Fabrication of one-dimensional heterostructured TiO2@SnO2 with enhanced photocatalytic activity†
Xin
Xu‡
a,
Guorui
Yang‡
b,
Jin
Liang
a,
Shujiang
Ding
*a,
Chengli
Tang
b,
Honghui
Yang
b,
Wei
Yan
bc,
Guidong
Yang
*d and
Demei
Yu
*a
aState Key Laboratory for Mechanical Behavior of Materials, MOE Key Laboratory for Nonequilibrium Synthesis and Modulation of Condensed Matter and Department of Applied Chemistry, School of Science, Xi'an Jiaotong University, Xi'an 710049, China. E-mail: dingsj@mail.xjtu.edu.cn; dmyu@mail.xjtu.edu.cn
bDepartment of Environmental Science & Engineering, Xi'an Jiaotong University, Xi'an 710049, China
cState Key Laboratory of Multiphase Flow in Power Engineering, Xi'an Jiaotong University, Xi'an 710049, China
dDepartment of Chemical Engineering, School of Chemical Engineering and Technology, Xi'an Jiaotong University, Xi'an, 710049, China. E-mail: guidongyang@mail.xjtu.edu.cn
First published on 15th October 2013
Abstract
TiO2@SnO2 nanosheets@nanotubes heterostructures were successfully prepared by a facile two-step method: prefabricated SnO2@PNT coaxial nanocables based on the in situ growth of SnO2 in the sulfonated gel matrix of polymeric nanotubes, and then the assembly of TiO2 nanoclusters that consist of ultrathin nanosheets through a solvothermal process. These heterostructures were characterized for the morphological, structural and optical properties by scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), UV-visible (UV-vis) and diffuse reflectance spectroscopy (DRS). The photocatalytic investigations showed that the TiO2@SnO2 heterostructures possessed enhanced photocatalytic efficiency in the photodegradation of Rhodamine B (RhB) and photocatalytic H2 evolution from water splitting under ultraviolet (UV) light irradiation, compared with the pristine TiO2 nanosheets, SnO2 nanotubes, the mechanically mixed two samples and P25. The enhanced photocatalytic performance can be ascribed to the beneficial microstructure and synergistic effects of coupled TiO2@SnO2 nanosheets@nanotubes heterostructures.
1. Introduction
Photocatalysis has attracted a great deal of attention as a promising candidate for the elimination of organic contaminants in water and air, since the pioneering work by Honda and Fujishima.1 Titanium dioxide (TiO2) has and continues to be one of the most widely studied semiconductors for photocatalytic applications due to its advantages, such as a suitable band gap for redox reactions, long-term stability, low cost, non-toxicity and so on.2,3 However, as a single component semiconductor photocatalyst, TiO2 has an inherent drawback that the photogenerated electron/hole (e−/h+) pairs recombine fast (∼10 ns)4 and hence only a fraction of e−/h+ pairs are available for the photoreaction. To date, this disadvantage can be overcome by means of semiconductor coupling.5 With this approach, the assembled semiconductors are selected suitably so that efficient charge transfer occurs between them. This condition spatially separates the photoexcited electrons and holes onto the different constituents and reduces the rate of e−/h+ pair recombination.6In recent years, various coupled semiconductor systems based on TiO2 have been developed, such as SnO2/TiO2,7–10 ZnO/TiO2,11 WO3/TiO2,12 CdS/TiO2,13 Fe2O3/ZnO14 and so on. In particular, the SnO2/TiO2 system with high photocatalytic activity has attracted extensive interest. SnO2 possesses a high electron mobility (∼100–200 cm2 V−1 s−1),15 indicating a faster transport of photoexcited electrons.16 Moreover, the conduction-band edge of SnO2 is more positive than that of TiO2, leading to the migration of photoexcited electrons from the conduction band of TiO2 to that of SnO2,17–20 consequently the recombination of charge carriers can be suppressed, thereby resulting in a higher photocatalytic performance.21–23 Aerosol assisted chemical vapour deposition was also introduced to synthesize a TiO2–SnO2 composite film, and the as-prepared TiO2–SnO2 film exhibited enhanced photocatalytic activity over the degradation of dye compared to the TiO2 film and SnO2 film.22 Gao et al. demonstrated that the power conversion efficiency value of the SnO2–TiO2 core–shell structured DSSCs is above five times higher than that of SnO2 nanotubes.24 Zhang et al. reported a novel-structured TiO2/SnO2 hybrid nanofiber photocatalyst with enhanced photocatalytic activity prepared via the electrospinning method, the heterostructures promote the photogenerated e−/h+ pairs separation.25
To date, numerous types of coupled SnO2/TiO2 systems, such as particle,26 composite film,27 stacks,15 core–shell heterostructures28 and so on, have been successfully obtained. However the particle-like morphology photocatalyst is very inconvenient for recycling due to its extreme dispersive property and it may cause secondary pollution. On the other hand, the film-type photocatalyst increases the recovery efficiency at the expense of the specific surface area, resulting in a low photocatalytic activity. One-dimensional (1D) structures, such as nanowires, nanobelts and nanotubes, have attracted tremendous attention within the last decade. One promising approach to overcoming these drawbacks simultaneously is to use 1D photocatalysts, which provide a relatively large specific surface area as compared with nanoparticles, and are recycled easily due to the large length to diameter ratio. Among the huge variety of 1D nanostructures, semiconducting nanotubes are particularly interesting, not only for fundamental research due to the unique hollow structural and physical properties relative to their bulk counterparts, but also for their fascinating potential in optoelectronic and electronic devices. More importantly, it has been proven that the 1D nanomaterials effectively support the direct growth of secondary nanostructures by heterogeneous nucleation because of their porous surface and large surface area.29–31
In this work, we employ polymeric nanotubes (PNTs) with uniform size, synthesized by the cationic polymerization of divinylbenzene using immiscible initiator nanodroplets of boron trifluoride etherate complex as hard templates after sulfonation.32 The synthetic process is illustrated in Scheme 1. Firstly, we prefabricated SnO2@PNT coaxial nanocables based on the in situ growth of SnO2 in the sulfonated gel matrix of polymeric nanotubes.33–37 Then the SnO2@PNT coaxial nanocables were assembled by TiO2 nanoclusters that consist of ultrathin nanosheets after a solvothermal process. After being calcined in air, the hierarchically structured TiO2@SnO2 composite was successfully obtained. Moreover, the uniform PNTs used in this work have three main advantages as templates: (i) the positively charged precursor ions can be easily adsorbed to the surface of the sulfonated PNTs due to the electrostatic interaction with the negative functional groups (–SO3); (ii) PNTs can be easily removed through calcination at a lower temperature, by which the nanostructure of the metal oxide can be well preserved; (iii) the produced gas and osmotic pressure during the calcination can be released through the inner channels and open ends of the PNTs.33
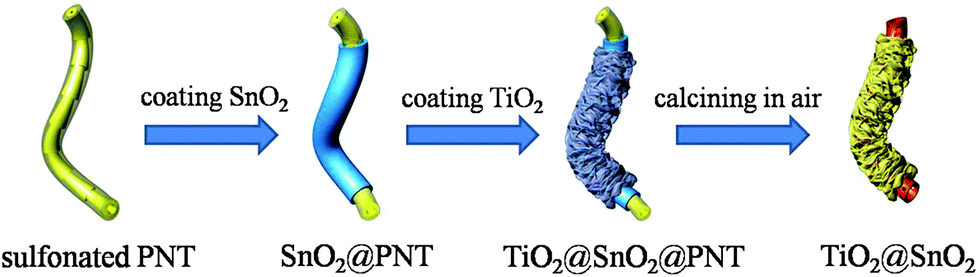 | ||
| Scheme 1 Schematic illustration of the synthetic procedure of the TiO2@SnO2 composite. | ||
2. Experimental
2.1 Material synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, w/w) and the mixture was ultrasonicated for 10 min to disperse them. After stirring for 24 h at 40 °C the yellow precipitate was collected by centrifugation and washed thoroughly with ethanol.
:30, w/w) and the mixture was ultrasonicated for 10 min to disperse them. After stirring for 24 h at 40 °C the yellow precipitate was collected by centrifugation and washed thoroughly with ethanol.
2.2 Characterization
The product morphology was examined using field-emission scanning electron microscopy (FESEM; HITACHI, su-8010) and transmission electron microscopy (TEM; JEOL, JEM-2100). Crystallographic information of the samples was collected using powder X-ray diffraction (XRD; SHIMADZU, Lab X XRD-6000). The chemical states of the products were studied using the X-ray photoelectron spectroscopy (XPS) measurement performed on an Axis Ultra, Kratos (UK) at monochromatic Al Kα radiation (150 W, 15 kV and 1486.6 eV). The vacuum in the spectrometer was 10–9 Torr. Binding energies were calibrated relative to the C 1s peak (284.6 eV). Thermogravimetric analysis (Perkin-Elmer TGA 7) was carried out under a flow of air with a temperature ramp of 20 °C min−1 from room temperature to 600 °C. The specific surface area and pore size distribution of the products were measured using a BET analyzer (ASAP 2020M) at 77 K.2.3 Photocatalytic tests
The photocatalytic performance of the as-fabricated products was studied by degrading Rhodamine B (RhB) in simulated wastewater under UV irradiation. The photocatalytic experiments were conducted in a homemade quartz photochemical reactor with a quartz jacket and cooled off by circulating water. In the experiment, a 500 W xenon lamp with a cutoff filter (λ < 420 nm) was employed as the ultraviolet light source; the distance between the xenon lamp and the front surface of the reactor was fixed to be 8 cm. For comparison, the mechanically mixed SnO2 nanotubes with TiO2 nanosheets (the weight ratio of SnO2: TiO2 = 1:4, which is similar to that of TiO2@SnO2 composite), and which is labeled as SnO2 + TiO2, was studied as a reference.| %RhB concentration = C/C0 × 100% |
3. Results and discussion
Fig. 1 shows the morphology of the acid-treated PNTs and SnO2@PNT composite. It can be seen from the scanning electron microscope (SEM) image of the acid-treated PNTs that these templates are about 100–250 nm in diameter (Fig. 1A). The transmission electron microscopy (TEM) image indicates that the acid-treated PNTs possess an extremely smooth surface (Fig. 1B). The morphology of the SnO2@PNT is shown in Fig. 1C and D. It can be observed that the diameter of the acid-treated PNTs increases to approximately 125–275 nm (Fig. 1C), the surface of the SnO2@PNT composite becomes much rougher than the PNTs before coating with SnO2, and the thickness of the SnO2 layer is approximately 15–25 nm (Fig. 1D). | ||
| Fig. 1 (A) SEM image and (B) TEM image of the sulfonated PNTs; (C) SEM image and (D) TEM image of the SnO2@PNT composite. | ||
Fig. 2 shows the SEM and TEM images of the TiO2@SnO2@PNT and TiO2@SnO2 composites. The TiO2@SnO2@PNT composite displays a tubular structure with a diameter of around 500–600 nm (Fig. 2A). The TEM image shows that the SnO2@PNT nanotubes are uniformly assembled by TiO2 nanoclusters that consist of ultrathin nanosheets (Fig. 2B), and the heights of these TiO2 nanosheets are about 225–475 nm through calculation. A hierarchical structure of the TiO2@SnO2@PNT composite's surface can be clearly revealed from the TEM image (Fig. 2C). The formation of the TiO2 nanosheets could be explained by the dual function of diethylenetriamine (DETA): catalyzing the hydrolysis of titanium (IV) is opropoxide (TIP) to form TiO2 nanosheets and assisting the self-assembly of the formed TiO2 nanostructures on the surface of SnO2@PNT, possibly through electrostatic interactions.38,39Fig. 2D shows the SEM image of the TiO2@SnO2 composite obtained after calcining the TiO2@SnO2@PNT composite at 450 °C for 2 h. The diameter of the hierarchically structured TiO2@SnO2 composite is approximately 450–550 nm, smaller than the TiO2@SnO2@PNT composite, possibly because of the partial collapse of the hollowed TiO2@SnO2 structure during the annealing process. The calcined TiO2@SnO2 composite maintains its tubular and sheet-like structures after removing the PNT templates. The TEM image of the calcined TiO2@SnO2 composite displays that the sample has an extra porous structure of the inside nanotubes, and the thickness of the TiO2 nanosheets is approximately 10 nm (Fig. 2E and F). It is noteworthy that the ultrasonic process before TEM observation was unable to cause TiO2 nanosheets to fall off from the SnO2 nanotubes, which may suggest that a compact interface was established between the TiO2 and SnO2. As shown in the HRTEM image of Fig. S1,† we can see the clear interface between the SnO2 and TiO2. Moreover, the XPS data further confirmed the composition of the heterostructured TiO2@SnO2 composite (Fig. S2†).
 | ||
| Fig. 2 (A) SEM image; (B and C) TEM images of TiO2@SnO2@PNT composite; (D) SEM image; (E and F) TEM images of TiO2@SnO2 composite after calcining the TiO2@SnO2@PNT composite at 450 °C for 2 h. | ||
The chemical composition of the different samples was characterized by using X-ray diffraction (XRD; Fig. 3A). It is clear from the patterns of curve I and curve II that pure TiO2 nanosheets and pristine SnO2 nanotubes can be assigned to anatase TiO2 (JCPDS card no. 21-1272) and rutile SnO2 (JCPDS card no. 41-1445). All the diffraction peaks of curve I and curve II can be discovered from curve III clearly, indicating that SnO2 and TiO2 successfully formed on the surface of acid-treated PNTs. The weight fraction of SnO2 and TiO2 in the hybrid material was determined by thermogravimetric analysis (TGA), with the results shown in Fig. 3B. The SnO2@PNT and TiO2@SnO2@PNT composites show significant weight loss at about 450 °C, which is attributed to the combustion of the acid-treated PNT templates. After reaching 600 °C, the SnO2@PNT and TiO2@SnO2@PNT composites show a total weight loss of 79.3% and 44.0%, respectively. The mass ratio of SnO2 and TiO2 calculated from the two curves is about 1:4.
 | ||
| Fig. 3 (A) XRD patterns of the TiO2 nanosheets (I), SnO2 nanotubes (II) and TiO2@SnO2 composite and (B) TGA analysis of the pure sulfonated PNTs (I), SnO2@PNT composite (II) and TiO2@SnO2@PNT composite (III). | ||
Fig. 4A shows the solid diffuse reflection UV-vis spectrum (DRS) of the SnO2 nanotubes, TiO2 nanosheets and TiO2@SnO2 composite. A strong absorption edge at ca. 395 nm appears in the absorption spectrum of the TiO2@SnO2 composite, while the onset of the absorption of the TiO2 nanosheets and SnO2 nanotubes were 385 nm and 375 nm. According to the formula of the band gap (Eg = 1240/λ),13 the band-gap values of TiO2@SnO2 composite, TiO2 nanosheets and SnO2 nanotubes were calculated to be 3.15 eV, 3.21 eV and 3.42 eV. The absorption edge of the TiO2@SnO2 composite was red-shifted compared to the TiO2 nanosheets and SnO2 nanotubes, which reflects that the electronic structure and optical properties of the bare TiO2 or SnO2 have been modified. This is presumably because the TiO2 was doped by a fraction of the Sn on the interface between the TiO2 and SnO2 during the final annealing process,40 or the effective coupling of SnO2 and TiO2,11 and results in the red-shifting of the absorption edge in the TiO2@SnO2 composite.
 | ||
| Fig. 4 (A) DRS of the SnO2 nanotubes, TiO2 nanosheets and TiO2@SnO2 composite, (B) degradation profiles of RhB over different samples: SnO2 nanotubes (□), TiO2 nanosheets (○), SnO2 + TiO2 mixture (■), P25 (●), TiO2@SnO2 composite (*), without photocatalyst (◑) and without UV light (◐). | ||
Fig. 4B shows the degradation profiles of the Rhodamine B (RhB) wastewater under ultraviolet light (UV) with various photocatalysts. As one can see, the degradation of RhB by the pure SnO2 nanotubes is the least significant among all the catalysts, and only about 82.0% of RhB was degraded in 60 min. This is primarily ascribed to the wide band gap of SnO2, leading to a limited absorption of photons. In comparison, the samples of TiO2 nanosheets and mechanically mixed SnO2 nanotubes with TiO2 nanosheets exhibit higher activities, with a degradation rate of 88.9% and 90.3% for RhB, respectively. The improved activity can be assigned to the narrow band gap of the TiO2. The degradation efficiency of P25 reaches 94.5%, which is higher than the TiO2 nanosheets due to the coexistence and optimal ratio of rutile and anatase TiO2. The TiO2@SnO2 composite exhibits the best performance of the photodegradation of RhB among the as-prepared samples, and almost 100% of the RhB molecules decomposed in 60 min. To investigate the degradation kinetics of RhB in solution and quantitatively compare the photocatalytic performances of these samples, the pseudo first-order equation was adopted to describe the experimental data because of the low initial concentrations of the substrates as follows: ln(C0/C) = kt, where k is a reaction rate constant. As can be seen in Fig. S3,† the order of the k values, which is derived from the plots of ln(C0/C) versus irradiation time (t), is summarized as follows: TiO2@SnO2 composite > P25 > TiO2 + SnO2 mixture > TiO2 nanosheets > SnO2 nanotubes. This is in good agreement with the results presented in Fig. 4B. In addition to organic pollutants removal, photocatalytic H2 generation was also observed over the SnO2@TiO2 heterostructures. As illustrated in Fig. 5, the H2 evolution over the SnO2@TiO2 heterostructures was significantly higher than that of the bare SnO2 nanotubes, pristine TiO2 nanosheets, the mechanically mixed two samples and P25 throughout the 6 h of UV light irradiation, which is similar to the photocatalytic efficiency. These outcomes demonstrated that the SnO2@TiO2 heterostructures obtained in this work possess excellent properties in both photo-oxidation and photo–reduction reactions.
 | ||
| Fig. 5 Photocatalytic H2 production efficiency of samples TiO2@SnO2, TiO2 + SnO2, TiO2 nanosheets, SnO2 nanotubes and P25. | ||
The reasons for the high photocatalytic performance of the sample TiO2@SnO2 could be explained in detail as follows: (i) SnO2@TiO2 heterostructures have a large specific surface area (124 m2 g−1) compared with SnO2 nanotubes (102 m2 g−1).33 As is known, nanomaterials with larger specific surface areas could offer more active sites and absorb more oxygen, which leads to a higher photocatalytic performance.9 (ii) The band alignment between SnO2 and TiO2 is generally classified as a type II structure (staggered band alignment between these two different materials).41 In this band-gap configuration, when photogenerated e−/h+ pairs are generated in SnO2 nanotubes and TiO2 nanosheets, the electrons on the TiO2 particle surface transfer swiftly to the conduction band of SnO2via interfaces; similarly, the holes on the SnO2 surface migrate to TiO2 owing to the different valence band edge. The probability of the recombination of the photogenerated e−/h+ pairs is retarded at the same time. When the heterojunctions between SnO2 and TiO2 are formed, the Fermi energies in the boundary between the SnO2 and TiO2 phases should be matched.42 This leads to the potential of the conduction band and valence band of SnO2 to be more negative.43,44 (see Fig. S2 E in the ESI†) Therefore, the conduction band position of SnO2 will meet the requirements of the hydrogen evolution reaction.45 Moreover, the nanosheets/nanotubes-type structure possesses a large interfacial region, which can improve the charge carrier separation to a large extent.46 (iii) The nanosheets-nanotubes structures could improve the photon utilization efficiency. When the surfaces of the TiO2@SnO2 composite was irradiated with light, some of the incident photons were directly absorbed by SnO2 and TiO2, some were reflected and scattered, multiplied by the nanotubes and nanosheets. Hence, along with the increase in effective optical path length, the photon–matter interaction length multiplies, leading to a higher light harvesting based on the Beer–Lambert Law.47–49 (iv) The unique 1D TiO2@SnO2 heterostructures obtained in this work could prevent the photo induced agglomeration and result in low boundary density,50 thereby leading to the decrease of the recombination centre. This could be proven by the SEM images of the sample after catalytic reaction (Fig. S4†). (v) The TiO2@SnO2 composite has an obvious red shift of ca. 10–20 nm in the absorption edge, compared with the pure SnO2 nanotubes and TiO2 nanosheets. This larger absorption edge would lead to the enhancement of the photocatalytic performance of the TiO2@SnO2 composite.51
From the viewpoint of practical application, it is crucial that the as-prepared photocatalyst maintains a high activity and stability for long-term use. As Fig. 6 illustrates, the TiO2@SnO2 composite photocatalyst possessed a sustainable photocatalytic performance in H2 generation after 4 cycles. There was insignificant loss of the H2 evolution rate, which indicates that the TiO2@SnO2 heterostructures were highly stable and reusable. The SEM images of the TiO2@SnO2 after the photocatalytic process display that the TiO2@SnO2 still retain the original morphology even after four recycles without any detected agglomeration or damage (Fig. S4†). Moreover, at the end of the reaction when the mixing was stopped, the TiO2@SnO2 composite photocatalyst can be easily separated from the solution by sedimentation, thus indicating it has good separation characteristics, which may be due to the large length to diameter ratio of the TiO2@SnO2 lD heterostructures.
 | ||
| Fig. 6 Reuse cycles of photocatalytic H2 generation over the sample TiO2@SnO2 composite; every 6 h the reaction system was bubbled with N2 for 30 min to remove the H2 inside. | ||
4. Conclusions
In summary, a TiO2@SnO2 composite has been successfully prepared by a facile two-step method. SEM, TEM, XRD and XPS analysis revealed the successful coating of TiO2 nanosheets on the surface of SnO2 nanotubes. In comparison with the uncoated SnO2 nanotubes, pure TiO2 nanosheets, the mechanically mixed two samples and P25, the TiO2@SnO2 composite exhibited the best photocatalytic performance for the decomposition of RhB and photocatalytic H2 production over water splitting under UV light irradiation. In particular, 1D TiO2@SnO2 composites processed several excellent properties including a relatively large specific surface area for supplying abundant active sites, suitable band engineering for charge carriers separation and unique 1D heterostructures for resistance to photo induced agglomeration. We assumed that the synergistic effects of the aforementioned factors contributed to the enhance photocatalytic performance of the 1D TiO2@SnO2 heterostructures.Acknowledgements
This research was supported partially by the National Natural Science Foundation of China (no. 51273158, 21303131); Natural Science Basis Research Plan in Shaanxi Province of China (no. 2012JQ6003); Fundamental Research Funds for the Central Universities of China (2011JDGZ15, xjj2012092, 2012jdhz34) and Natural science fund of Jiangsu Province, China (SBK201022919).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- H. B. Wu, H. H. Hng and X. W. Lou, Adv. Mater., 2012, 24, 2567–2571 CrossRef CAS PubMed.
- J. S. Chen, C. P. Chen, J. Liu, R. Xu, S. Z. Qiao and X. W. Lou, Chem. Commun., 2011, 47, 2631–2633 RSC.
- R. Sasikala, A. Shirole, V. Sudarsan, T. Sakuntala, C. Sudakar, R. Naik and S. R. Bharadwaj, Int. J. Hydrogen Energy, 2009, 34, 3621–3630 CrossRef CAS PubMed.
- L. Xu, E. M. P. Steinmiller and S. E. Skrabalak, J. Phys. Chem. C, 2012, 116, 871–877 CAS.
- W. L. Yang, L. Zhang, Y. Hu, Y. J. Zhong, H. B. Wu and X. W. Lou, Angew. Chem., Int. Ed., 2012, 51, 11501–11504 CrossRef CAS PubMed.
- H. A. J. L. Mourão, W. A. Junior and C. Ribeiro, Mater. Chem. Phys., 2012, 135, 524–532 CrossRef PubMed.
- K. Vinodgopal, I. Bedja and P. V. Kamat, Chem. Mater., 1996, 8, 2180–2187 CrossRef CAS.
- H. Tada, A. Hattori, Y. Tokihisa, K. Imai, N. Tohge and S. Ito, J. Phys. Chem. B, 2000, 104, 4585–4587 CrossRef CAS.
- Y. A. Cao, X. T. Zhang, W. S. Yang, H. Du, Y. B. Bai, T. J. Li and J. N. Yao, Chem. Mater., 2000, 12, 3445–3448 CrossRef CAS.
- P. F. Du, L. X. Song, J. Xiong, N. Li, Z. Q. Xi, L. C. Wang, D. L. Jin, S. Y. Guo and Y. F. Yuan, Electrochim. Acta, 2012, 78, 392–397 CrossRef CAS PubMed.
- B. Lu, X. Li, T. Wang, E. Xie and Z. Xu, J. Mater. Chem. A, 2013, 1, 3900–3906 CAS.
- C. L. Li, J. Yuan, B. Y. Han and W. F. Shangguan, Int. J. Hydrogen Energy, 2011, 36, 4271–4279 CrossRef CAS PubMed.
- Y. Liu, L. Yu, Y. Hu, C. F. Guo, F. M. Zhang and X. W. Lou, Nanoscale, 2012, 4, 183–187 RSC.
- F. S. Cai, Z. H. Yuan, Y. Q. Duan and L. J. Bie, Thin Solid Films, 2011, 519, 5645–5648 CrossRef CAS PubMed.
- C. Gao, X. Li, B. Lu, L. Chen, Y. Wang, F. Teng, J. Wang, Z. Zhang, X. Pan and E. Xie, Nanoscale, 2012, 4, 3475–3481 RSC.
- H.-J. Wang and S.-C. Lee, Mater. Trans., 2009, 50, 2329–2334 CrossRef CAS.
- S. Y. Chang, S. F. Chen and Y. C. Huang, J. Phys. Chem. C, 2011, 115, 1600–1607 CAS.
- Q. Gu, J. L. Long, Y. G. Zhou, R. S. Yuan, H. X. Lin and X. X. Wang, J. Catal., 2012, 289, 88–99 CrossRef CAS PubMed.
- S. N. Chai, G. H. Zhao, P. Q. Li, Y. Z. Lei, Y. N. Zhang and D. M. Li, J. Phys. Chem. C, 2011, 115, 18261–18269 CAS.
- S. Shah, M. C. Benson, L. M. Bishop, A. M. Huhn, R. E. Ruther, J. C. Yeager, Y. Z. Tan, K. M. Louis and R. J. Hamers, J. Mater. Chem., 2012, 22, 11561–11567 RSC.
- S. Ponja, S. Sathasivam, N. Chadwick, A. Kafizas, S. M. Bawaked, A. Y. Obaid, S. Al-Thabaiti, S. N. Basahel, I. P. Parkin and C. J. Carmalt, J. Mater. Chem. A, 2013, 1, 6271–6278 CAS.
- L. Xu, E. M. P. Steinmiller and S. E. Skrabalak, J. Phys. Chem. C, 2012, 116, 871–877 CAS.
- C. T. Gao, X. D. Li, B. G. Lu, L. L. Chen, Y. Q. Wang, F. Teng and J. T. Wang, Nanoscale, 2012, 4, 3475–3481 RSC.
- R. Zhang, H. Wu, D. D. Lin and W. Pan, J. Am. Ceram. Soc., 2009, 92, 2463–2466 CrossRef CAS.
- M. Hirano, H. Dozono and T. Kono, Mater. Res. Bull., 2011, 46, 1384–1390 CrossRef CAS PubMed.
- E. M. El-Maghraby, Phys. B, 2010, 405, 2385–2389 CrossRef CAS PubMed.
- A. M. Nie, J. B. Liu, Q. Q. Li, Y. C. Cheng, C. Z. Dong, W. Zhou, P. Wang, Q. X. Wang, Y. Yang, Y. H. Zhu, Y. W. Zeng and H. T. Wang, J. Mater. Chem., 2012, 22, 10665–10671 RSC.
- Z. Y. Zhang, C. L. Shao, X. H. Li, Y. Y. Sun, M. Y. Zhang, J. B. Mu, P. Zhang, Z. C. Guo and Y. C. Liu, Nanoscale, 2013, 5, 606–618 RSC.
- L. Zhang, G. Q. Zhang, H. B. Wu, L. Yu and X. W. Lou, Adv. Mater., 2013, 25, 2589–2593 CrossRef CAS PubMed.
- G. Q. Zhang, L. Yu, H. E. Hoster and X. W. Lou, Nanoscale, 2013, 5, 877–881 RSC.
- W. Ni, F. X. Liang, J. G. Liu, X. Z. Qu, C. L. Zhang, J. L. Li, Q. Wang and Z. Z. Yang, Chem. Commun., 2011, 47, 4727–4729 RSC.
- X. Xu, J. Liang, H. Zhou, D. M. Lv, F. X. Liang, Z. L. Yang, S. J. Ding and D. M. Yu, J. Mater. Chem. A, 2013, 1, 2995–2998 CAS.
- Z. W. Niu, Z. Z. Yang, Z. B. Hu, Y. F. Lu and C. C. Han, Adv. Funct. Mater., 13, 949–954 CrossRef CAS.
- M. Yang, J. Ma, Z. W. Niu, X. Dong, H. F. Xu, Z. K. Meng, Z. G. Jin, Y. F. Lu, Z. B. Hu and Z. Z. Yang, Adv. Funct. Mater., 2005, 15, 1523–1528 CrossRef CAS.
- Z. Z. Yang, Z. W. Niu, Y. F. Lu, Z. B. Hu and C. C. Han, Angew. Chem., Int. Ed., 2003, 42, 1943–1945 CrossRef CAS PubMed.
- M. Yang, J. Ma, Z. Z. Yang and Y. F. Lu, Angew. Chem., Int. Ed., 2005, 44, 6727–6730 CrossRef CAS PubMed.
- P. Si, S. J. Ding, J. Yuan, X. W. Lou and D. H. Kim, ACS Nano, 2011, 5, 7617–7626 CrossRef CAS PubMed.
- S. J. Ding, J. S. Chen and X. W. Lou, Adv. Funct. Mater., 2011, 21, 4120–4125 CrossRef CAS.
- E. J. Wang, T. He, L. S. Zhao, Y. M. Chen and Y. A. Cao, J. Mater. Chem., 2011, 21, 144–150 RSC.
- Z. Y. Liu, D. D. Sun, P. Guo and J. O. Leckie, Nano Lett., 2007, 7, 1081–1085 CrossRef CAS PubMed.
- G. R. Yang, Q. Zhang, W. Chang and W. Yan, J. Alloys Compd., 2013, 580, 29–36 CrossRef CAS PubMed.
- Y. Wen, B. T. Liu, W. Zeng and Y. H. Wang, Nanoscale, 2013, 5, 9739–9746 RSC.
- D. F Hou, X. L. Hu, P. Hu, W. Zhang, M. F. Zhang and Y. H. Huang, Nanoscale, 2013, 5, 9764–9772 RSC.
- Y. Xu and M. A. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CAS.
- J. Nayak, S. N. Sahu, J. Kasuya and S. Nozaki, Appl. Surf. Sci., 2008, 254, 7215–7218 CrossRef CAS PubMed.
- M. Zhou, J. Bao, M. S. Tao, R. Zhu, Y. Q. Zeng, Z. W. Wei and Y. Xie, Chem. Commun., 2012, 48, 3439–3441 RSC.
- C. Q. Zhu, B. G. Lu, Q. Su, E. Q. Xie and W. Lan, Nanoscale, 2012, 4, 3060–3064 RSC.
- B. G. Lu, C. Q. Zhu, Z. X. Zhang, W. Lan and E. Q. Xie, J. Mater. Chem., 2012, 22, 1375–1379 RSC.
- P. S. Archana, R. Jose, C. Vijila and S. Ramakrishna, J. Phys. Chem. C, 2009, 113, 21538–21542 CAS.
- S. S. Lee, H. W. Bai, Z. Y. Liu and D. D. Sun, Int. J. Hydrogen Energy, 2012, 37, 10575–10584 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ta12863f |
| ‡ Xin Xu and Guorui Yang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |