivity and carrier mobility in charge transfer complexes are relative high, advantageous for longer spin-diffusion in spin valves. A caveat, however is that the metallic CT salts; e.g. TTF:TCNQ and BEDT-TTF:TCNQ, with their metallic conductivities do not exhibit a semiconductor energy gap that may be a necessary requirement for spin valves.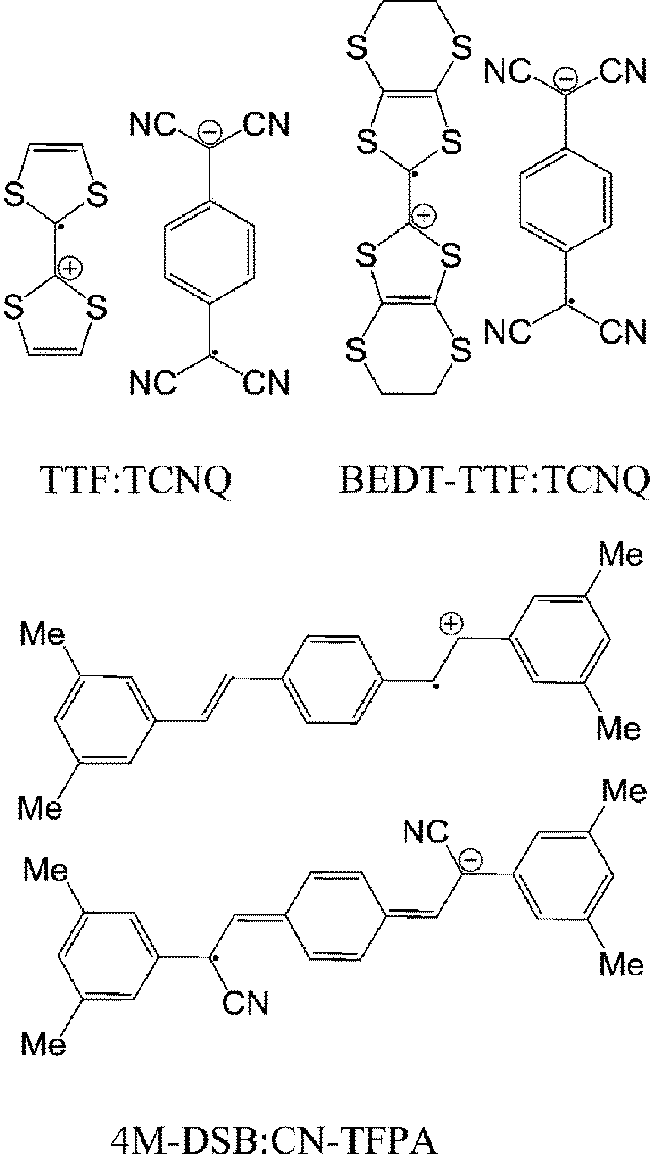
High mobility semiconducting polymers
The main attractions for polymeric FETs are low cost, mechanical flexibility and solution-processibility.58 The advent of organic semiconductors in OFETs has witnessed remarkable improvement in terms of carrier mobility and lifetime of the devices, as a result of combined efforts in syntheses and optimization of device fabrication. Up to date, the mobility of polymeric semiconductors has improved significantly, with a reported hole mobility up to 10.5 cm2 V−1 s−1,59 comparable to the state of the art, rubrene single crystal.The semiconducting polymers, P3HT and DOO-PPV, have already been investigated in spin valves under the same spin-polarized electrodes (LSMO/Co).21,24 The spin-diffusion length of P3HT is 80 nm, six times higher than that exhibited by DOO-PPV. It is difficult to draw a conclusion based on these results. But a partial answer to why P3HT is better than DOO-PPV can be gleaned from the electronic properties of the materials. The Fermi-level of LSMO is 4.8 eV, and the HOMO levels of P3HT and DOO-PPV are −4.9 and −5.3 eV, respectively. Therefore, spin injection from the electrode to P3HT should be easier than to DOO-PPV. More importantly, the hole-mobility of P3HT is much higher than DOO-PPV, 10−1versus 10−7 cm2 V−1 s−1. Hence, we naively suggest that longer spin-diffusion length can be obtained by a careful choice of polymeric semiconductors, based on their energy levels and carrier mobility but one should not forget that morphology and hyperfine interaction also play important roles in the devices.
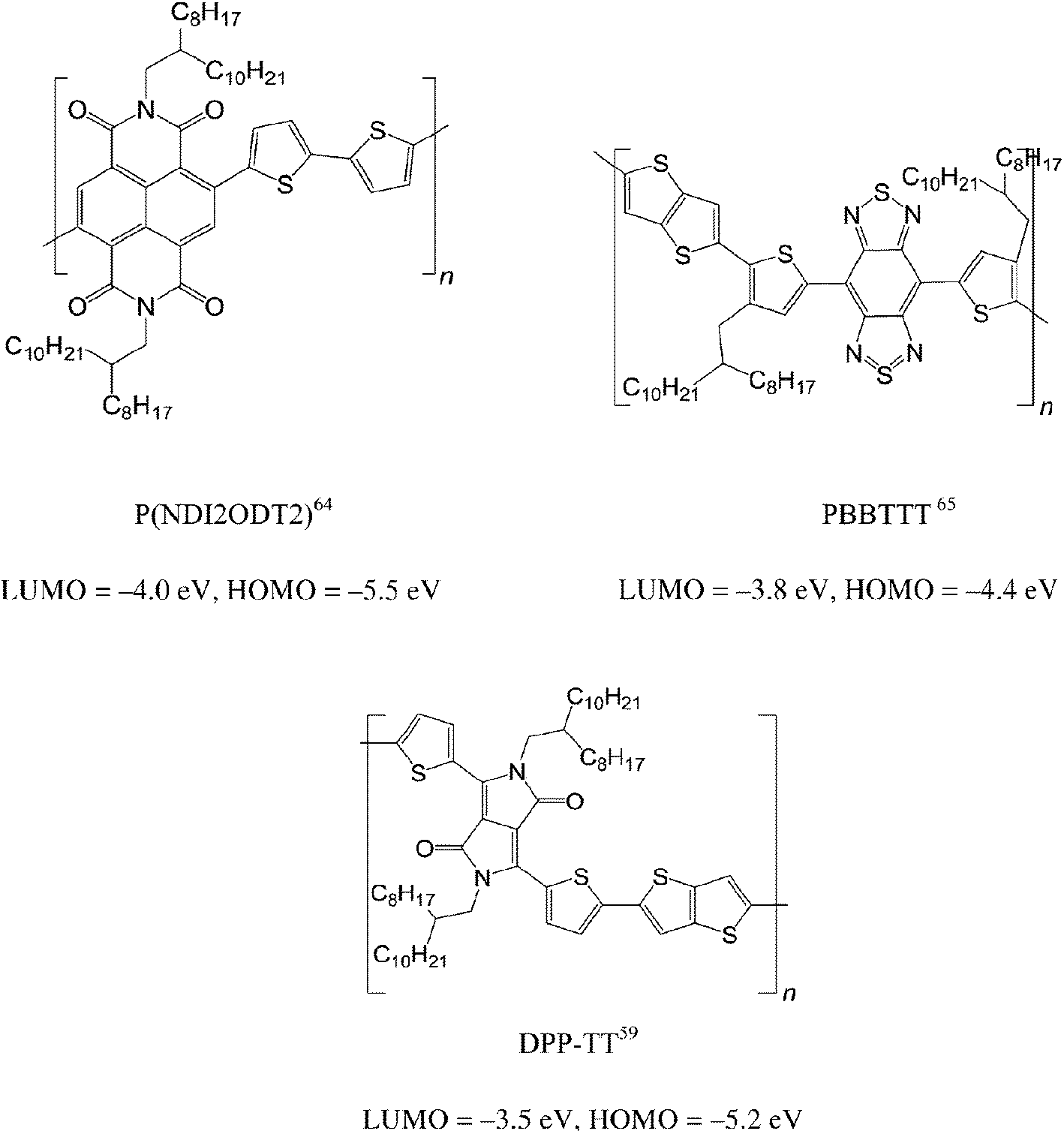
Recent progress in polymeric OFETs was reviewed, not only in terms of materials syntheses but also fundamental physics.60–63 Herein, we introduce examples of state of the art polymeric semiconductors with high carrier mobility. All these high performance polymers are alternating donor (D) and acceptor (A) copolymers, allowing the tuning of the LUMO (A) and HOMO (D) tuning. Something that would be impossible with homopolymers such as P3HT. In 2009, Yan et al. introduced a stable n-type polymer P(NDI2OD-T2), where the naphthalene diimide core acts as strong acceptor with an electron mobility up to 0.85 cm2 V−1 s−1.64 The HOMO and LUMO energy levels for this polymer are −4.0 and −5.5 eV, respectively. Since then, carrier mobilities of semiconducting polymers have been enhanced. In 2012, Fan et al. synthesized polymer PBBTTT [donor: bi(thiophen-2-yl)-thieno[3,2-b]thiophene (TT) and acceptor: benzobisthiadiazole bithiophene-thienothiophene (BBT)] with both hole and electron mobilities close to 1.0 cm2 V−1 s−1.65 Furthermore, the same group demonstrated a hole mobility improvement of the polymer PBBTTT to 2.5 cm2 V−1 s−1 by changing the donor unit of TT to quaterthiophene.66 The LUMO level of BBT-based polymers is around −3.8 eV, while the HOMO level can be tuned from −4.4 to −4.7 eV by changing the donor part. Polymer DPP-TT (acceptor: diketopyrrolopyrrole (DPP) and donor: TT) was demonstrated to have a record high hole mobility of 10.5 cm2 V−1 s−1, the highest value for polymer semiconductors and comparable to some of the best single crystal small molecules.59 Moreover, the same polymer has displayed ambipolar transport with balanced hole and electron mobility of 1.5 cm2 V−1 s−1.67 The HOMO and LUMO energy levels for the polymer are −3.5 and −5.2 eV, respectively. Summarizing, these D–A polymers have shown possible advantages over homopolymers for a spin valve application. It will be very exciting to see how the polymer with a hole mobility of 10.5 cm2 V−1 s−1 influences the spin-diffusion length in spin valve. Nevertheless, n-type and ambipolar polymers have yet to be studied in spin valves.
Zwitterions
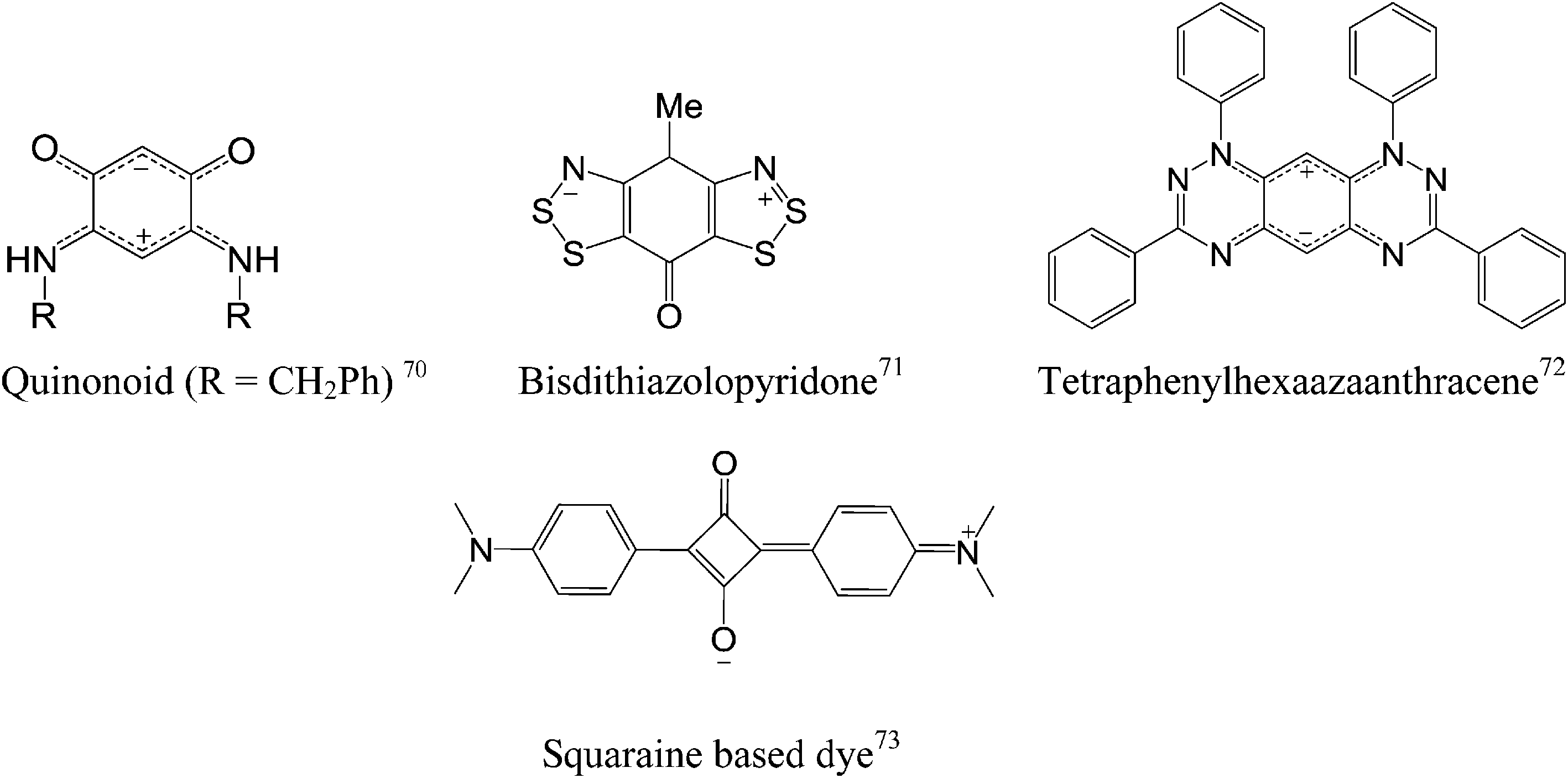
Recently, organic polymers possessing a large dipole moment have been used to change the work function of OFET contacts by decreasing the injection barrier between electrodes and the active layer in the devices.68 Moreover, a monolayer of small dipolar molecules displayed a similar function at the interface of electrodes and the active layer.69A zwitterion is an overall neutral molecule with a positive and a negative electrical charge at different locations within the molecule. Amino acids are the best-known examples of zwitterions. Only a handful of aromatic zwitterions have been found and studied such as quinonoid,70 bisdithiazolopyridone,71 tetraphenylhexaazaanthracene72 and squaraine.73 Among these zwitterions, only squaraine zwitterions have found their place in organic electronics such as solar cells.73 The rest of the mentioned molecules were less explored in organic electronic applications. The distinguishing feature of aromatic zwitterions is their large intrinsic dipole moment, close to 10 D.74 To put things in perspective, the dipole moment of potassium bromide is 10.4 D.
In quinonoid zwitterions, the positive and negative charges are delocalized as a nitrogen cyanine and an oxygen cyanine, respectively. Routaboul et al. have demonstrated film formation of quinonoid zwitterions on a gold surface and suggested a highly conductive interface with a metallic electrode can be formed, resulting in improved efficiency of carrier injection into an organic film.70 A study has also shown that quinonoid zwitterions, as a thin film, have electronic properties close to that of a semimetal.75
It is clear that molecules with large dipole moments have beneficial effects in organic electronics. For organic spin valves, incorporation of zwitterions should have a dramatic impact on spin injection from the ferromagnetic electrode to the spacer.
Spin-charge conversion: Inverse Spin-Hall Effect
The Spin-Hall Effect (SHE) is a transport phenomenon, predicted by M.I. Dyakonov and V.I. Perel,76 refers to the generation of a spin current transverse to an applied electric current in the absence of applied magnetic fields.77–79 In 2004, the Spin-Hall Effect was observed for the first time experimentally by Kato et al.80 Two years later, a phenomenon called Inverse Spin-Hall Effect (ISHE) that converts a pure spin current into an electric-charge current was successfully demonstrated.81 The importance of the ISHE observation is to link the forthcoming spintronics technology with the old fashioned electronics based on charge currents. The principle of ISHE is that the spin–orbit interaction causes a flow of spins to induce an electric field EISHE perpendicular to both the spin polarization σ and the flow direction of the spin current js: EISHE‖js × σ.81–83 The basic ISHE device structure is shown in Scheme 2.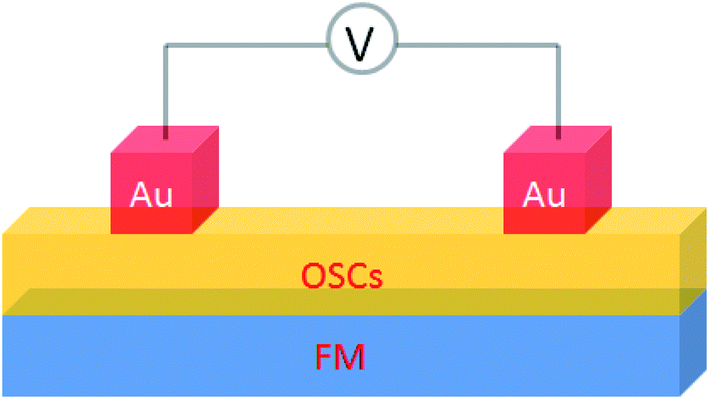 | ||
| Scheme 2 Device structure for spin-charge conversion. Spin-current is generated by the microwave excitation in the ferromagnetic electrode and then pumped into the OSCs. Finally, charge-current is detected at the electrodes as a result of ISHE. | ||
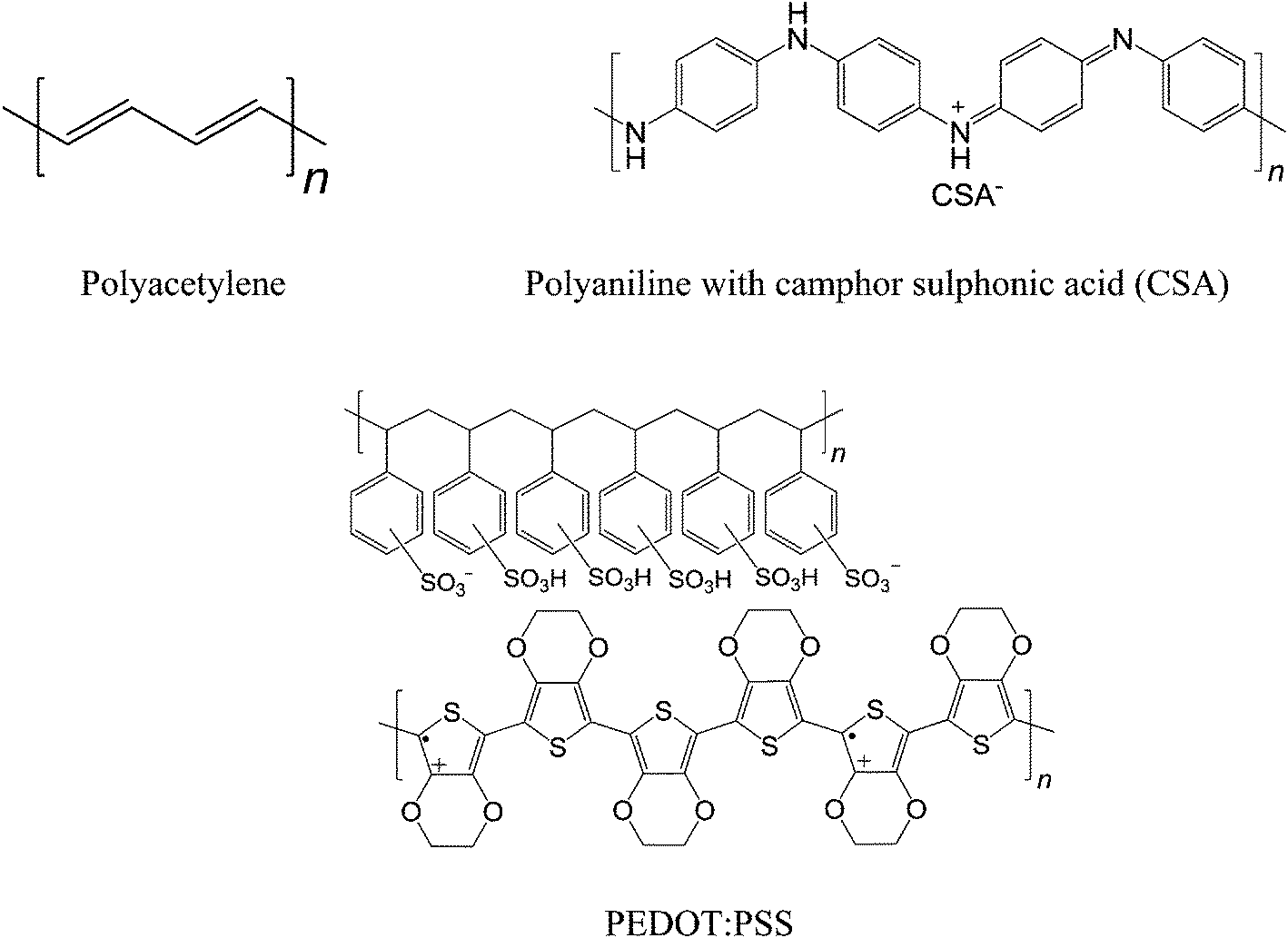
After the discovery of the ISHE phenomenon, the field developed into one of the hottest areas in spintronics. Scientists have focused on their ISHE studies in inorganic materials. Until very recently, Ando et al. have successfully observed the ISHE with polymeric semiconductor poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS), which displays higher spin-current to charge-current conversion efficiency and longer spin lifetimes in comparison to inorganic materials.14 Again, long spin lifetimes in organic materials are due to their low spin–orbit coupling and weak hyperfine interaction. The most popular conducting polymer for organic electronics is PEDOT:PSS, which has conductivity on the order of 103 S cm−1.84 Moreover, conducting polymers such as doped polyacetylene and polyaniline exhibit conductivities in the range of 103 to 104 S cm−1.85–87 Recently, Lee et al. have shown metallic transport in polyaniline.88 Polymers with high conductivity should find their place in the application to spin-charge conversion. Nevertheless, high conductive charge transfer complexes and high mobility polymeric semiconductors are expected to be promising candidates for spin-charge conversion.
Conclusion
Spintronics is a very attractive field for scientists. In the past 10 years, organic compounds have emerged as promising materials for spintronics due to their weaker spin–orbit and hyperfine couplings, unlike their inorganic counterparts. Spin valves have been the center for organic materials to study their spin-transporting properties. Nevertheless, a fascinating phenomenon of spin-charge conversion has shown a great potential to combine the forthcoming spintronics with conventional electronic based on electric currents. In this article we addressed the issues from a chemist's perspective to present the reader with a variety of interesting organic materials such as charge transfer complexes, high conductive/mobility polymers and zwitterions that are potentially applicable to this fast growing field.Acknowledgements
Yonghao Zheng was supported Department of Energy (Grant# DE-FG02-08ER46535) and WPI AIMR-CNSI joint center at University of California, Santa Barbara.References
- S. A. Wolf, D. D. Awschalom, R. A. Buhrman, J. M. Daughton, S. von Molnár, M. L. Roukes, A. Y. Chtchelkanova and D. M. Treger, Science, 2001, 294, 1488–1495 CrossRef CAS PubMed.
- S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC.
- D. Carsten and D. Vladimir, Rep. Prog. Phys., 2010, 73, 096401 CrossRef.
- R. A. J. Janssen and J. Nelson, Adv. Mater., 2013, 25, 1847–1858 CrossRef CAS PubMed.
- T. W. Kelley, P. F. Baude, C. Gerlach, D. E. Ender, D. Muyres, M. A. Haase, D. E. Vogel and S. D. Theiss, Chem. Mater., 2004, 16, 4413–4422 CrossRef CAS.
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS PubMed.
- L. E. Hueso, J. M. Pruneda, V. Ferrari, G. Burnell, J. P. Valdes-Herrera, B. D. Simons, P. B. Littlewood, E. Artacho, A. Fert and N. D. Mathur, Nature, 2007, 445, 410–413 CrossRef CAS PubMed.
- V. A. Dediu, L. E. Hueso, I. Bergenti and C. Taliani, Nat. Mater., 2009, 8, 707–716 CrossRef CAS PubMed.
- N. A. Morley, A. Rao, D. Dhandapani, M. R. J. Gibbs, M. Grell and T. Richardson, J. Appl. Phys., 2008, 103, 07F306 CrossRef.
- T. D. Nguyen, G. Hukic-Markosian, F. Wang, L. Wojcik, X.-G. Li, E. Ehrenfreund and Z. V. Vardeny, Nat. Mater., 2010, 9, 345–352 CrossRef CAS PubMed.
- Z. H. Xiong, D. Wu, Z. Valy Vardeny and J. Shi, Nature, 2004, 427, 821–824 CrossRef CAS PubMed.
- F. J. Wang, C. G. Yang, Z. V. Vardeny and X. G. Li, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 245324 CrossRef.
- M. Gobbi, F. Golmar, R. Llopis, F. Casanova and L. E. Hueso, Adv. Mater., 2011, 23, 1609–1613 CrossRef CAS PubMed.
- K. Ando, S. Watanabe, S. Mooser, E. Saitoh and H. Sirringhaus, Nat. Mater., 2013, 12, 622–627 CrossRef CAS PubMed.
- X. Zhang, S. Mizukami, T. Kubota, Q. Ma, M. Oogane, H. Naganuma, Y. Ando and T. Miyazaki, Nat. Commun., 2013, 4, 1392 CrossRef PubMed.
- J. S. Moodera, L. R. Kinder, T. M. Wong and R. Meservey, Phys. Rev. Lett., 1995, 74, 3273–3276 CrossRef CAS PubMed.
- D. D. Awschalom and M. E. Flatte, Nat. Phys., 2007, 3, 153–159 CrossRef CAS.
- S. Sanvito, Nat. Phys., 2010, 6, 562–564 CrossRef CAS.
- G. Schmidt, D. Ferrand, L. W. Molenkamp, A. T. Filip and B. J. van Wees, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, R4790–R4793 CrossRef CAS.
- M. Julliere, Phys. Lett. A, 1975, 54, 225–226 CrossRef.
- T. D. Nguyen, G. Hukic-Markosian, F. Wang, L. Wojcik, X.-G. Li, E. Ehrenfreund and Z. V. Vardeny, Synth. Met., 2011, 161, 598–603 CrossRef CAS.
- J. H. Shim, K. V. Raman, Y. J. Park, T. S. Santos, G. X. Miao, B. Satpati and J. S. Moodera, Phys. Rev. Lett., 2008, 100, 226603 CrossRef CAS PubMed.
- S. Mooser, J. F. K. Cooper, K. K. Banger, J. Wunderlich and H. Sirringhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235202 CrossRef.
- S. Majumdar, H. S. Majumdar, R. Laiho and R. Österbacka, J. Alloys Compd., 2006, 423, 169–171 CrossRef CAS.
- F. J. Wang, Z. H. Xiong, D. Wu, J. Shi and Z. V. Vardeny, Synth. Met., 2005, 155, 172–175 CrossRef CAS.
- S. Pramanik, C. G. Stefanita, S. Patibandla, S. Bandyopadhyay, K. Garre, N. Harth and M. Cahay, Nat. Nanotechnol., 2007, 2, 216–219 CrossRef CAS PubMed.
- H. Park, D.-S. Shin, H.-S. Yu and H.-B. Chae, Appl. Phys. Lett., 2007, 90, 202103 CrossRef.
- Y. Yang, F. Arias, L. Echegoyen, L. P. F. Chibante, S. Flanagan, A. Robertson and L. J. Wilson, J. Am. Chem. Soc., 1995, 117, 7801–7804 CrossRef CAS.
- K. Itaka, M. Yamashiro, J. Yamaguchi, M. Haemori, S. Yaginuma, Y. Matsumoto, M. Kondo and H. Koinuma, Adv. Mater., 2006, 18, 1713–1716 CrossRef CAS.
- Y. P. Liu, H. Idzuchi, Y. Fukuma, O. Rousseau, Y. Otani and W. S. Lew, Appl. Phys. Lett., 2013, 102, 033105 CrossRef.
- V. Dediu, M. Murgia, F. C. Matacotta, C. Taliani and S. Barbanera, Solid State Commun., 2002, 122, 181–184 CrossRef CAS.
- X. Gong, M. Tong, F. G. Brunetti, J. Seo, Y. Sun, D. Moses, F. Wudl and A. J. Heeger, Adv. Mater., 2011, 23, 2272–2277 CrossRef CAS PubMed.
- A. K. Pandey and J.-M. Nunzi, Appl. Phys. Lett., 2007, 90, 263508 CrossRef.
- H. Tatsuo and T. Jun, Sci. Technol. Adv. Mater., 2009, 10, 024314 CrossRef.
- S. Z. Bisri, T. Takenobu, T. Takahashi and Y. Iwasa, Appl. Phys. Lett., 2010, 96, 183304 CrossRef.
- M. Popinciuc, H. T. Jonkman and B. J. van Wees, J. Appl. Phys., 2006, 100, 093714–093718 CrossRef.
- S. Yoo, B. Domercq and B. Kippelen, Appl. Phys. Lett., 2004, 85, 5427–5429 CrossRef CAS.
- S. Lee, B. Koo, J. Shin, E. Lee, H. Park and H. Kim, Appl. Phys. Lett., 2006, 88, 162109 CrossRef.
- I. Kaur, W. Jia, R. P. Kopreski, S. Selvarasah, M. R. Dokmeci, C. Pramanik, N. E. McGruer and G. P. Miller, J. Am. Chem. Soc., 2008, 130, 16274–16286 CrossRef CAS PubMed.
- G. Giri, E. Verploegen, S. C. B. Mannsfeld, S. Atahan-Evrenk, D. H. Kim, S. Y. Lee, H. A. Becerril, A. Aspuru-Guzik, M. F. Toney and Z. Bao, Nature, 2011, 480, 504–508 CrossRef CAS PubMed.
- W. Xu, G. J. Szulczewski, P. LeClair, I. Navarrete, R. Schad, G. Miao, H. Guo and A. Gupta, Appl. Phys. Lett., 2007, 90, 072506 CrossRef.
- M. Neghabi, A. Behjat, B. B. Fatemeh Mirjalili and L. Zamani, Curr. Appl. Phys., 2013, 13, 302–306 CrossRef.
- I. H. Campbell, T. W. Hagler, D. L. Smith and J. P. Ferraris, Phys. Rev. Lett., 1996, 76, 1900–1903 CrossRef CAS PubMed.
- I. H. Campbell, D. L. Smith, C. J. Neef and J. P. Ferraris, Appl. Phys. Lett., 1999, 74, 2809–2811 CrossRef CAS.
- M. R. Bryce, Chem. Soc. Rev., 1991, 20, 355–390 RSC.
- M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891–4946 CrossRef CAS PubMed.
- U. Geiser and J. A. Schlueter, Chem. Rev., 2004, 104, 5203–5242 CrossRef CAS PubMed.
- J. Ferraris, D. O. Cowan, V. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95, 948–949 CrossRef CAS.
- Y. Yamashita, M. Tomura and M. B. Zaman, Chem. Commun., 1998, 1657–1658 RSC.
- J. Li, Y. Xiong, Q. Wu, S. Wang, X. Gao and H. Li, Eur. J. Org. Chem., 2012, 2012, 6136–6139 CrossRef CAS.
- M. J. Cohen, L. B. Coleman, A. F. Garito and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 1974, 10, 1298–1307 CrossRef CAS.
- H. Alves, A. S. Molinari, H. Xie and A. F. Morpurgo, Nat. Mater., 2008, 7, 574–580 CrossRef CAS PubMed.
- B. Mukherjee and M. Mukherjee, Langmuir, 2011, 27, 11246–11250 CrossRef CAS PubMed.
- Y. Takahashi, T. Hasegawa, Y. Abe, Y. Tokura, K. Nishimura and G. Saito, Appl. Phys. Lett., 2005, 86, 063504 CrossRef.
- Y. Takahashi, T. Hasegawa, Y. Abe, Y. Tokura and G. Saito, Appl. Phys. Lett., 2006, 88, 073504 CrossRef.
- M. Sakai, H. Sakuma, Y. Ito, A. Saito, M. Nakamura and K. Kudo, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 045111 CrossRef.
- S. K. Park, S. Varghese, J. H. Kim, S.-J. Yoon, O. K. Kwon, B.-K. An, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2013, 135, 4757–4764 CrossRef CAS PubMed.
- G. Malliaras and R. Friend, Phys. Today, 2005, 58, 53–58 CrossRef CAS.
- J. Li, Y. Zhao, H. S. Tan, Y. Guo, C.-A. Di, G. Yu, Y. Liu, M. Lin, S. H. Lim, Y. Zhou, H. Su and B. S. Ong, Sci. Rep., 2012, 2, 754 Search PubMed.
- L. Biniek, B. C. Schroeder, C. B. Nielsen and I. McCulloch, J. Mater. Chem., 2012, 22, 14803–14813 RSC.
- A. Facchetti, Chem. Mater., 2010, 23, 733–758 CrossRef.
- D. Natali and M. Caironi, Adv. Mater., 2012, 24, 1357–1387 CrossRef CAS PubMed.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dotz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed.
- J. Fan, J. D. Yuen, M. Wang, J. Seifter, J.-H. Seo, A. R. Mohebbi, D. Zakhidov, A. Heeger and F. Wudl, Adv. Mater., 2012, 24, 2186–2190 CrossRef CAS PubMed.
- J. Fan, J. D. Yuen, W. Cui, J. Seifter, A. R. Mohebbi, M. Wang, H. Zhou, A. Heeger and F. Wudl, Adv. Mater., 2012, 24, 6164–6168 CrossRef CAS PubMed.
- Z. Chen, M. J. Lee, R. Shahid Ashraf, Y. Gu, S. Albert-Seifried, M. Meedom Nielsen, B. Schroeder, T. D. Anthopoulos, M. Heeney, I. McCulloch and H. Sirringhaus, Adv. Mater., 2012, 24, 647–652 CrossRef CAS PubMed.
- Y. Zhou, C. Fuentes-Hernandez, J. Shim, J. Meyer, A. J. Giordano, H. Li, P. Winget, T. Papadopoulos, H. Cheun, J. Kim, M. Fenoll, A. Dindar, W. Haske, E. Najafabadi, T. M. Khan, H. Sojoudi, S. Barlow, S. Graham, J.-L. Brédas, S. R. Marder, A. Kahn and B. Kippelen, Science, 2012, 336, 327–332 CrossRef CAS PubMed.
- S. Khodabakhsh, B. M. Sanderson, J. Nelson and T. S. Jones, Adv. Funct. Mater., 2006, 16, 95–100 CrossRef CAS.
- L. Routaboul, P. Braunstein, J. Xiao, Z. Zhang, P. A. Dowben, G. Dalmas, V. Da Costa, O. Félix, G. Decher, L. G. Rosa and B. Doudin, J. Am. Chem. Soc., 2012, 134, 8494–8506 CrossRef CAS PubMed.
- S. M. Winter, R. J. Roberts, A. Mailman, K. Cvrkalj, A. Assoud and R. T. Oakley, Chem. Commun., 2010, 46, 4496–4498 RSC.
- K. Hutchison, G. Srdanov, R. Hicks, H. Yu, F. Wudl, T. Strassner, M. Nendel and K. N. Houk, J. Am. Chem. Soc., 1998, 120, 2989–2990 CrossRef CAS.
- C. Qin, W.-Y. Wong and L. Han, Chem.–Asian J., 2013, 8, 1706–1719 CrossRef CAS PubMed.
- P. Braunstein, O. Siri, J.-P. Taquet, M.-M. Rohmer, M. Bénard and R. Welter, J. Am. Chem. Soc., 2003, 125, 12246–12256 CrossRef CAS PubMed.
- L. G. Rosa, J. Velev, Z. Zhang, J. Alvira, O. Vega, G. Diaz, L. Routaboul, P. Braunstein, B. Doudin, Y. B. Losovyj and P. A. Dowben, Phys. Status Solidi B, 2012, 249, 1571–1576 CrossRef CAS PubMed.
- M. I. Dyakonov and V. I. Perel, Phys. Lett. A, 1971, 35, 459–460 CrossRef.
- J. E. Hirsch, Phys. Rev. Lett., 1999, 83, 1834–1837 CrossRef CAS.
- S. Zhang, Phys. Rev. Lett., 2000, 85, 393–396 CrossRef CAS PubMed.
- S. Murakami, N. Nagaosa and S.-C. Zhang, Science, 2003, 301, 1348–1351 CrossRef CAS PubMed.
- Y. K. Kato, R. C. Myers, A. C. Gossard and D. D. Awschalom, Science, 2004, 306, 1910–1913 CrossRef CAS PubMed.
- E. Saitoh, M. Ueda, H. Miyajima and G. Tatara, Appl. Phys. Lett., 2006, 88, 182509 CrossRef.
- T. Kimura, Y. Otani, T. Sato, S. Takahashi and S. Maekawa, Phys. Rev. Lett., 2007, 98, 156601 CrossRef CAS PubMed.
- S. O. Valenzuela and M. Tinkham, Nature, 2006, 442, 176–179 CrossRef CAS PubMed.
- Y. H. Kim, C. Sachse, M. L. Machala, C. May, L. Müller-Meskamp and K. Leo, Adv. Funct. Mater., 2011, 21, 1076–1081 CrossRef CAS.
- Y. S. Negi and P. V. Adhyapak, J. Macromol. Sci., Polym. Rev., 2002, 42, 35–53 CrossRef.
- C. K. Chiang, M. A. Druy, S. C. Gau, A. J. Heeger, E. J. Louis, A. G. MacDiarmid, Y. W. Park and H. Shirakawa, J. Am. Chem. Soc., 1978, 100, 1013–1015 CrossRef CAS.
- W.-S. Huang, B. D. Humphrey and A. G. MacDiarmid, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 2385–2400 RSC.
- K. Lee, S. Cho, S. Heum Park, A. J. Heeger, C.-W. Lee and S.-H. Lee, Nature, 2006, 441, 65–68 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |