Controllable synthesis of graphene supported MnO2 nanowires via self-assembly for enhanced water oxidation in both alkaline and neutral solutions
Weiyong
Yuan
a,
Pei Kang
Shen
b and
San Ping
Jiang
*a
aFuels and Energy Technology Institute & Department of Chemical Engineering, Curtin University, Perth, WA 6102, Australia. E-mail: s.jiang@curtin.edu.au; Fax: +61 89266 1138; Tel: +61 8 9266 9804
bAdvanced Energy Materials Laboratory, School of Physics and Engineering, Sun Yat-sen University, Guangzhou 510275, China
First published on 21st October 2013
Abstract
The sluggish water oxidation reaction represents a significant challenge in water splitting for energy storage using hydrogen. We herein report the synthesis of MnO2 nanowires with the ultrasmall diameter and aspect ratio as high as 125 on graphene using a novel in situ polymer-mediated self-assembly approach in aqueous solution and under ambient conditions. The self-assembly process is simple and controllable by the concentration and pH of the polymer solution, in which the polymer serves as a soft template to direct the growth of MnO2 nanowires and also stabilize the structure, forming a unique graphene supported MnO2 nanowire, G@MnO2 NW. This nanostructure shows the most significant improvement of the catalytic activity compared to the graphene supported MnO2 nanoparticle and commercial Pt/C toward water oxidation under both alkaline and neutral conditions, and demonstrates for the first time a remarkable effect of the shape of MnO2 nanostructures on water oxidation catalysis. For example, at 0.7 V, it produces a current density of 5.9 mA cm−2, 14.8 times that of the graphene supported MnO2 nanoparticle (4.0 mA cm−2) and 8.4 times that of Pt/C (0.7 mA cm−2) in alkaline solution. Furthermore, it displays the highest turnover frequency reported among all the Mn oxides used for water oxidation catalysis. The G@MnO2 NW shows great potential as a water oxidation catalyst for energy storage applications.
Introduction
Due to increasing energy consumption and concern about global warming, renewable and clean energy forms such as solar and wind energy have attracted much attention in recent years.1–3 However, they are intermittent and undulating, requiring an efficient energy storage system.1–3 Hydrogen has a high energy density and produces no carbon emission during conversion, making it highly promising for energy storage.2,4,5 One of the most promising strategies for realizing the storage of energy in hydrogen is water splitting.1,5,6 Unfortunately, water oxidation is rather sluggish, involving a four-electron process and a massive molecular rearrangement.7,8 Therefore, it is highly desirable to design high-performance catalysts for significantly increasing its reaction rate, while reducing the overpotential.Nature has provided an efficient water oxidation catalyst, the μ-oxo bridged tetrameric Mn cluster (Mn4CaO5), which is located in photosystem II.9,10 It has been found in nearly all the photosynthetic organisms and no elements other than Mn have been identified in the shared reaction center.7,11 This exclusive use of Mn by nature inspires scientists to fabricate water oxidation catalysts containing the earth-abundant and cheap Mn element. Molecular complexes of Mn have been studied for active catalysts.12,13 However, they normally consist of organic components, which are thermodynamically unstable in the presence of O2/air, and easily oxidized during the water oxidation process.8,14,15 In comparison, Mn oxides are much more stable, but their catalytic activity is still low particularly under neutral conditions.5,6,15 Thus, there is a great need to improve the catalytic activity of Mn oxides for water oxidation.
Currently, most Mn oxides for catalyzing the water oxidation reaction are used as homogeneous catalysts in aqueous solutions with additional sacrificial agents or as heterogeneous catalysts in dense film form deposited on a bulk substrate.2,7,10 For the former one, particles are easily aggregated and precipitated from solution, greatly reducing their surface area and charge transport;7,16 while for the latter, the dense structures lead to a low surface area and slow ion diffusion.17,18 In addition, Mn oxides are not conductive,19,20 severely restricting the catalytic activity for the systems involving electron transport. Nanostructuring could greatly increase the surface area while shortening the path for charge transport. It has been found that when the size approached nanoscale, the water oxidation catalytic activity drastically increases.16,21 However, nanoparticles, particularly when their size is very small, are easy to aggregate.7,16 Furthermore, the remarkable effect of shapes on some nanoscale properties has not been recognized for the catalysis of water oxidation.20,21
Using a nanoscale support is a promising approach to stabilize the nanostructures for good dispersion, and also generate synergistic effects.22,23 Recently, graphene has emerged as an attractive support because of its ultrahigh specific surface area, excellent charge transport property, and superior chemical stability.24,25 Therefore, graphene can be used as a potential nanoscale support of Mn oxide nanostructures. However, it is still a great challenge to create a facile, mild, and controllable approach for synthesizing graphene supported MnO2 nanostructures with ultrasmall size and uniform distribution. However, there appears to be no report on using graphene supported Mn oxide nanostructures as catalysts for water oxidation.
Self-assembly can not only maintain the intrinsic properties of individual components, but also introduce synergistic effects between them.26–29 In this work, we reported a novel in situ and facile self-assembly approach to directly synthesize MnO2 nanostructures on pristine graphene. The controllability was investigated and the self-assembly mechanism was proposed based on the experimental results. The obtained nanostructures were then explored as catalysts for water oxidation.
Experimental section
Materials
Graphene (>98.3%, oxygen content 6–8%, Sinocarbon Materials Technology Co., Ltd.), poly(ethyleneimine) (PEI, 50% in water, Mw 750![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma-Aldrich), and KMnO4 (Analytical Reagent, Thermo Fisher Scientific) were used without treatment. The commercial Pt/C catalyst (30% loading) was purchased from Alfa Aesar. The deionized (DI) water (18.2 MΩ cm) produced using a Milli-Q water purification system (Millipore) was used in all experiments.
000, Sigma-Aldrich), and KMnO4 (Analytical Reagent, Thermo Fisher Scientific) were used without treatment. The commercial Pt/C catalyst (30% loading) was purchased from Alfa Aesar. The deionized (DI) water (18.2 MΩ cm) produced using a Milli-Q water purification system (Millipore) was used in all experiments.
Synthesis of nanostructured MnO2 on graphene
5 mg graphene was dispersed in 10 ml PEI solution by ultrasonicating for 1 h. Then 3.6 mg KMnO4 was added into the mixed solution, which was stirred for another 2 h. The molar ratios of N:Mn were varied between 1:5 and 2:1 by changing the concentration of PEI and the pH of PEI solution was controlled at 8.8, 5.4, and 3.0. Finally, the obtained sample was filtered, washed with DI water thoroughly, and dried at 70 °C for 3 h. The synthesis of nanostructured MnO2 on graphene was also carried out without the use of PEI. In this case, 5 mg G was dispersed in 10 ml H2O by ultrasonicating for 1 h. Then 3.6 mg KMnO4 was added into the mixed solution, which was stirred for another 2 days. Finally, the obtained sample was filtered, washed with DI water thoroughly, and dried at 70 °C for 3 h.
Characterization
The nanostructures of catalysts were examined using field emission scanning electron microscopy (FE-SEM) on a NEOL 40 EsB electron microscope and transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) on a JEOL 3000F FEGTEM. The crystal structures were investigated with an X-ray diffractometer (XRD, Bruker D8 Advance Diffractometer).Electrochemical measurements
The electrochemical tests were carried out on a CHI 4402D potentiostat (CH Instrument, USA) in a three-electrode configuration with saturated calomel electrode (SCE) as the reference electrode and Pt foil as the counter electrode. For the preparation of the working electrode, 5 μl of catalyst solutions with Nafion (mass ratio of electrocatalyst to Nafion = 4) was dropped on the clean GC electrode and then dried. The loading of MnO2 or Pt is 15 μg cm−2. The catalytic activity was tested via linear sweep voltammetry (LSV) at a scan rate of 1 mV s−1 in 1 M KOH solution and 0.25 M K2SO4 solution, and chronoamperometry under a series of different potentials in both of the above solutions.Results and discussion
Fig. 1 shows the FE-SEM and TEM images of pristine graphene and nanostructured MnO2 supported on graphene. The surface of graphene is smooth and featureless (Fig. 1A and D). Interestingly, after PEI directed self-assembly, high-density MnO2 nanowires were uniformly grown on graphene (Fig. 1B and E, denoted as G@MnO2 NWs). The average diameter of nanowires is ∼4.0 nm, and the length is as long as ∼500 nm, achieving an aspect ratio of ∼125. To the best of our knowledge, the MnO2 NW synthesized in the present study has the smallest diameter and the highest aspect ratio among those reported.20,21,30 The synthesis was also carried out under the same conditions except that no PEI was added. It took a much longer time to complete (see the Experimental part), and instead of MnO2 nanowires, MnO2 nanoparticles with an average diameter of ∼3.5 nm were obtained and the distribution on graphene was uniform (Fig. 1C and F, denoted as G@MnO2 NPs). This indicates that KMnO4 has been reduced during the synthesis process and the much faster synthesis process of G@MnO2 NWs as compared to that of G@MnO2 NPs implies that PEI accelerates the reduction process. Therefore, the presence of PEI promotes the formation of MnO2 nanowires, while in the absence of PEI, KMnO4 would be reduced by graphene, forming MnO2 nanoparticles. | ||
| Fig. 1 FE-SEM image of graphene (A), G@MnO2 NWs (B), and G@MnO2 NPs (C), and TEM image of graphene (D), G@MnO2 NWs (E), and G@MnO2 NPs (F). | ||
The XRD patterns of the obtained products are shown in Fig. 2. For all the three samples, the peak at ∼26.2° is due to the π–π stacking between graphene nanosheets, which is hardly avoidable for pristine graphene with an ultralow oxygen content used in this work.31,32 Besides this peak, for both G@MnO2 NWs and G@MnO2 NPs, four additional characteristic peaks located at 12.4°, 25.7°, 36.6°, and 65.9° are observed, which can be well indexed to (001), (002), (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 11), and (005) of a layered birnessite-type MnO2 (δ-MnO2) (JCPDS no. 80-1098).10,33,34 In addition, after rescaling the XRD pattern from 30° to 75° (Fig. 2, inset), several more peaks are found, which can also be indexed to δ-MnO2, further demonstrating the formation of the δ-MnO2 crystal structure.10,34 The very weak and broad XRD peaks of MnO2 structures are due to their ultrasmall size, which is in agreement with FESEM and TEM results (Fig. 1).
11), and (005) of a layered birnessite-type MnO2 (δ-MnO2) (JCPDS no. 80-1098).10,33,34 In addition, after rescaling the XRD pattern from 30° to 75° (Fig. 2, inset), several more peaks are found, which can also be indexed to δ-MnO2, further demonstrating the formation of the δ-MnO2 crystal structure.10,34 The very weak and broad XRD peaks of MnO2 structures are due to their ultrasmall size, which is in agreement with FESEM and TEM results (Fig. 1).
 | ||
| Fig. 2 XRD patterns of G, G@MnO2 NPs, and G@MnO2 NWs. | ||
HRTEM was used to provide insight into the atomic level structure of G@MnO2 NWs and G@MnO2 NPs (Fig. 3). Nearly all the MnO2 nanowires exhibit a layered structure with an interlayer spacing of ∼0.67 nm (Fig. 3B), in agreement with the layered birnessite-type MnO2, the lattice spacing between (001) planes of which is calculated to be 0.73 nm.33,35 The slightly smaller lattice spacing from HRTEM is ascribed to the collapse of the layered structure due to the loss of water between layers.33,35 In contrast, no layered structure is observed for MnO2 nanoparticles, and the lattice spacing is ∼0.22 nm (Fig. 3D), corresponding to that between (11) faces of δ-MnO2.33,36 Since the size of water molecules is ∼0.28 nm,37,38 the highly exposed layered structure of MnO2 nanowires allows water molecules to easily penetrate into the interlayer space, thus having more active sites for the catalysis of water oxidation.
 | ||
| Fig. 3 HRTEM images of G@MnO2 NWs (A and B) and G@MnO2 NPs (C and D). (B) and (D) are higher resolution images of (A) and (C). | ||
The effect of PEI concentration on the formation and distribution of MnO2 nanowires on graphene was investigated by changing the concentration ratio of PEI to KMnO4 (Fig. 4). When the ratio of N to Mn is 1:5, large aggregated MnO2 clusters are randomly assembled on graphene (Fig. 4A), indicating that the content of PEI is not enough to either stabilize their surface to prevent aggregation or cover the surface of graphene uniformly for their homogeneous deposition. As the ratio increases to 3:5 (Fig. 4B), much smaller MnO2 nanospheres with better dispersion are formed. This is due to the fact that an increased PEI content generates more seeds and also stabilizes nanoparticles more effectively to inhibit their growth and aggregation.39,40 The distribution of nanospheres on graphene is also more uniform, suggesting that the surfaces of graphene are more uniformly modified by PEI. It is notable that a few nanospheres are linked together to form chain-like structures. The linear chain of PEI could serve as a soft template to direct the growth and assembly of nanospheres.39,40 Interestingly, as the ratio further increases to 1:1 (Fig. 4C), nearly all the nanoparticles are connected together to form nanowires. In addition, these nanowires are distributed homogeneously all over the graphene surface. This result clearly demonstrates that PEI acts as a template to direct the self-assembly of nanoparticles into nanowires and also as a stabilizer to prevent them from aggregation. However, with further increasing the ratio to 2:1 (Fig. 4D), nanowires disappear again. It is most likely that too much PEI makes the self-assembly of MnO2 nanostructures difficult due to the competitive interactions between PEI, PEI modified MnO2, and graphene.41,42
 | ||
| Fig. 4 FE-SEM images of graphene supported MnO2 nanostructures obtained at N to Mn ratios of (A) 1:5, (B) 3:5, (C) 1:1, and (D) 2:1. The arrows in B indicate the chain-like structures linked by nanospheres. | ||
PEI is a weak polyelectrolyte with pH-responsive charge density, and therefore the synthesis pH was tuned, while keeping the ratio of N to Mn at 1 to 1, to further investigate the mechanism. At pH 8.8, which is the pH value of the original solution, ultrathin and long nanowires with high density and good dispersion are formed on graphene (Fig. 5A). However, when decreasing the synthesis pH to 5.4 (Fig. 5B), severe aggregation occurs. The higher charge density and content of protonized amine groups at this pH could result in a weaker hydrophobic interaction between PEI and graphene.43,44 Thus, less PEI with more intermolecular aggregations will be assembled on graphene, inducing the aggregation between nanowires. At a low pH of 3.0 (Fig. 5C), much larger aggregates are observed which are likely to be formed in the solution rather than on the graphene support.
 | ||
| Fig. 5 FE-SEM images of graphene supported MnO2 nanostructures obtained at PEI pH of (A) 8.8, (B) 5.4, and (C) 3.0. | ||
The possible mechanism for in situ self-assembly of MnO2 nanowires on graphene is proposed based on the experimental results (Scheme 1). MnO2 preferentially nucleates on the PEI modified graphene surface, and then grows and self-assembles along the molecular chains. On the other hand, PEI chains can be assembled on MnO2 nanostructures to prevent their aggregation. This biomimetic soft-template approach has the following advantages: it can be carried out at room temperature, in aqueous solution, in one pot and one step, and is a fast synthesis process; therefore it has great potential to be extended for the fabrication of other graphene supported nanostructures for a variety of energy conversion and storage applications.39,40 The prepared MnO2 nanowires have an ultrasmall diameter and ultrahigh aspect ratio, thus providing high charge mobility, fast ion diffusion, and large surface area.20,45,46 In addition, the accessible layered structure with interlayer spacing larger than the water molecule could offer more active sites.7,16,21 The intimate integration of ultrathin MnO2 nanowires with pristine graphene could be favourable to enhancing the charge transport and increasing the stability and dispersion of MnO2 nanostructures, while reducing the aggregation between graphene nanosheets.18,20,24 All these unique advantages suggest promising potential of this nanostructure as an effective catalyst for water oxidation.
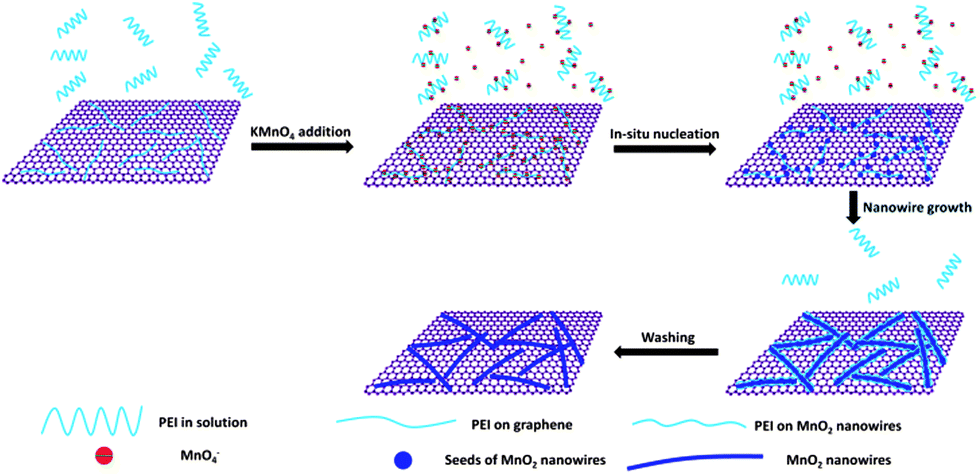 | ||
| Scheme 1 Possible self-assembly mechanism for G@MnO2 NWs. | ||
The G@MnO2 NW was then investigated as a catalyst for water oxidation under both alkaline and neutral conditions using electrochemical techniques (Fig. 6). In the alkaline solution, LSV results (Fig. 6A) show a distinct trend: the onset potential of the G@MnO2 NW is ∼0.51 V, 50 mV lower than that of G@MnO2 NPs (∼0.56 V) and 150 mV lower than that of Pt/C (∼0.66 V); the peak potential of the G@MnO2 NW is 0.70 V, similar to G@MnO2 NPs (0.69 V), but is 160 mV lower than that of the commercial Pt/C catalyst (0.86 V); and the peak current density of the G@MnO2 NW is 5.9 mA cm−2, ∼14.8 times that of G@MnO2 NPs (4.0 mA cm−2) and 1.8 times that of the commercial Pt/C catalyst (3.3 mA cm−2). In addition, the G@MnO2 NW produces a current density of 3.3 mA cm−2 at a potential of 0.6 V, 260 mV lower than that measured on the Pt/C catalyst. At the peak potential of the G@MnO2 NW (0.7 V), the current density is 8.4 times that of commercial Pt/C (0.7 mA cm−2). Under the neutral conditions (Fig. 6B), the onset potential of the G@MnO2 NW is ∼0.69 V, 440 mV lower than that of G@MnO2 NPs (∼1.13 V) and 430 mV lower than that of the commercial Pt/C catalyst (∼1.12 V). Furthermore, the current density of the G@MnO2 NW at 1.3 V is 0.48 mA cm−2, 2.2 times that of G@MnO2 NPs and 2.5 times that of the commercial Pt/C catalyst. This is probably the first report on such a significant improvement of catalytic activity compared to the commercial Pt/C catalyst under both alkaline and neutral conditions, and on such a remarkable effect of the nanostructure shape on water oxidation performance. Our result demonstrates the great potential of the G@MnO2 NW as a highly efficient catalyst for water oxidation.
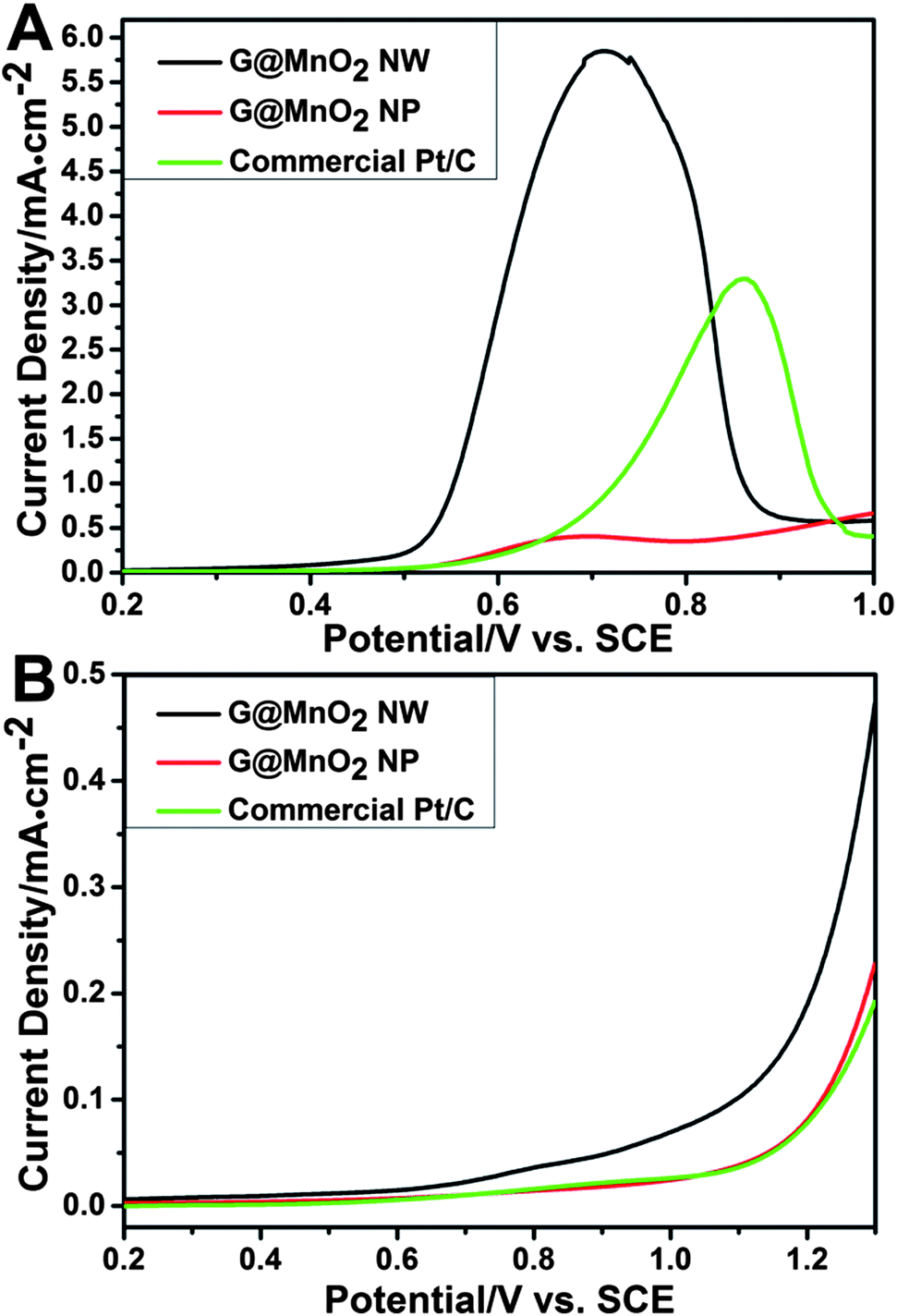 | ||
| Fig. 6 LSV curves of different samples in 1 M KOH (A) and 0.25 M K2SO4 (B) solution. | ||
The catalytic activity was further investigated using chronoamperometry, in which the current density was monitored under a sequence of potential steps (Fig. 7). In the alkaline solution (Fig. 7A), the difference between current densities of tested samples is very small at the potential of 0.3 V, but becomes increasingly obvious with the increase of the potential; the G@MnO2 NW apparently has a much higher current density and durability than the G@MnO2 NP and commercial Pt/C catalyst, while the commercial Pt/C catalyst has the lowest stability. On the other hand, in the neutral solution, significant currents are observed above 0.9 V and similarly the difference becomes more significant at higher potentials. The G@MnO2 NW shows higher current density and durability than the G@MnO2 NP and commercial Pt/C catalyst. The Pt/C catalyst has the lowest activity for the water oxidation reaction. The high durability of graphene supported MnO2 nanostructures could be ascribed to the strong binding of MnO2 nanostructures to graphene and good chemical stability of graphene. It is noteworthy that no structural changes are observed from FE-SEM and TEM images after the chronoamperometry testing up to a potential of 0.6 V in 1 M KOH solution and 1.3 V in 0.25 M K2SO4 solution (data not shown). However, it has been found that a further increase of the potential caused the detachment of the catalysts from the glass carbon electrode due to the production of a large amount of bubbles, resulting in the drop in catalytic currents.
 | ||
| Fig. 7 Chronoamperometric curves of different samples under a series of potentials in 1 M KOH (A) and 0.25 M K2SO4 (B) solution. | ||
To further evaluate the catalytic activity, the turnover frequency (TOF) per Mn atom was estimated from choronoamperometry (Fig. 7) according to the following equation:47

 | ||
| Fig. 8 TOF values of the G@MnO2 NW, G@MnO2 NP, and commercial Pt/C catalyst versus the overpotential in 1 M KOH (A) and 0.25 M K2SO4 (B) solution. | ||
Conclusion
In conclusion, we have synthesized novel graphene supported MnO2 nanowires with the smallest diameter and highest aspect ratio via in situ self-assembly at room temperature, under ambient pressure, in aqueous solution, and in the one pot synthesis process. The self-assembly process is controllable by the PEI concentration and the pH. For example, under conditions of a molar ratio of N:Mn of 1:1 and pH of 8.8, ultrathin and long MnO2 NWs with aspect ratio as high as 125 were synthesized on graphene. The polymer serves as a soft template to direct the growth of MnO2 nanowires and also stabilize them for good dispersion. This nanostructure shows the most significant improvement of the catalytic activity compared to commercial Pt/C toward water oxidation under both alkaline and neutral conditions, and demonstrates for the first time a remarkable effect of the shape of MnO2 nanostructures on water oxidation catalysis. Furthermore, it exhibits the highest TOF value (0.017 S−1 at 0.44 V vs. SCE in alkaline solution and 00.0073 S−1 at 0.73 V vs. SCE in neutral solution) reported among all the Mn oxides used in water oxidation catalysis. Thus, the G@MnO2 NW reported in this study will have great potential as a catalyst for water oxidation for various clean energy applications.
Acknowledgements
This project is supported by the Australian Research Council (ARC DP120102325 and DP120104932), Australia and the Major (Regional) Joint Research Project of the National Natural Science Foundation of China (51210002). The authors acknowledge the facilities, and scientific and technical assistance of the Electron Microscope Laboratories of Curtin University and Centre for Microscopy Characterisation and Analysis of The University of Western Australia.Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253 CrossRef CAS PubMed.
- F. E. Osterloh and B. A. Parkinson, MRS Bull., 2011, 36, 17 CrossRef CAS.
- H. G. Park and J. K. Holt, Energy Environ. Sci., 2010, 3, 1028 CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60 CrossRef CAS PubMed.
- A. Sartorel, M. Carraro, F. M. Toma, M. Prato and M. Bonchio, Energy Environ. Sci., 2012, 5, 5592 CAS.
- K. E. deKrafft, C. Wang, Z. Xie, X. Su, B. J. Hinds and W. Lin, ACS Appl. Mater. Interfaces, 2012, 4, 608 CAS.
- M. Najafpour, T. Ehrenberg, M. Wiechen and P. Kurz, Angew. Chem., Int. Ed., 2010, 49, 2233 CrossRef CAS PubMed.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 18153 CrossRef CAS PubMed.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 1519 CrossRef CAS PubMed.
- D. G. H. Hetterscheid and J. N. H. Reek, Angew. Chem., Int. Ed., 2012, 51, 9740 CrossRef CAS PubMed.
- G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, R. Pryadun, J. Sheats, L. Spiccia and G. Swiegers, Acc. Chem. Res., 2009, 42, 1935 CrossRef CAS PubMed.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572 RSC.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474 CrossRef CAS PubMed.
- F. Jiao and H. Frei, Chem. Commun., 2010, 46, 2920 RSC.
- P. Balaya, Energy Environ. Sci., 2008, 1, 645 CAS.
- A. Zabet-Khosousi and A.-A. Dhirani, Chem. Rev., 2008, 108, 4072 CrossRef CAS PubMed.
- L. Wei, C. Li, H. Chu and Y. Li, Dalton Trans., 2011, 40, 2332 RSC.
- Z.-S. Wu, W. Ren, D.-W. Wang, F. Li, B. Liu and H.-M. Cheng, ACS Nano, 2010, 4, 5835 CrossRef CAS PubMed.
- V. B. R. Boppana and F. Jiao, Chem. Commun., 2011, 47, 8973 RSC.
- Y. Shao, J. Liu, Y. Wang and Y. Lin, J. Mater. Chem., 2009, 19, 46 RSC.
- Y. Zhou, K. Neyerlin, T. S. Olson, S. Pylypenko, J. Bult, H. N. Dinh, T. Gennett, Z. Shao and R. O'Hayre, Energy Environ. Sci., 2010, 3, 1437 CAS.
- P. V. Kamat, J. Phys. Chem. Lett., 2011, 2, 242 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752 CrossRef CAS PubMed.
- W. Yuan, Z. Lu and C. M. Li, J. Mater. Chem., 2011, 21, 5148 RSC.
- W. Yuan, Z. Lu, H. Wang and C. M. Li, Adv. Funct. Mater., 2012, 22, 1932 CrossRef CAS.
- Z. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2010, 5, 15 CrossRef CAS PubMed.
- Y. Xiang, S. F. Lu and S. P. Jiang, Chem. Soc. Rev., 2012, 41, 7291 RSC.
- R. Zou, Z. Zhang, L. Yu, Q. Tian, Z. Chen and J. Hu, Chem.–Eur. J., 2011, 17, 13912 CrossRef CAS PubMed.
- K. Zhang, L. Zhang, X. S. Zhao and J. Wu, Chem. Mater., 2010, 22, 1392 CrossRef CAS.
- S. Wang, Y. Zhang, N. Abidi and L. Cabrales, Langmuir, 2009, 25, 11078 CrossRef CAS PubMed.
- H. T. Zhu, J. Luo, H. X. Yang, J. K. Liang, G. H. Rao, J. B. Li and Z. M. Du, J. Phys. Chem. C, 2008, 112, 17089 CAS.
- Y. Li, J. Wang, Y. Zhang, M. N. Banis, J. Liu, D. Geng, R. Li and X. Sun, J. Colloid Interface Sci., 2012, 369, 123 CrossRef CAS PubMed.
- W. Xiao, D. Wang and X. Lou, J. Phys. Chem. C, 2010, 114, 1694 CAS.
- M. A. Cheney, R. Jose, A. Banerjee, P. K. Bhowmik, S. Qian and J. M. Okoh, J. Nanomater., 2009, 940462 Search PubMed.
- J. S. D'Arrigo, Am. J. Physiol., 1978, 235, C109 CAS.
- J. Sekulic, D. H. A. Blank, J. ten Elshof, C. Abadal, J. Sekulić and S. Chowdhury, Microporous Mesoporous Mater., 2003, 65, 197 CrossRef PubMed.
- S.-F. Chen, J.-H. Zhu, J. Jiang, G.-B. Cai and S.-H. Yu, Adv. Mater., 2010, 22, 540 CrossRef CAS PubMed.
- S. I. Stupp and P. V. Braun, Science, 1997, 277, 1242 CrossRef CAS.
- N. Zacharia, D. DeLongchamp, M. Modestino and P. Hammond, Macromolecules, 2007, 40, 1598 CrossRef CAS.
- W. Yuan, Z. Lu and C. Li, J. Mater. Chem., 2011, 21, 5148 RSC.
- H. Mohwald, M. Schneider, M. Brinkmann and H. Möhwald, Macromolecules, 2003, 36, 9510 CrossRef.
- F. Alarcon, E. Perez and A. Gama Goicochea, Soft Matter, 2013, 9, 3777 RSC.
- C. Xu, Y. Zhao, G. Yang, F. Li and H. Li, Chem. Commun., 2009, 7575 RSC.
- A. Vomiero, I. Concina, E. Comini, C. Soldano, M. Ferroni, G. Faglia and G. Sberveglieri, Nano Energy, 2012, 1, 372 CrossRef CAS PubMed.
- L. Tong, M. Gothelid and L. Sun, Chem. Commun., 2012, 48, 10025 RSC.
| This journal is © The Royal Society of Chemistry 2014 |