 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermoresponsive PNIPAAM bottlebrush polymers with tailored side-chain length and end-group structure†
Xianyu
Li
a,
Hadi
ShamsiJazeyi
a,
Stacy L.
Pesek
a,
Aditya
Agrawal
a,
Boualem
Hammouda
b and
Rafael
Verduzco
*a
aDepartment of Chemical and Biomolecular Engineering, Rice University, 6100 Main Street, Houston, Texas 77005, USA. E-mail: rafaelv@rice.edu
bNational Institute of Standards and Technology, 100 Bureau Drive, Gaithersburg, Maryland 20899, USA
First published on 15th January 2014
Abstract
We explore the phase behaviour, solution conformation, and interfacial properties of bottlebrush polymers with side-chains comprised of poly(N-isopropylacrylamide) (PNIPAAM), a thermally responsive polymer that exhibits a lower critical solution temperature (LCST) in water. PNIPAAM bottlebrush polymers with controlled side-chain length and side-chain end-group structure are prepared using a “grafting-through” technique. Due to reduced flexibility of bottlebrush polymer side-chains, side-chain end-groups have a disproportionate effect on bottlebrush polymer solubility and phase behaviour. Bottlebrush polymers with a hydrophobic end-group have poor water solubilities and depressed LCSTs, whereas bottlebrush polymers with thiol-terminated side-chains are fully water-soluble and exhibit an LCST greater than that of PNIPAAM homopolymers. The temperature-dependent solution conformation of PNIPAAM bottlebrush polymers in D2O is analyzed by small-angle neutron scattering (SANS), and data analysis using the Guinier–Porod model shows that the bottlebrush polymer radius decreases as the temperature increases towards the LCST for PNIPAAM bottlebrush polymers with relatively long 9 kg mol−1 side-chains. Above the LCST, PNIPAAM bottlebrush polymers can form a lyotropic liquid crystal phase in water. Interfacial tension measurements show that bottlebrush polymers reduce the interfacial tension between chloroform and water to levels comparable to PNIPAAM homopolymers without the formation of microemulsions, suggesting that bottlebrush polymers are unable to stabilize highly curved interfaces. These results demonstrate that bottlebrush polymer side-chain length and flexibility impact phase behavior, solubility, and interfacial properties.
Introduction
Bottlebrush polymers are branched macromolecules with novel and potentially useful bulk and interfacial properties. For example, as a result of their large size and highly extended backbone conformation, brush block copolymers self-assemble into polymer photonic crystals with large (100 nm or greater) domains.1,2 Bottlebrush polymers are also of interest for interfacially-active materials due to dense crowding of the polymeric side-chains. This property can be used in the design of new nanomaterials for drug delivery,3–5 surfactants,6 organic electronics,7 or as responsive surface coatings.8 While the size and conformation of the bottlebrush polymer backbone has been analyzed in detail, the conformation and flexibility of the side-chains is poorly understood. Characterizing the conformation of bottlebrush polymer side-chains and backbone simultaneously is challenging due to the dense-crowding of side-chains and proximity of the bottlebrush polymer backbone. The side-chain flexibility and length may be relevant for applications that rely on the interfacial properties of bottlebrush polymers.PNIPAAM is a well-known thermoresponsive polymer which exhibits a lower critical solution temperature (LCST) of 32 °C.9,10 In aqueous solution, a reversible coil-to-globule phase transition and chain aggregation is induced by the dehydration of polymer chains when heating the solution above LCST. For bottlebrush polymers with PNIPAAM side chains, we hypothesized that collapse of the side-chains above the LCST may result in backbone extension and lead to, potentially, a larger single-molecule size above the LCST and a greater aspect ratio. The high grafting density of PNIPAAM side-chains may also lead to a change in the LCST temperature and a two-stage collapse, as has been observed for end-tethered PNIPAAM polymer brush films11 as opposed to surface-tethered PNIPAAM networks.12
Here, we report the solution conformation, solubility, phase behaviour, and interfacial properties of PNIPAAM bottlebrush polymers. We prepared a series of PNIPAAM bottlebrush polymers with different side-chain molecular weights and side-chain end-groups. We find that side-chain end-groups have a disproportionate effect on bottlebrush polymer solubility and phase behaviour, likely due to stretching and reduced flexibility of bottlebrush polymer side-chains.13,14 Both PNIPAAM bottlebrush solubility and LCST in water is affected by the end-group structure, and a stronger effect is seen for shorter side-chains. SANS measurements reflect a collapse of the side-chains with increasing temperature, with a greater collapse for longer PNIPAAM side-chains. PNIPAAM bottlebrush polymers with relatively long 9 kg mol−1 side-chains form lyotropic liquid crystal phases at temperatures higher than the LCST. Finally, bottlebrush polymers are shown to be interfacially active and can reduce the interfacial tension (IFT) between chloroform and water to values comparable to PNIPAAM homopolymer. These results demonstrate that the solution properties of bottlebrush polymers depend in large part on side-chain length and flexibility, and thermoresponsive side-chains lead to lyotropic liquid crystal ordering. Bottlebrush polymers with thiol-terminated PNIPAAM side-chains are soluble in water and may be useful for biomedical applications15,16 or for the preparation of polymer–gold nanoparticle composite materials.17,18
Results and discussion
Solubility and lower-critical solution temperature of PNIPAAM bottlebrush polymers
Bottlebrush polymers were prepared in a two-step “grafting through” synthetic approach (Scheme 1), similar to that reported previously for poly(styrene) bottlebrush polymers.8,19 First, norbornene functionalized PNIPAAM (NB-PNIPAAM-CTA) macromonomers were prepared by reversible addition–fragmentation chain transfer (RAFT) from an exo-norbornene-functionalized chain-transfer agent (NB-CTA). Bottlebrush polymers were synthesized via ROMP in the presence of Grubbs' third generation catalyst ((H2IMeS)(3-Br-Py)2(Cl2)RuCHPh) at room temperature, resulting in PNIPAAM bottlebrush polymer with CTA-terminated side-chains P(PNIPAAM-CTA). This approach was repeated for three different PNIPAAM side-chain lengths, as shown in Table 1. Size-exclusion chromatography indicates successful polymerization in all cases, but the presence of unreacted homopolymers was unavoidable (see ESI, Fig. S1†). In measurements described below, we systematically compare bottlebrush polymer samples with those of pure PNIPAAM homopolymers to distinguish potential effects arising from the homopolymer impurities.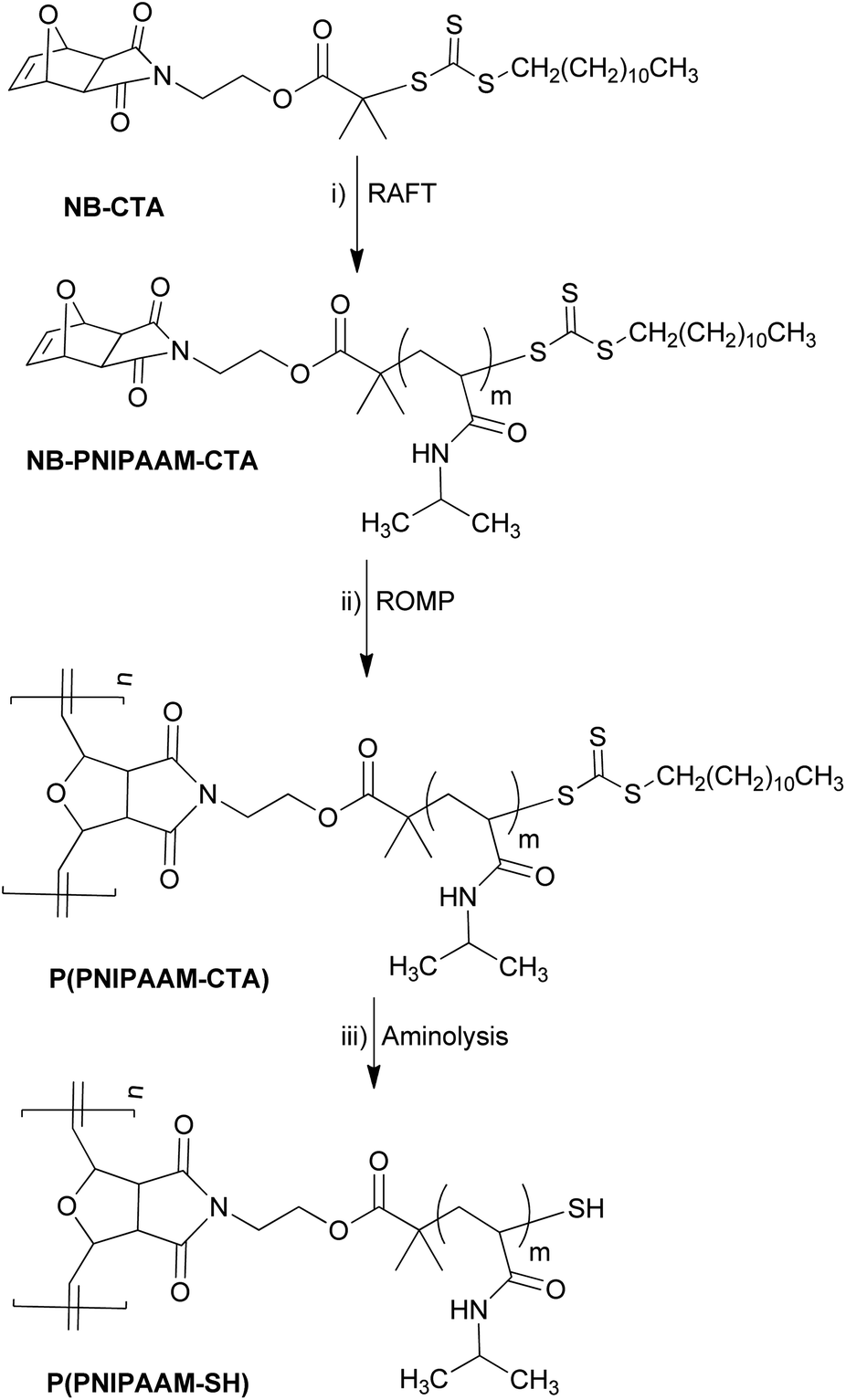 | ||
| Scheme 1 Synthetic route for the preparation of bottlebrush polymers with poly(NIPAAM) side-chains. (i) AIBN, 1,4-dioxane, N-isopropylacrylamide, 65 °C, (ii) CH2Cl2, ((H2IMeS)(3-Br-Py)2(Cl2)RuCHPh) (iii) 1,4-dioxane, hexylamine, tributylphosphine. | ||
| Bottlebrush polymers | Side-chain Mwa (kg mol−1) | Overall Mwb (kg mol−1) | Backbone DP | PDIa |
|---|---|---|---|---|
| a Measured by size-exclusion chromatography relative to monodisperse polystyrene standards. b Measured by size-exclusion chromatography with multi-angle laser light scattering detection. | ||||
| P(4K-NIPAAM-CTA) | 3.9 | 154 | 39 | 1.06 |
| P(4K-NIPAAM-SH) | 3.7 | 146 | 39 | 1.14 |
| P(6K-NIPAAM-CTA) | 5.6 | 204 | 36 | 1.06 |
| P(6K-NIPAAM-SH) | 5.4 | 197 | 36 | 1.19 |
| P(9K-NIPAAM-CTA) | 9.0 | 292 | 33 | 1.14 |
| P(9K-NIPAAM-SH) | 8.8 | 286 | 33 | 1.27 |
After ROMP, the PNIPAAM bottlebrush polymer side-chains are terminated by a trithiocarbone chain-transfer agent (CTA) with a hydrophobic dodecyl tail. While CTA-terminated PNIPAAM homopolymers are fully water soluble, PNIPAAM bottlebrush polymers with 3.9 and 5.6 kg mol−1 side-chains are insoluble in water, and PNIPAAM bottlebrush polymers with 9.0 kg mol−1 side-chains are only partially water-soluble. In order to improve the water-solubility of the bottlebrush polymers, the CTA end-group was removed using a mild aminolysis reaction,20 resulting in PNIPAAM bottlebrush polymers with thiol-terminated side-chains. Both UV-VIS absorbance measurements and 1H NMR spectroscopy show quantitative removal of the CTA end-group (ESI, Fig. S2–S5†). Removal of the CTA end-group reduces the molecular weight of the side-chains and the bottlebrush polymers, but the backbone degree of polymerization (DP) remains constant. We observe only a small increase in the polydispersity (PDI) after removal of the trithiocarbonate CTA, and all bottlebrush polymers are water soluble at room temperature. The characteristics of CTA-terminated and thiol-terminated bottlebrush polymers are presented in Table 1.
As shown schematically in Fig. 1, solubility can be affected by side-chain stretching and side-chain length. Bottlebrush polymers are expected to have highly stretched, relatively inflexible side-chains due to strong steric interactions. As a result, side-chain end-groups may have a disproportionate impact on solubility. Further, side-chains become more flexible with increasing side-chain length. The solubility of bottlebrush polymers with longer side-chains should therefore be less sensitive to end-group structure. The solubility behavior of PNIPAAM bottlebrush polymers is consistent with this physical picture. Although the CTA is a relatively small content of the side-chains (roughly 9, 6, and 4% mass fraction for 4, 6, and 9 kg mol−1 side-chains, respectively), bottlebrush polymers with CTA-terminated PNIPAAM side-chains exhibit poor solubility in water. Only P(9K-PNIPAAM-CTA) is partially soluble in water compared with the insolubility of P(4K-PNIPAAM-CTA) and P(6K-PNIPAAM-CTA). After removal of the hydrophobic CTA end-group, all bottlebrush polymers are fully water soluble. By comparison, PNIPAAM-CTA homopolymers are fully water soluble.
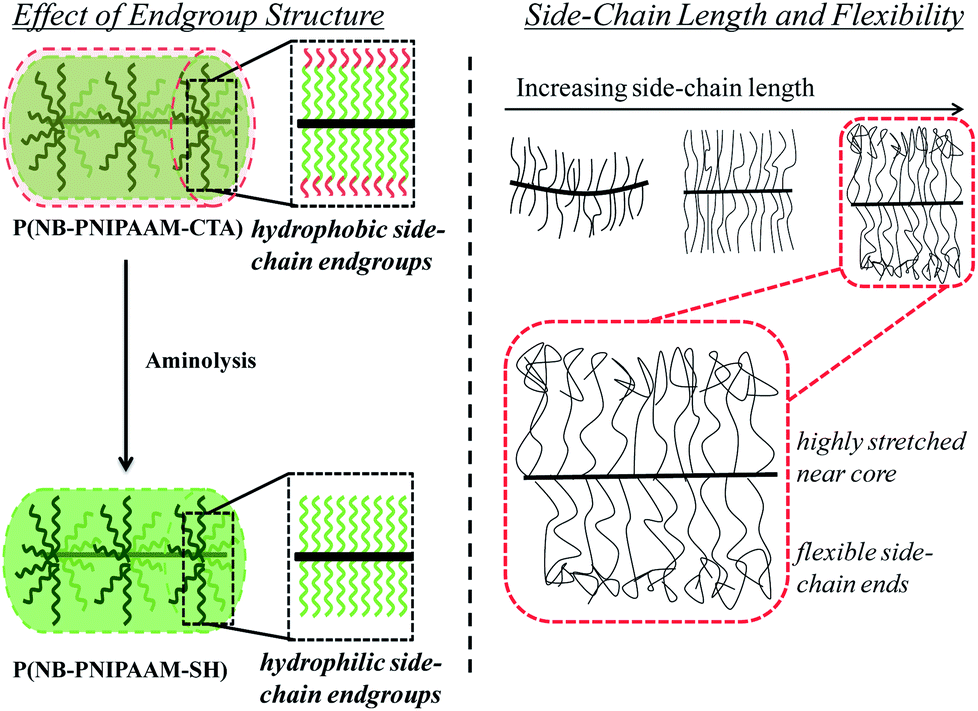 | ||
| Fig. 1 Schematic for the solution conformation of bottlebrush polymers and the role of side-chain end-group and length. | ||
Thiol-terminated PNIPAAM bottlebrush polymers exhibit an LCST in water that can be easily observed visually, as shown for 1 wt% 9K-NIPAAM-SH in Fig. 2. Prior work has shown the LCST of PNIPAAM can be affected by crowding and by the incorporation of different comonomers.21,22 In general, increasing the hydrophilicity of PNIPAAM increases the LCST while introducing hydrophobic comonomers de-stabilizes the homogeneous phase and reduces the LCST.23 Differential scanning calorimetry (DSC) was used to measure the LCST for PNIPAAM-CTA homopolymers of all bottlebrush polymers, including those with pendant CTAs. PNIPAAM-CTA bottlebrush polymers are partially water soluble, and a thermoreversible LCST can be detected by DSC for all samples. DSC measurements show that 9 kg mol−1 NB-PNIPAAM-CTA homopolymer has an LCST near 32 °C, similar to that of linear PNIPAAM.9 Shorter PNIPAAM-CTA homopolymers have a reduced CTA, with the greatest reduction measured for 4 kg mol−1 PNIPAAM-CTA. Thus, the presence of the CTA destabilizes the water-soluble phase, as expected due to the hydrophobicity of the CTA. Further, the impact of the CTA end-group is more significant for shorter side-chains, with P(4K-NIPAAM-CTA) exhibiting an LCST near 25 °C. For all bottlebrush polymers, the LCST increases upon removal of the CTA. The LCST increases by more than 6 °C on aminolysis of P(4K-NIPAAM-CTA) and 1 °C for P(9K-NIPAAM-CTA). The glass transition temperatures (Tg) are insensitive to side-chain endgroup structure. LCST and Tg values are shown for all polymers and the corresponding macromonomers in Table 2. We observe a significant difference between the LCST of bottlebrush polymers and NIPAAM macromonomers.
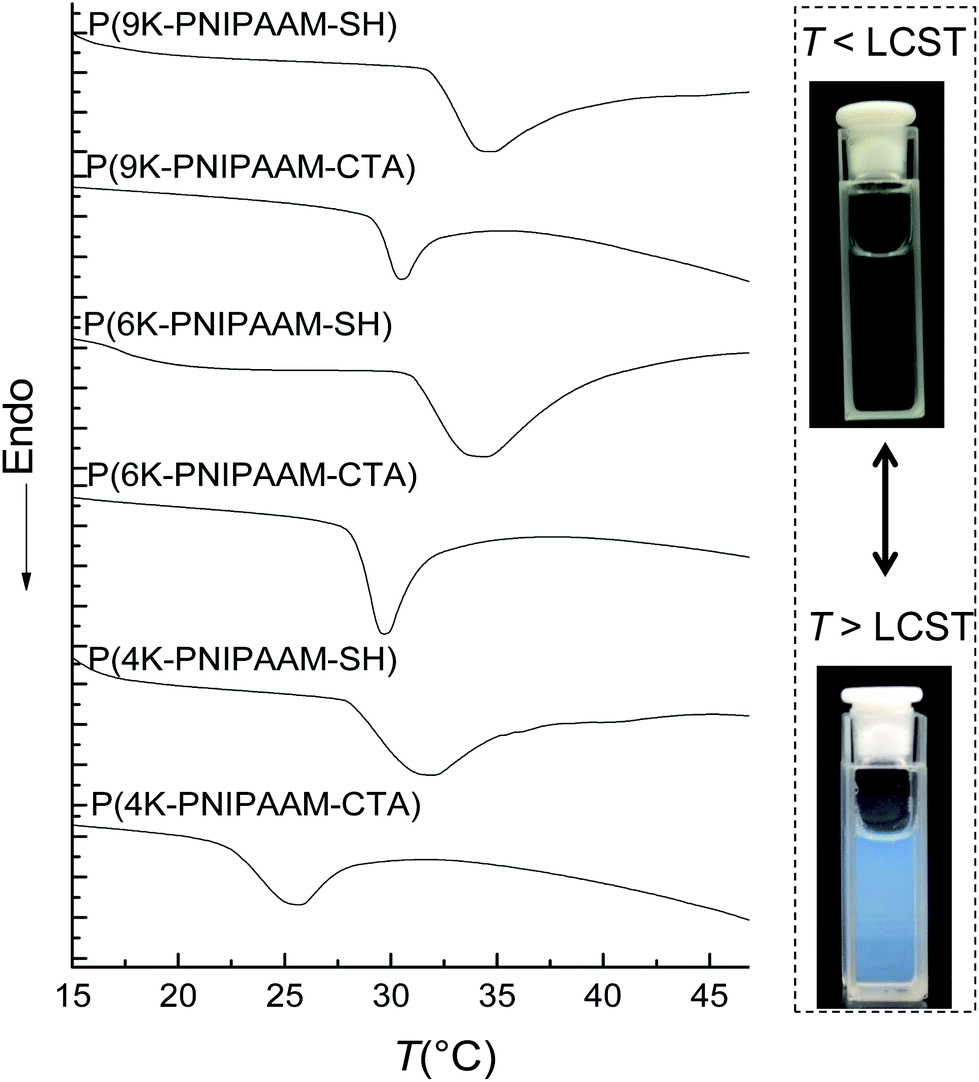 | ||
| Fig. 2 (Left) DSC thermograms of PNIPAAM bottlebrush polymer in water and (right) photographs of 1 wt% P(9K-NIPAAM-SH) at room temperature and 34 °C. The second heating cycle of a heat/cool/heat sequence is shown, using a heating rate of 5 °C min−1. The data show a reduced LCST for bottlebrush polymers with CTA terminated side-chains and lower molecular-weight side-chains. | ||
| Sample | LCST (°C) | T g (°C) |
|---|---|---|
| 4K-PNIPAAM-CTA | 27.3 | 142 |
| P(4K-NIPAAM-CTA) | 25.5 | 145 |
| P(4K-NIPAAM-SH) | 31.8 | 145 |
| 6K-PNIPAAM-CTA | 31.0 | 144 |
| P(6K-NIPAM-CTA) | 29.8 | 145 |
| P(6K-NIPAM-SH) | 34.3 | 144 |
| 9K-PNIPAAM-CTA | 32.3 | 144 |
| P(9K-NIPAAM-CTA) | 33.3 | 145 |
| P(9K-PNIPAAM-SH) | 34.3 | 146 |
The LCST can be also determined by turbidity measurements (see ESI, Fig. S6†). This method gave LCST values in good agreement with those measured by DSC. The transmittance of bottlebrush polymer solutions decreased slowly with temperature over a wide temperature range above the LCST while PNIPAAM homopolymers exhibit a much sharper drop in transmission above the LCST.
Solution conformation of bottlebrush polymers
Small angle neutron scattering (SANS) measurements were carried out to quantitatively analyze the size and conformation in solution, with particular interest in detecting collapse of the side-chains on approaching the LCST. We expected that bottlebrush polymers would exhibit a collapse of the PNIPAAM side-chains on approaching the LCST which might, due to steric interactions between side-chains, result in greater backbone extension. Due to increased flexibility with increasing molecular weight of the side-chains (shown schematically in Fig. 1), we would also expect a greater change in the side-chain length with temperature for bottlebrush polymers with longer PNIPAAM side-chains. SANS measurements were carried out on the NG3 instrument at the National Institute of Standards and Technology (NIST). Samples were loaded into a 1 mm thick cell and sealed by two quartz discs. Samples were equilibrated for 10 min at each temperature. Three bottlebrush samples with 4 K, 6 K, and 9 K side-chains were measured at 7 different temperatures, ranging from 10 °C to 50 °C.At temperatures below the LCST, SANS traces for P(4K-NIPAAM-SH) and P(6K-NIPAAM-SH) exhibit a clear plateau at low q (<0.01 Å−1) with a small amount of aggregation as indicated by a small upturn at very low q (<0.007 Å−1). The shape of the SANS trace is consistent with a spherical conformation of the bottlebrush polymers.19 By comparison, the SANS trace for P(9K-NIPAAM-SH) does not exhibit a plateau at low q and instead increases with decreasing q. This suggests an extended bottlebrush polymer conformation in solution, as has been observed for bottlebrush polymers with long backbone lengths19 and other rod-like macromolecules. For all samples, the scattering intensity increases with temperature at low-q. This increase is modest below the LCST, but near the LCST the intensity is rapidly increases with temperature (Fig. 3).
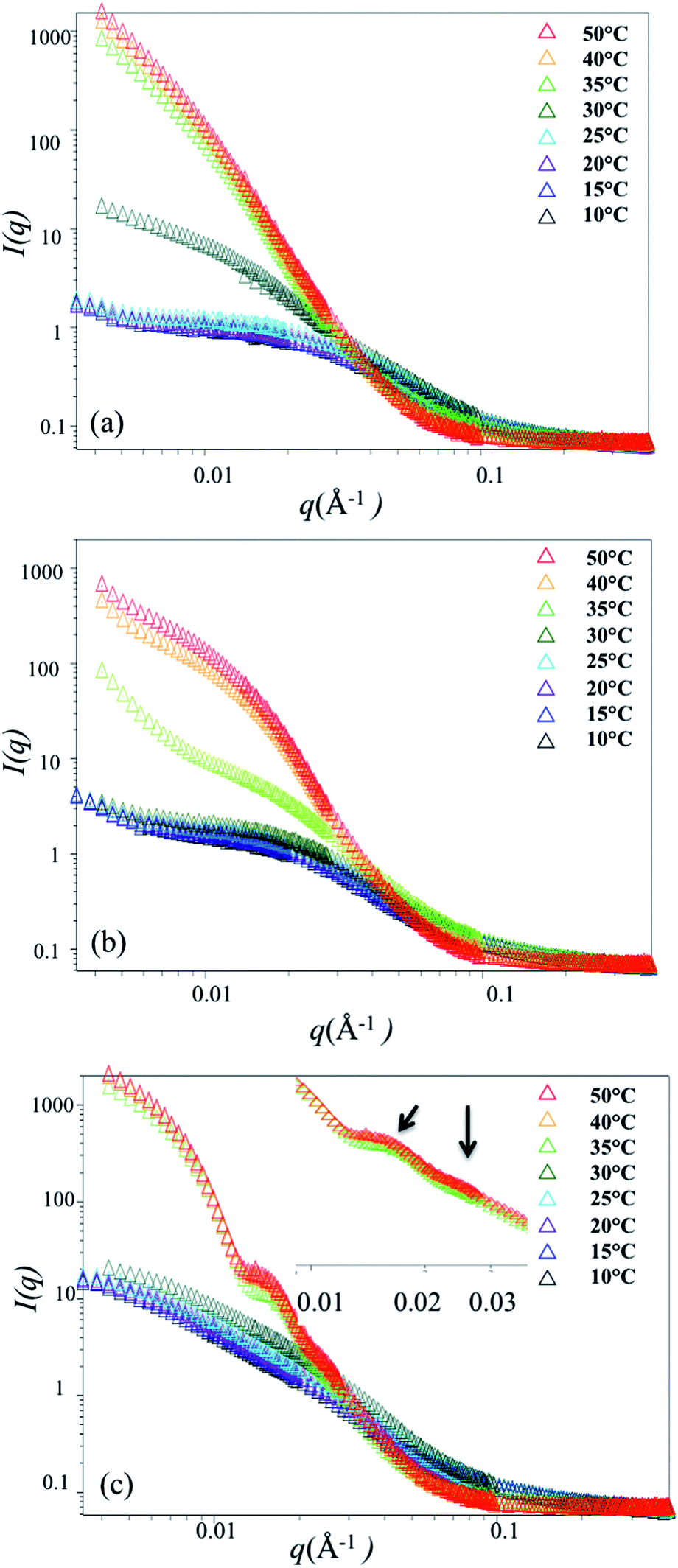 | ||
| Fig. 3 SANS data for 1 wt% solutions in water: (a) P(4K-PINPAAM-SH), (b) P(6K-PNIPAAM-SH), and (c) P(9K-PNIPAAM-SH). The inset shows SANS trace at 35 °C, 40 °C and 50 °C. Error bars are smaller than the symbols shown. | ||
A quantitative estimate of bottlebrush polymer size and anisotropy can be obtained by fitting the data to a Guinier–Porod model19 or a cylindrical model.19,24,25 Here, we restrict our analysis to the Guinier–Porod model since recent work has demonstrated that this model is more appropriate for bottlebrush polymers with relatively short (DP < 100) backbones. The Guinier–Porod model makes no assumptions about the bottlebrush polymer shape, which may be spherical or cylindrical in solution. As shown in Fig. 4, SANS traces for samples at temperatures below the LCST can be fit using the Guinier–Porod model. Model fitting provides an estimate of both the radius of the bottlebrush polymers R and anisotropy through the dimension parameter s (see ESI, Table S1†).
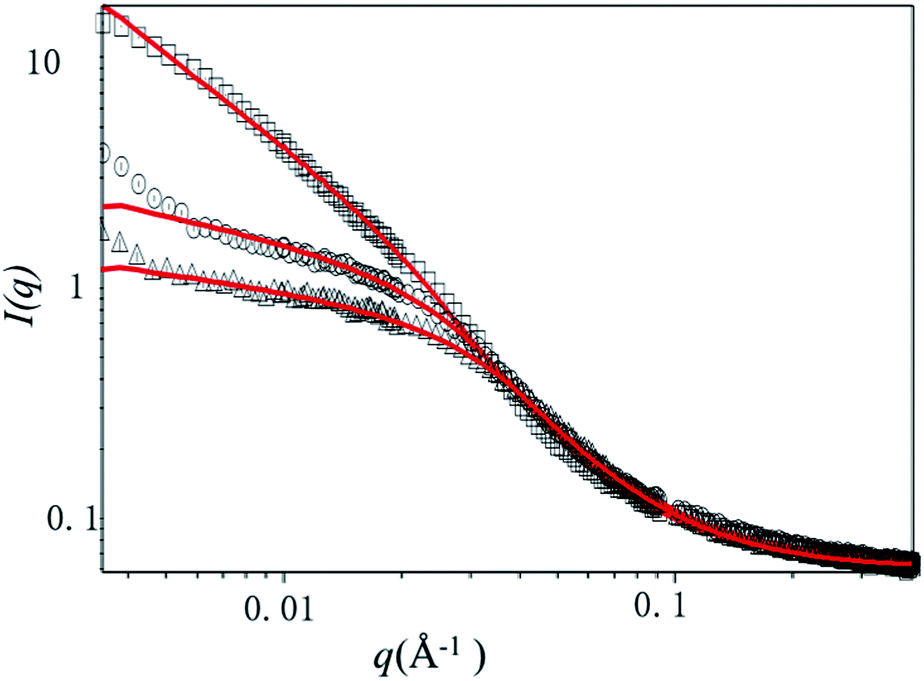 | ||
| Fig. 4 Representative SANS data and Guinier–Porod model fits. SANS data for P(9K-PNIPAAM-SH) (square), P(6K-PNIPAAM-SH) (circle), and P(4K-PNIPAAM-SH) (triangle) at 10 °C are shown along with corresponding Guinier–Porod model fits in red. | ||
Comparing three bottlebrush polymer samples at the lowest temperature studied, 10 °C, the radius increases with side-chain molecular weight, as expected, from 3.9 nm (P(4K-PNIPAAM-SH)) to 6.0 nm (P(9K-PNIPAAM-SH)). With increasing temperature, the radius of P(9K-PNIPAAM-SH) continuously decreases with temperature while that of P(4K-PNIPAAM-SH) and P(6K-PNIPAAM-SH) are roughly constant with temperature (Fig. 5). The dimension parameter s is relatively insensitive to temperature, but all samples show a slight increase with temperature. The dimension parameter is near 0.2 for both P(4K-PNIPAAM-SH) and P(6K-PNIPAAM-SH), indicating a slightly extended ellipsoidal globule, but 1 for P(9K-PNIPAAM-SH), reflecting a more extended shape in solution.
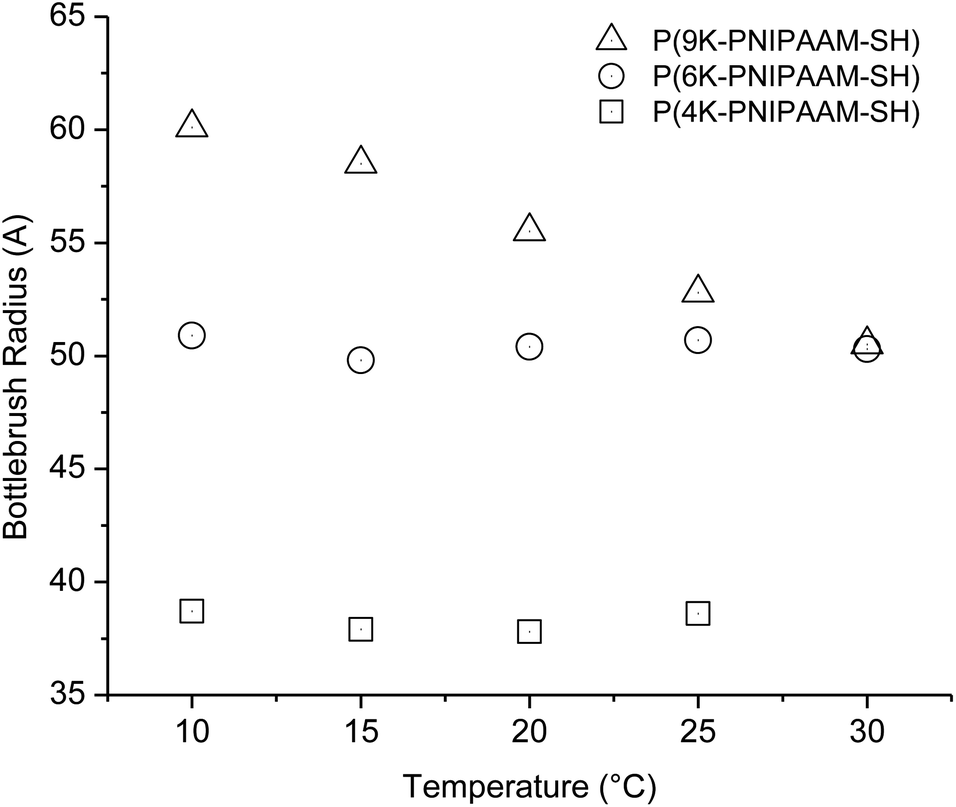 | ||
| Fig. 5 Bottlebrush polymer radius obtained from Guinier–Porod fitting for 1 wt% PNIPAAM bottlebrush polymers in D2O. | ||
Above the LCST, bottlebrush polymers become insoluble and scattering at low-q is dominated by large aggregates for all bottlebrush polymer samples. Consistent with DSC and turbidity measurements, aggregates are detected at 30 °C for P(4K-PNIPAAM-SH) but only above 30 °C for P(6K-PNIPAAM-SH) and P(9K-PNIPAAM-SH). Data for P(4K-PNIPAAM-SH) and P(6K-PNIPAAM-SH) can be fit to a Guinier–Porod model at temperatures above the LCST. This analysis indicates the radius increases above the LCST. By contrast, the shape of the SANS trace for P(9K-PNIPAAM-SH) is qualitatively different than P(4K-PNIPAAM-SH) and P(6K-PNIPAAM-SH), and the SANS data for P(9K-PNIPAAM-SH) solutions cannot be fit by a Guinier–Porod model. Notably, the SANS trace P(9K-PNIPAAM-SH) exhibits two peaks at q values of 0.0147 Å−1 and 0.0246 Å−1. The ratio of these peaks is close to √3, which is characteristic of cylindrical packing. Small changes in the peak positions are observed with temperature. The peaks shift to lower-q from with increasing temperature from 35 °C to 40 °C, but little change is seen on further heating to 50 °C.
The SANS data reflect significant conformational changes on heating up to and beyond the LCST. In the case of P(4K-PNIPAAM-SH) and P(6K-PNIPAAM-SH), the side-chain length is invariant with temperature below the LCST. Only a small decrease in the dimension parameter s is observed with increasing temperature. Above the LCST, SANS traces are dominated by scattering from large aggregates, and featureless traces reflect disordered aggregates. In the case of P(9K-PNIPAAM-SH), a significant reduction in bottlebrush polymer radius is observed, roughly 15%, when heating from 10 to 35 °C. Prior work has observed that end-tethered PNIPAAM brushes do not collapse on crossing the LCST due to steric interactions between chains.26,27 The results presented here indicate that significant side-chain collapse is only present for PNIPAAM bottlebrush polymers with long (∼9 kg mol−1) side-chains.
Above the LCST, P(9K-PNIPAAM-SH) forms structured aggregates, as reflected by the presence of peaks in the SANS traces. The positions of these peaks indicate cylindrically packed bottlebrush polymers, as shown schematically in Fig. 7 and changes in the peak positions on heating from 35 to 40 °C potentially reflect further collapse of the side-chains in this temperature range.
Early work with bottlebrush polymers reported the formation of lyotropic liquid crystal phases.28,29 However, more recent studies have failed to observe lyotropic liquid crystal phases in bottlebrush polymers prepared using a “grafting-from” technique.24,30 Lyotropic liquid crystal phases are expected for ratios of the persistence length to bottlebrush polymer radius greater than 10.30,31 To determine if liquid crystal phases were present in any of the bottlebrush polymer solutions, 1% mass fraction solutions in glass capillaries were analyzed by polarizing optical microscopy under crossed polarizers.
All solutions are clear near room temperature and become visibly turbid above the LCST. As shown in Fig. 6, solution birefringence is observed in the case of P(9K-PNIPAAM-SH) at temperatures above the LCST. The birefringence reversibly appears and disappears on heating and cooling the sample past the LCST. This is consistent with lyotropic liquid crystal ordering at temperatures above the LCST. Together with the SANS data, these results indicate the formation of a lyotropic liquid crystal phase with cylindrical packing of P(9K-PNIPAAM-SH) bottlebrush polymers above the LCST. The temperature-dependent conformation of all bottlebrush Polymers is depicted schematically in Fig. 7.
 | ||
| Fig. 6 Micrographs of P(4K-PNIPAAM-SH) (left), P(6K-PNIPAAM-SH) (middle), and P(9K-PNIPAAM-SH) (right) viewed under crossed polarizers at temperatures below (top) and above (bottom) LCST. | ||
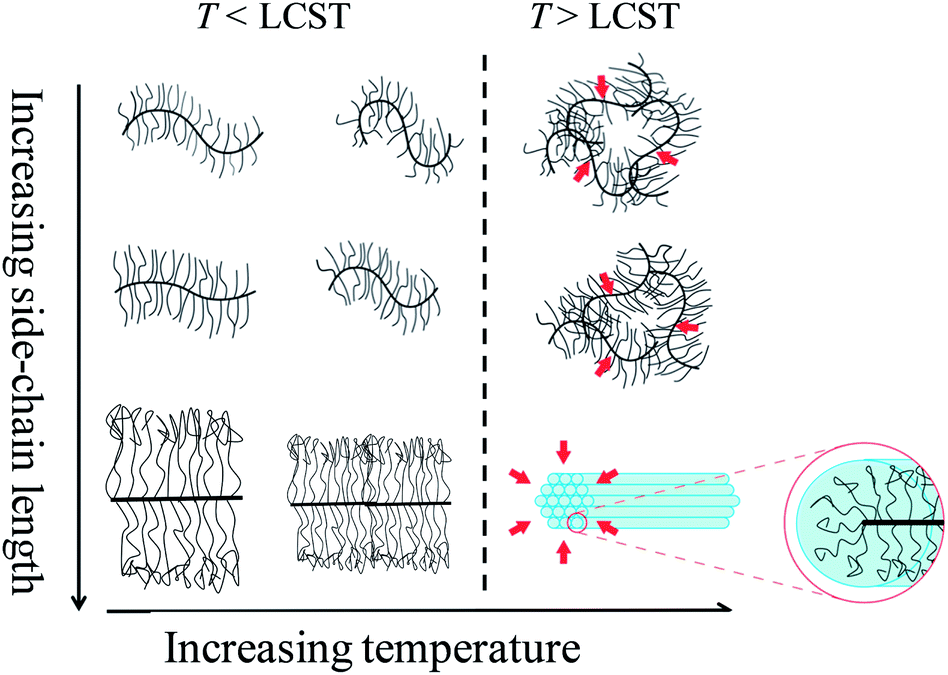 | ||
| Fig. 7 Schematic for changing bottlebrush polymer conformation with temperature. For short side-chains, the side-chain length is invariant with temperature. Above the LCST, bottlebrush polymers form disordered aggregates. For long side-chains, the side-chain length decreases with temperature, and above the LCST bottlebrush polymers exhibit lyotropic liquid crystal ordering due to the formation of structured aggregates. | ||
Interfacial properties of bottlebrush polymers
Prior work has shown that polymer-coated nanoparticles and branched polymers are interfacially active materials.8,32 Bottlebrush polymers have the advantage that the side-chain and backbone length can be precisely controlled, and the results above show that the side-chain flexibility can be tuned by changing the side-chain length. Here, we explored the reduction in interfacial tension (IFT) between water and chloroform with the addition of PNIPAAM bottlebrush polymers.To analyze the interfacial properties of PNIPAAM bottlebrush polymers, 1 wt% solutions of bottlebrush polymers in chloroform were prepared. These solution were mixed with water (50% v/v) and the mixture was equilibrated for at least 24 h. Visually, a noticeable change in the curvature at the interface between water and chloroform is observed with the addition of bottlebrush polymers (Fig. 8). Compared with a chloroform/water control, solutions containing bottlebrush polymer exhibited a noticeably flatter interface at temperatures below the LCST. When heated above the LCST, the interface curvature increased, indicating an increase in the interfacial tension. This suggests that PNIPAAM bottlebrush polymers reduce the IFT between chloroform and water below the LCST.
 | ||
| Fig. 8 Micrographs of water/chloroform two-phase mixtures with no polymer added (middle), with 0.1% mass fraction P(6K-PNIPAAM-SH) at room temperature (right), and with 0.1% mass fraction P(6K-PNIPAAM-SH) at T > LCST (left). | ||
Quantitative values for the IFT were obtained using the pendant drop method (Table 3). At room temperature, both linear PNIPAAM (3.4 ± 0.32 mN m−1) and bottlebrush PNIPAAM (4.99 ± 0.78 mN m−1) reduce the IFT by one order of magnitude. Above the LCST, the IFT increases for all solutions due to the reduced hydrophilicity of PNIPAAM. The IFT values for bottlebrush polymer and PNIPAAM homopolymer solutions are comparable, with PNIPAAM homopolymer solutions exhibiting lower IFT values at temperatures below the LCST. Since IFT is strongly dependent on the salinity of aqueous phase and bottlebrush polymers may be of interest for applications such as enhanced oil recovery, we explored the IFT between chloroform and brine.33,34 As shown in Table 3, IFT values are consistently lower in the presence of brine, but the trends are qualitatively the same as in water.
| System | IFT mN m−1 | |
|---|---|---|
| R.T. | 35 °C | |
| a All IFT values measured by the pendant drop method with 0.1 wt% polymer added. | ||
| No added polymer | 28.84 ± 1.53 | N/A |
| P(4K-PNIPAAM-SH), 0% NaCl | 6.89 ± 1.79 | 12.22 ± 0.31 |
| P(6K-PNIPAAM-SH), 0% NaCl | 4.99 ± 0.78 | 20.28 ± 1.14 |
| P(9K-PNIPAAM-SH), 0% NaCl | 11.75 ± 0.94 | 20.62 ± 1.59 |
| 6K-PNIPAAM-SH, 0% NaCl | 3.40 ± 0.32 | 18.86 ± 0.93 |
| P(4K-PNIPAAM-SH), 10% NaCl | 4.29 ± 1.16 | 8.54 ± 1.27 |
| P(6K-PNIPAAM-SH), 10% NaCl | 1.50 ± 0.65 | 26.44 ± 1.65 |
| P(9K-PNIPAAM-SH), 10% NaCl | 7.58 ± 0.28 | 11.58 ± 0.68 |
| 6K-PNIPAAM-SH, 10% NaCl | 2.21 ± 0.35 | 27.16 ± 1.36 |
A difference between homopolymer and bottlebrush polymer solutions is observed on vigorous shaking of the chloroform/water solutions. In homopolymer solutions, a thick emulsion forms that does not settle for up to several weeks, but bottlebrush polymer solutions remain clear (see Fig. S7†). This may indicate the inability for bottlebrush polymers to stabilize highly curved interfaces, as has been observed for bottlebrush polymers and nanoparticles at liquid–liquid interfaces.35 These results suggest that bottlebrush polymers may be useful as interfacially active materials, but further work is needed to understand the role of side-chain length and interface curvature.
Conclusions
Well-defined water-soluble PNIPAAM bottlebrushes were synthesized via a “grafting through” method, combining RAFT and ROMP polymerization techniques. Both side-chain end-groups and side-chain lengths impact solubility, phase behavior, lower-critical solution temperature in water, and interfacial properties. PNIPAAM bottlebrush polymers with 9 kg mol−1 side-chains exhibit a temperature dependent conformation below the LCST, and above the LCST they exhibit lyotropic liquid crystal ordering. PNIPAAM bottlebrush polymers reduce the interfacial tension between chloroform and water but do not stabilize the formation of microemulsions. These results demonstrate the role of side-chain conformation and flexibility on macroscopic and interfacial properties.Acknowledgements
Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund (grant # 52435-DNI7) for support of this research. X.L acknowledges support from the Rice University Kobayashi Fellowship. S. L. P. acknowledges support from the National Science Foundation Graduate Fellowship Program (grant # 0940902). We acknowledge SANS beam time obtained from the NIST Center for Neutron Research (U.S. Department of Commerce) which is supported in part by the National Science Foundation under Agreement no. DMR-0944772. The identification of commercial products does not imply endorsement by the National Institute of Standards and Technology nor does it imply that these are the best for the purpose.Notes and references
- G. M. Miyake, V. A. Piunova, R. A. Weitekamp and R. H. Grubbs, Angew. Chem., Int. Ed., 2012, 51, 11173 CrossRef CAS.
- M. B. Runge and N. B. Bowden, J. Am. Chem. Soc., 2007, 129, 10551–10560 CrossRef CAS PubMed.
- J. A. Johnson, Y. Y. Lu, A. O. Burts, Y.-H. Lim, M. G. Finn, J. T. Koberstein, N. J. Turro, D. A. Tirrell and R. H. Grubbs, J. Am. Chem. Soc., 2010, 133, 559–566 CrossRef PubMed.
- A. O. Burts, Y. Li, A. V. Zhukhovitskiy, P. R. Patel, R. H. Grubbs, M. F. Ottaviani, N. J. Turro and J. A. Johnson, Macromolecules, 2012, 45, 8310–8318 CrossRef CAS.
- Z. Li, J. Ma, C. Cheng, K. Zhang and K. L. Wooley, Macromolecules, 2010, 43, 1182–1184 CrossRef CAS.
- Y. Li, J. Zou, B. P. Das, M. Tsianou and C. Cheng, Macromolecules, 2012, 45, 4623–4629 CrossRef CAS.
- S.-k. Ahn, D. L. Pickel, W. M. Kochemba, J. Chen, D. Uhrig, J. P. Hinestrosa, J.-M. Carrillo, M. Shao, C. Do, J. M. Messman, W. M. Brown, B. G. Sumpter and S. M. Kilbey, ACS Macro Lett., 2013, 761–765 CrossRef CAS.
- X. Li, S. L. Prukop, S. L. Biswal and R. Verduzco, Macromolecules, 2012, 45, 7118–7127 CrossRef CAS.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A: Pure Appl.Chem., 1968, 2, 1441–1455 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- H. Yim, M. S. Kent, S. Satija, S. Mendez, S. S. Balamurugan, S. Balamurugan and G. P. Lopez, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72, 051801 CrossRef CAS.
- A. Vidyasagar, J. Majewski and R. Toomey, Macromolecules, 2008, 41, 919–924 CrossRef CAS.
- H.-P. Hsu, W. Paul, S. Rathgeber and K. Binder, Macromolecules, 2010, 43, 1592–1601 CrossRef CAS.
- T. M. Birshtein, O. V. Borisov, Y. B. Zhulina, A. R. Khokhlov and T. A. Yurasova, Polym. Sci. USSR, 1987, 29, 1293–1300 CrossRef.
- T. Kawano, Y. Niidome, T. Mori, Y. Katayama and T. Niidome, Bioconjugate Chem., 2009, 20, 209–212 CrossRef CAS PubMed.
- Y. Shen, M. Kuang, Z. Shen, J. Nieberle, H. Duan and H. Frey, Angew. Chem., Int. Ed., 2008, 47, 2227–2230 CrossRef CAS PubMed.
- J. Ye, Y. Hou, G. Zhang and C. Wu, Langmuir, 2008, 24, 2727–2731 CrossRef CAS PubMed.
- G. Wei, F. Wen, X. Zhang, W. Zhang, X. Jiang, P. Zheng and L. Shi, J. Colloid Interface Sci., 2007, 316, 53–58 CrossRef CAS PubMed.
- S. L. Pesek, X. Li, B. Hammouda, K. Hong and R. Verduzco, Macromolecules, 2013, 46, 6998–7005 CrossRef CAS.
- M. A. Harvison and A. B. Lowe, Macromol. Rapid Commun., 2011, 32, 779–800 CrossRef CAS PubMed.
- R. Elisabetta, et al. , Biomed. Mater., 2010, 5, 035012 CrossRef PubMed.
- K. Rahimian-Bajgiran, N. Chan, Q. Zhang, S. M. Noh, H.-i. Lee and J. K. Oh, Chem. Commun., 2013, 49, 807–809 RSC.
- Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- B. Zhang, F. Gröhn, J. S. Pedersen, K. Fischer and M. Schmidt, Macromolecules, 2006, 39, 8440–8450 CrossRef CAS.
- S. Rathgeber, T. Pakula, A. Wilk, K. Matyjaszewski and K. L. Beers, J. Chem. Phys., 2005, 122, 124904–124913 CrossRef PubMed.
- X. Zhu, C. Yan, F. M. Winnik and D. Leckband, Langmuir, 2006, 23, 162–169 CrossRef PubMed.
- K. N. Plunkett, X. Zhu, J. S. Moore and D. E. Leckband, Langmuir, 2006, 22, 4259–4266 CrossRef CAS PubMed.
- M. Wintermantel, K. Fischer, M. Gerle, R. Ries, M. Schmidt, K. Kajiwara, H. Urakawa and I. Wataoka, Angew. Chem., Int. Ed., 1995, 34, 1472–1474 CrossRef CAS.
- Y. Tsukahara, Y. Ohta and K. Senoo, Polymer, 1995, 36, 3413–3416 CrossRef CAS.
- S. Rathgeber, T. Pakula, A. Wilk, K. Matyjaszewski, H.-i. Lee and K. L. Beers, Polymer, 2006, 47, 7318–7327 CrossRef CAS PubMed.
- P. van der Schoot, J. Chem. Phys., 1996, 104, 1130–1139 CrossRef CAS PubMed.
- D. Maillard, S. K. Kumar, B. Fragneaud, J. W. Kysar, A. Rungta, B. C. Benicewicz, H. Deng, L. C. Brinson and J. F. Douglas, Nano Lett., 2012, 12, 3909–3914 CrossRef CAS PubMed.
- K. Schaller, S. Fox, D. Bruhn, K. Noah and G. Bala, Appl. Biochem. Biotechnol., 2004, 115, 827–836 CrossRef.
- X. Li, E. Boek, G. C. Maitland and J. P. M. Trusler, J. Chem. Eng. Data, 2012, 57, 1078–1088 CrossRef CAS.
- V. Garbin, J. C. Crocker and K. J. Stebe, Langmuir, 2011, 28, 1663–1667 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sm52614c |
| This journal is © The Royal Society of Chemistry 2014 |