 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHexagonal closest-packed spheres liquid crystalline phases stabilised by strongly hydrated counterions†
Connie K.
Liu
and
Gregory G.
Warr
*
School of Chemistry F11, The University of Sydney, NSW, 2006 Australia. E-mail: gregory.warr@sydney.edu.au; Fax: +612 9351 3329; Tel: +612 9351 2106
First published on 16th October 2013
Abstract
The sequence and structure of lyotropic liquid crystals formed in C12–C16 alkyltrimethylammonium surfactants with hydrolysable and multivalent phosphate (PO43−, HPO42− and H2PO4−), oxalate (HC2O4− and C2O42−), and carbonate (HCO3−/CO32−) counterions were determined using a concentration gradient method coupled with polarising optical microscopy and small angle X-ray scattering. In addition to the discrete cubic (I1, space group Pm3n) and hexagonal (H1, p6m) phases, almost all of these surfactants also formed the (previously) rare hexagonally closest-packed spheres (HCPS, P63/mmc) phase at compositions between the Pm3n cubic and L1 micellar phases. This structure has not been previously observed in cationic surfactants, but is readily achieved by using strongly hydrated counterions to stabilise spherical micelles at high concentrations.
Introduction
The structure of liquid crystalline phases formed in aqueous solutions by surfactant self-assembly above the critical micelle concentration (CMC), depends on the packing of the surfactant molecules into micelles. The hydrocarbon chain length (lc) and volume (v), together with the effective head group area (a0) determine the value of the packing parameter, v/a0lc, and the propensity of a surfactant to form spherical micelles (v/a0lc ≤ 1/3), cylinders (1/3 < v/a0lc ≤ 1/2), or bilayers (1/2 < v/a0lc ≤ 1) under different solution conditions.1 As the surfactant concentration increases, self-assembly that usually begins with spherical micelles evolves into structures with lower curvature. Micelles adopt liquid-like order in isotropic solution (L1) transition first into discrete cubic (I1), then hexagonal (H1), bicontinuous cubic (V1) and lamellar (Lα) phases, as typified by the well-known behavior of dodecyltrimethylammonium chloride (DTAC).2The optically isotropic, “discrete micellar” cubic phase is found at compositions between the micellar solution and hexagonal phase. Here micelles are usually arranged into 3D cubic symmetries including body-centered cubic (BCC, Im3m), face-centered cubic (FCC, Fm3m, Fd3m) and tetrahedrally close-packed (Pm3n) structures. Pm3n is by far the most common of these phases, and since it was first reported in DTAC2 it has been found in numerous other cationic,3 anionic,4 ternary,5 non-ionic surfactant6,7 and phospholipid8,9 systems.10 Also sometimes counted as an I1 phase is the hexagonally close-packed spheres (HCPS, P63/mmc) structures, however this is only rarely observed, and then often classified separately.11–13
The discrete micellar cubic phase is something of a puzzle. Often described in texts as an ordered array of spherical micelles, it curiously does not have either the fcc or hcp symmetry of a closest-packed lattice. Three structures have been proposed for the Pm3n cubic phase, as illustrated in Fig. 1. Two of these suggest that at least some of the micelles on the cubic lattice are non-spherical. From NMR studies on the Pm3n cubic phase formed in the palmitoyllysophosphatidylcholine (PLPC)-water system, Fontell et al. and Eriksson et al. proposed a unit cell of 8 identical, slightly elongated micelles where those on the cube vertices are able to rotate freely in all directions while those on the cube faces have only one axis of rotation.14,15 From X-ray diffraction, freeze fracture electron microscopy and electron density mapping of the Pm3n cubic phase of DTAC, Vargas, Luzzati, and Delacroix proposed a unit cell containing two spherical and 6 disc shaped micelles.9,16 As the NMR results for PLPC yield a time-average structure close to the DTAC X-ray structure, it is possible that these two non-spherical micelle models are essentially equivalent (see Fig. 1).
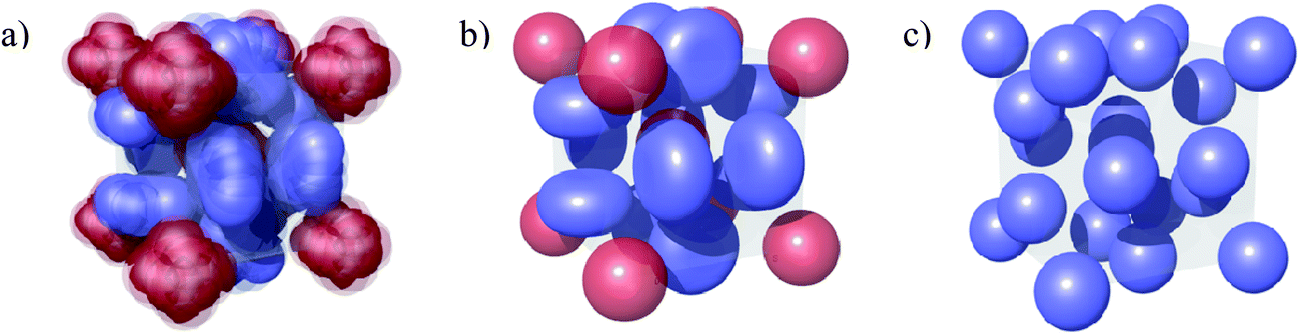 | ||
| Fig. 1 Comparison of proposed Pm3n unit cell structures comprised of: (a) 8 identical, slightly elongated rods illustrating free rotation of (red) micelles at the corner and in the centre of the unit cell and restricted rotation of (blue) micelles on its faces;14,15 (b) 2 spherical (red) and 6 disc-shaped (blue) micelles;9,16 (c) 8 identical spherical micelles.17 | ||
The third explanation of the observed symmetry, due to Imai et al. and based on studies of the C12EO8–water system,17 is that the spherical micelles in this phase pack so as to minimise the total interfacial contact area between neighbours, rather than to maximise packing density (whence fcc or hcp). This effect may be specific to the compression of the hydrophilic chains of polyoxyethylene in the micelle corona of these nonionic surfactants.
In contrast, the hexagonally closest-packed spheres phase (HCPS), which might be expected on packing density grounds, is rare. First identified in the binary non-ionic surfactant, C12EO8–water system at 31.5–39 wt% surfactant, and below 18 °C6 electron density mapping has confirmed that micelles are spheres with their polar ethylene oxide moieties in nearest-neighbour contact.12 Since then, the HCPS phase has only been found in one other binary system, comprising anionic choline carboxylate–water mixtures in a phase sequence that included the Pm3n cubic, hexagonal, bicontinuous cubic and lamellar phases at higher concentrations.18
HCPS has also been observed in two other, multicomponent systems. Soni et al. identified the HCPS phase in the triblock copolymer system poly(ethyleneoxide)20–poly(propylene oxide)70–poly(ethyleneoxide)20, (Pluronic® P123) with ethanol and water5 and also recently, inverse micelles adopting the HCPS arrangement was found by Sherman et al. in a mixture of dioleoylphosphatidylcholine, dioleoylglycerol, cholesterol and water.19 HCPS has also been reported as surface structures in silica films formed from the cationic surfactant CTAB,20 and in copolymer gels of methacrylate (MAA) and N-isopropylacrylamide (NIPAM), P(MAA/NIPAM), associated with tetradecyl- and dodecyltrimethylammonium surfactant cations.21
The primary surfactant packing condition that favours the formation of the HCPS phase over Pm3n (or hexagonal) structures is thus the preservation of spherical micelles at high surfactant concentration. As a secondary condition, steric interactions, or any interactions which might favour minimizing contact area between neighbouring micelles, are best avoided. This suggested to us that ionic surfactants with weakly binding counterions which would resist sphere-to-cylinder transformation by added electrolyte should stabilize the HCPS phase. We have recently shown22 that resiliently-spherical micelles are formed by cationic surfactants with hydrolysable phosphate (PO43−, HPO42− and H2PO4−), oxalate (HC2O4− and C2O42−) and carbonate (HCO3−/CO32−) counterions. Although weakly binding in the monovalent state, these anions remain strongly hydrated even when hydrolysed into their divalent or trivalent forms and are strongly electrostatically bound to the cationic micelles, and exhibit negligible micelle growth even at high common-ion electrolyte concentrations. This makes them excellent candidates for formation of HCPS phases.
Materials and methods
Alkyltrimethylammonium surfactants with hydrolysable carbonate, phosphate and oxalate counterions were prepared and characterized as described previously.22 These solutions will undergo some degree of anion speciation by hydrolysis, but are identified throughout according to the parent salt e.g. (DTA)3PO4, DTAHC2O4, etc. Although prepared as carbonate salts, these surfactants exist primarily as bicarbonates in solution due to atmospheric CO2. Polarizing optical microscopy and small angle X-ray scattering (SAXS) were both used to identify the sequence and structure of lyotropic phases formed in binary surfactant–water mixtures.In polarising optical microscopy, a Leica DM LB microscope fitted with cross polarisers, quarter-wave plate and a DC 300 digital camera was used. All experiments were performed at room temperature (23 °C). Concentration gradients were prepared by placing a small amount of solid surfactant sample onto a microscope slide, a cover-slip pressed on top and a few drops of water are added under the edge of the cover-slip. Water is drawn under the cover-slip through capillary action towards the surfactant sample creating a concentration gradient forming the full sequence of phases present in the surfactant–water mixture. Phases form separate bands which are distinguished by refractive index discontinuities. The phases can then be identified by their viscosity, birefringence and characteristic optical textures.11,23,24
Small angle X-ray scattering experiments were performed on the SAXS/WAXS beamline at The Australian Synchrotron, Victoria, Australia. The high intensity of a synchrotron X-ray beam allows for much higher resolution of scattering peaks and the detection of weak reflections. Concentration gradient samples were prepared by packing solid surfactant into the bottom of a 1.5 mm diameter glass capillary (Charles Supper Company). Water was then added and allowed to penetrate the surfactant and establish a concentration gradient over a 12–24 hour period prior to being scanned with the synchrotron X-ray beam. This experimental set up was inspired by a similar methodology developed by Ricoul et al. in the determination of ternary phase diagrams of cationic surfactant–glycolipid–water systems.25 The entire length of the capillary containing the sample was scanned at 0.80 mm (vertical) increments with synchrotron X-rays (λ = 0.6888 Å or 1.0332 Å). Typically, 40 points along the capillary was scanned in order to ensure that all the phases formed were sampled. The scattered X-rays were collected on a Dectris-Pilatus 1 M detector over a q-range of 0.008–0.7 Å−1. Spectra were collected for 10 or 30 s and measurements were performed at ambient temperature in the instrument hutch, 27 °C. The resulting characteristic diffraction patterns were indexed and the space group symmetry and consequently the structure of liquid crystalline phases are identified.6,11
Individual SAXS patterns are obtained for discrete points along a similarly-established gradient but within a sample capillary, allowing each phase formed to be examined. However, as phase boundaries formed within a capillary are not necessarily horizontal, and the beam itself is 0.250 mm (horizontal) × 0.150 mm (vertical) (FWHM), two adjacent phases were sometimes sampled simultaneously. Phases that occur at a narrow concentration range may be completely missed by the scanning synchrotron X-ray beam. Thus, all SAXS scans are interpreted in conjunction with the corresponding polarising optical microscopy image. The complete set of polarising optical microscopy flooding experiments may be found in the ESI.†
Results and discussion
Fig. 2 shows a representative polarising optical micrograph of a concentration gradient experiment on DTA2CO3, together with synchrotron SAXS patterns of a similar gradient sample. The two adjacent isotropic lyotropic phases denoted I′1 and I1 are separated by a refractive index discontinuity. The corresponding SAXS patterns, from bottom to top, confirm the structure sequence as micellar solution, then HCPS (P63/mmc), discrete cubic (Pm3n) and hexagonal phase (p6m), and confirm the existence of two distinct I1 structures; the more dilute corresponds to HCPS and the more concentrated to Pm3n. This sequence is unusual, and few systems with the adjacent discrete micellar phases have been found.6,7,18,26,27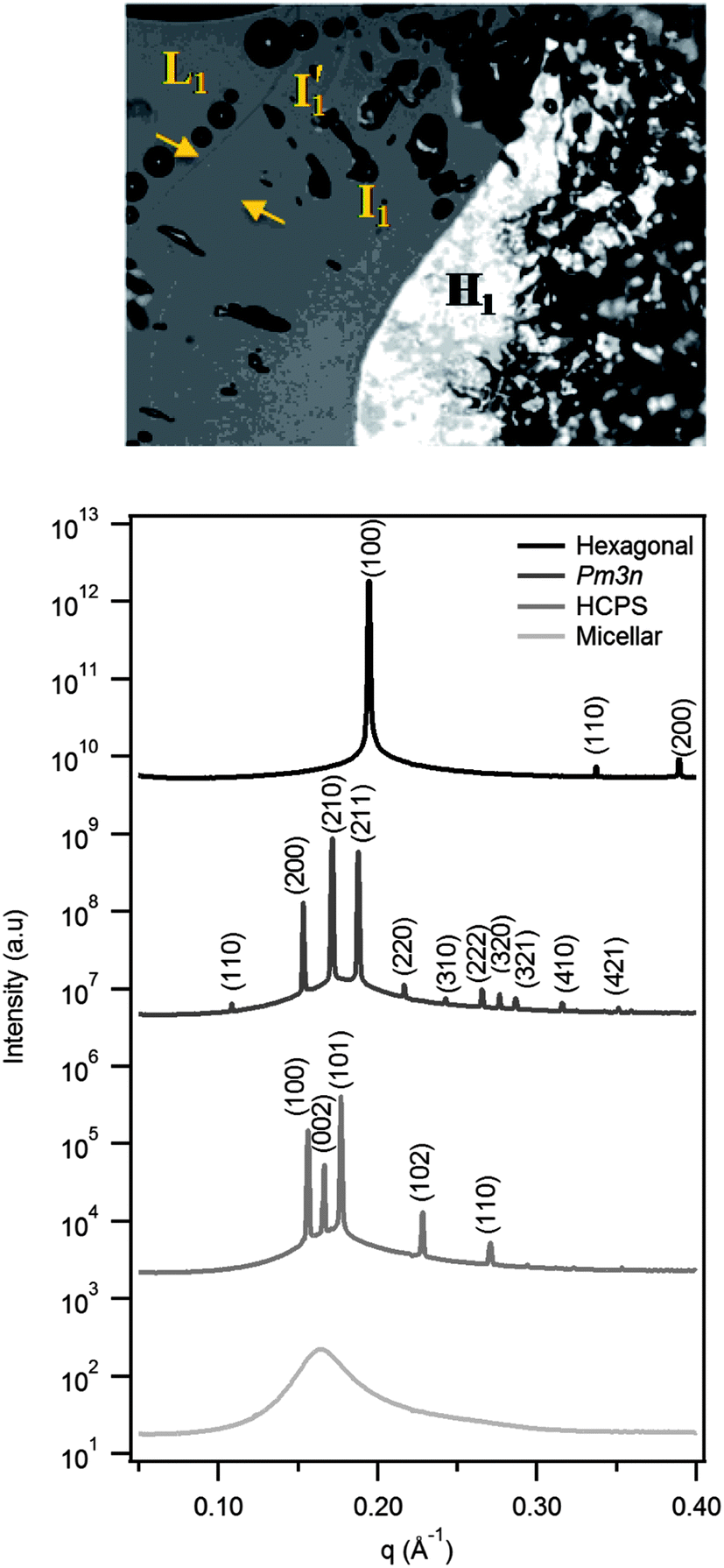 | ||
| Fig. 2 Polarising optical microscopy and corresponding synchrotron SAXS diffraction patterns showing the phase sequence for DTA2CO3 in a concentration gradient scan. SAXS patterns show increasing concentration from bottom to top. The phases and symmetry/space group are identified as micelles (L1), I′1 HCPS (P63/mmc), I1 cubic (Pm3n) and H1 hexagonal (p6m). Curves are offset for clarity. | ||
In fact all dodecyl-, tetradecyl- and hexadecyltrimethylammonium surfactants with hydrolysable phosphate, oxalate and carbonate counterions (except CTAHC2O4) showed two such adjacent, I1 phases no matter what the hydrolysis state of the counterion. (See ESI† for complete set of polarizing optical micrographs.) Table 1 summarises the phases observed, including comparative literature data for bromide, chloride and sulfate counterions. For the alkyltrimethylammonium halides and cetyltrimethylammonium sulfate (CTA2SO4), bicontinuous cubic, intermediate and lamellar phases have also been reported at higher temperatures and surfactant concentrations above the hexagonal phase.24,28–32
| Surfactant series | Counterion | L1 | HCPS | Pm3n | H1 |
|---|---|---|---|---|---|
| a [1] ref. 32, [2] ref. 24, [3] ref. 2, [4] ref. 28, [5] ref. 29, [6] ref. 31, [7] ref. 30. b Cubic phase identified but symmetry not confirmed (i.e. not necessarily Pm3n). | |||||
| DTA+ (C12) | Br− | X1 | — | — | X1 |
| Cl− | M2 | — | M2, X3 | M2, X4 | |
| Phosphates | PO43− | X | X | X | X |
| HPO42− | X | X | X | X | |
| H2PO4− | X | X | X | X | |
| Oxalates | C2O42− | X | X | X | X |
| HC2O4− | X | M | X | X | |
| Carbonate | HCO3− | X | X | X | X |
| TTA+ (C14) | Br− | M5 | — | — | M5 |
| Cl− | M2 | — | M2b | M2 | |
| Phosphates | PO43− | X | X | X | X |
| HPO42− | X | M | X | X | |
| H2PO4− | M | M | M | M | |
| Oxalates | C2O42− | X | M | X | M |
| HC2O4− | X | M | X | X | |
| Carbonate | HCO3− | X | X | X | X |
| CTA+ (C16) | Br− | X6 | — | — | X6 |
| Cl− | X4 | — | — | X4 | |
| SO42− | M7 | — | M7b | M7 | |
| Phosphates | PO43− | X | M | X | M |
| HPO42− | X | M | X | M | |
| H2PO4− | X | M | X | X | |
| Oxalates | C2O42− | X | X | X | M |
| HC2O4− | X | — | X | X | |
| Carbonate | HCO3− | X | M | M | X |
Fig. 3 shows the characteristic SAXS patterns of HCPS phases found in the three DTA+ phosphate surfactants, irrespective of the hydrolysis state of the counterion. For all the phosphate surfactants examined, both the HCPS and Pm3n discrete cubic phases are preserved as the chain length is increased from C12 to C16. The HCPS phase appears to form over a narrower composition range with longer alkyl chains however, as the X-ray scans either missed or showed overlapping SAXS patterns in this region in the majority of samples (see ESI† for an example).
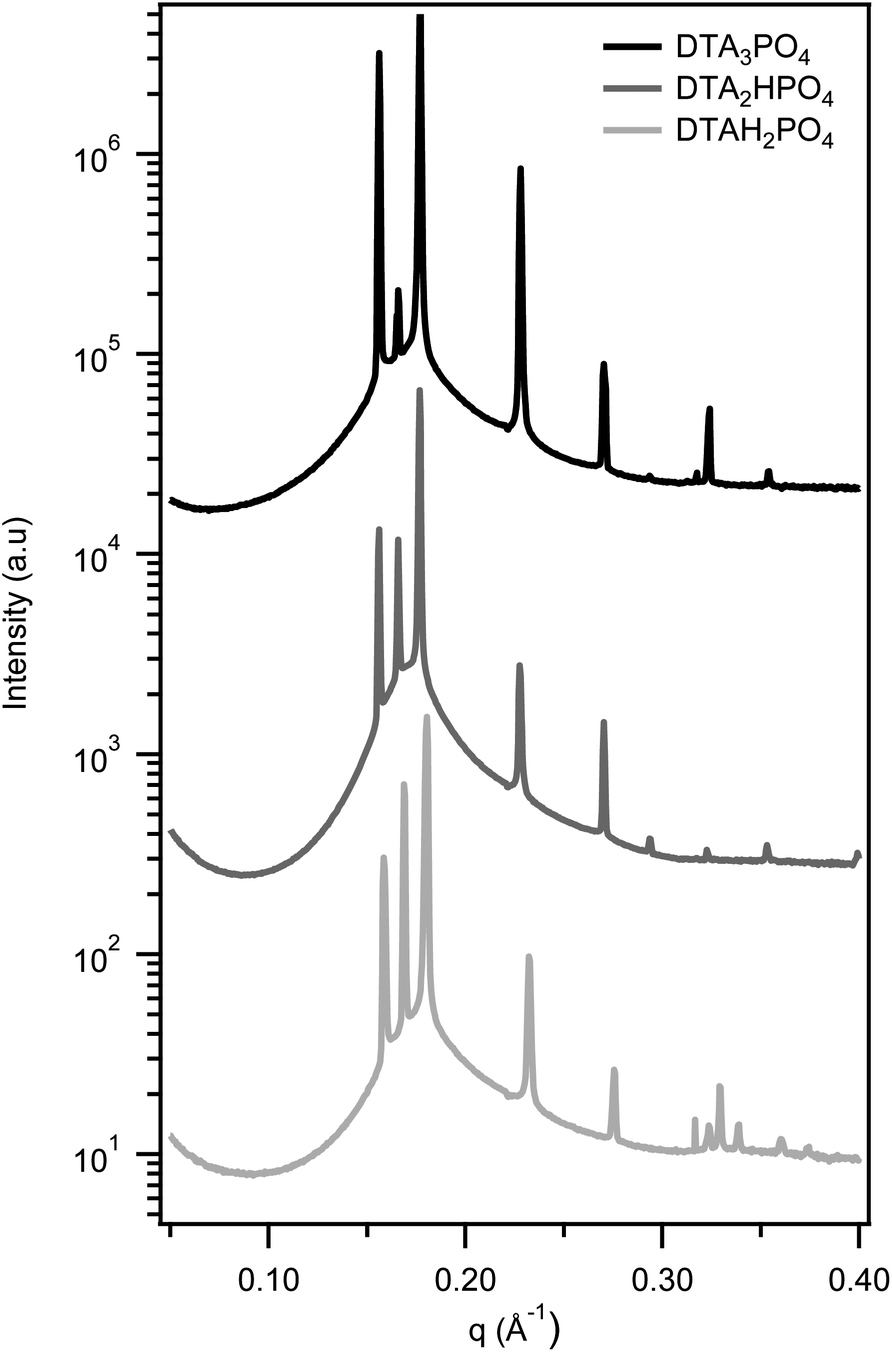 | ||
| Fig. 3 SAXS patterns showing the HCPS phase with P63/mmc symmetry is formed in all DTA+ phosphate surfactants. HCPS appears before the Pm3n cubic phase and after the micellar solution in all cases, irrespective of the hydrolysis state of the phosphate counterion. Curves are offset for clarity. | ||
In the HCPS phase, the constituent micelles are closest-packed, identical spheres.12,17 Its widespread occurrence in these surfactants is consistent with our recent small-angle neutron scattering investigation of the L1 phase of these systems, which showed the spherical micelles to be extremely stable and resistant to a sphere-to-rod transformation.22
Specific counterion effects are one of the best known ways of controlling surfactant self-assembly structure. Changing counterions changes the effective head group area a0 and therefore the surfactant packing parameter, v/a0lc. Conventionally, weaker counterion binding is associated with larger a0, and favours spherical micelles. This is exemplified by comparing chloride counterions with the more strongly binding bromide in DTAC2versus DTAB.32 By binding more strongly (and being less hydrated at the micelle surface), bromide more effectively screens repulsions between cationic headgroups.33–35 Thus it reduces a0 and increases the surfactant packing parameter, favouring the formation of rod-like micelles. Consequently, DTAB forms a hexagonal phase adjacent to its micellar phase at room temperature while DTAC first forms the Pm3n cubic phase.
An unusual feature of the counterions in the present study is that they can be strongly binding in their multivalent hydrolysis state(s), as measured by the residual micellar charge from e.g. conductivity.22 Both phosphate and oxalate ions are, however, known to remain strongly hydrated and to have large radii of hydration.36–39 Thus, despite binding strongly to micelles, these ions are unable to effectively screen the repulsions between neighbouring headgroups within a micelle. This results in the preservation of spherical micelles with a large head group area, a0. The formation of HCPS phases in all DTA+ phosphates, oxalates and bicarbonate, but not in DTAC, underscores that these counterions at all hydrolysis states behave like a more weakly binding ion than chloride. This is further highlighted by the persistence of the HCPS phase as the alkyl chain length is increased from C12 to C16, where even the Pm3n cubic phase is no longer formed by the chloride salt.
Only in CTAHC2O4 was the HCPS phase not detected. This suggests that the monovalent HC2O4− more effectively screens neighbouring head groups than its divalent C2O42− form. As with the phosphates, dilute solution measurements have shown that C2O42− neutralizes the micelle charge more effectively than HC2O4−, meaning that hydration of C2O42− must be the determining factor.22 Para and Warszynski have shown in a related system that HSO4− binds more strongly to a surface than SO42−, as the divalent ions are more strongly hydrated.40
The almost ubiquitous observation of small, spherical micelles in both the L1 and HCPS phases of these cationic surfactants, together with the recurring phase progression on to I1 (Pm3n) and H1 suggests to us that curvature changes drive changes in self-assembly structure. It is harder to rationalize a change in spherical micelle packing from maximum density in HCPS to area minimization in I1 at higher concentration, as proposed by Imai.17
In these systems the decreased availability of water to hydrate the counterions with increasing surfactant concentration may be the cause of changes in counterion binding, and of the accompanying reduction in a0 that leads to the formation of less curved aggregates. With increasing concentration we would thus expect a transition from spheres to aspherical micelles (either short rods or disks, as in Fig. 1(a) and (b)) in the Pm3n cubic phase, before elongated cylinders form in H1, as is observed.
Conclusions
The rare hexagonally close-packed spherical micelles (HCPS, P63/mmc space group) lyotropic liquid crystalline phase is not really so rare. It can be commonly found in alkyltrimethylammonium surfactants associated with strongly hydrated counterions such as the hydrolysable phosphate (PO43−, HPO42− and H2PO4−), oxalate (HC2O4− and C2O42−), and carbonate (HCO3−/CO32−). In this phase, micelles are spherical, consistent with the stability of spherical micelles of these surfactants in more dilute solution.The HCPS phase persists for alkyl chain lengths up to sixteen carbons for alkyltrimethylammonium phosphate, carbonate and oxalates regardless of the hydrolysis state of the counterion, except for hexadecyltrimethylammonium HC2O4−, which forms the Pm3n cubic phase as its most dilute lyotropic phase. With increasing surfactant concentration all the systems examined form the Pm3n cubic and then the H1 hexagonal phase.
Acknowledgements
This research was undertaken on the SAXS/WAXS beamline at the Australian Synchrotron, Victoria, Australia, and we thank Dr Nigel Kirby for his assistance. CKL acknowledges receipt of an Australian Postgraduate Award and an AINSE PGRA scholarship.References
- D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1981, 77, 601–629 RSC.
- R. R. Balmbra, J. S. Clunie and J. F. Goodman, Nature, 1969, 222, 1159–1160 CrossRef CAS.
- A. Svensson, L. Piculell, L. Karlsson, B. Cabane and B. Jonsson, J. Phys. Chem. B, 2003, 107, 8119–8130 CrossRef CAS.
- D. Kaneko, U. Olsson and K. Sakamoto, Langmuir, 2002, 18, 4699–4703 CrossRef CAS.
- S. S. Soni, G. Brotons, M. Bellour, T. Narayanan and A. Gibaud, J. Phys. Chem. B, 2006, 110, 15157–15165 CrossRef CAS PubMed.
- M. Clerc, J. Phys. II, 1996, 6, 961–968 CrossRef CAS.
- P. Sakya, J. M. Seddon, R. H. Templer, R. J. Mirkin and G. J. T. Tiddy, Langmuir, 1997, 13, 3706–3714 CrossRef CAS.
- P. Mariani, V. Luzzati and H. Delacroix, J. Mol. Biol., 1988, 204, 165–189 CrossRef CAS.
- R. Vargas, P. Mariani, A. Gulik and V. Luzzati, J. Mol. Biol., 1992, 225, 137–145 CrossRef CAS.
- K. Fontell, Colloid Polym. Sci., 1990, 268, 264–285 CAS.
- S. T. Hyde, in Handbook of Applied Surface and Colloid Chemistry, Volumes 1–2, ed. K. Holmberg, John Wiley & Sons, 2001, pp. 299–321 Search PubMed.
- X. Zeng, Y. Liu and M. Impéror-Clerc, J. Phys. Chem. B, 2007, 111, 5174–5179 CrossRef CAS PubMed.
- G. C. Shearman, A. I. I. Tyler, N. J. Brooks, R. H. Templer, O. Ces, R. V. Law and J. M. Seddon, Liq. Cryst., 2010, 37, 679–694 CrossRef CAS.
- K. Fontell, K. K. Fox and E. Hansson, Mol. Cryst. Liq. Cryst., 1985, 1, 9–17 CAS.
- P. O. Eriksson, G. Lindblom and G. Arvidson, J. Phys. Chem., 1987, 91, 846–853 CrossRef CAS.
- H. Delacroix, T. Gulikkrzywicki, P. Mariani and V. Luzzati, J. Mol. Biol., 1993, 229, 526–539 CrossRef CAS PubMed.
- M. Imai, I. Yoshida, T. Iwaki and K. Nakaya, J. Chem. Phys., 2005, 122, 044906 CrossRef CAS PubMed.
- R. Klein, G. J. T. Tiddy, E. Maurer, D. Touraud, J. Esquena, O. Tache and W. Kunz, Soft Matter, 2011, 7, 6973–6983 RSC.
- G. C. Shearman, A. I. I. Tyler, N. J. Brooks, R. H. Templer, O. Ces, R. V. Law and J. M. Seddon, J. Am. Chem. Soc., 2009, 131, 1678–1679 CrossRef CAS PubMed.
- S. Besson, T. Gacoin, C. Ricolleau, C. Jacquiod and J. P. Boilot, J. Mater. Chem., 2003, 13, 404–409 RSC.
- S. Zhou, C. Burger, F. Yeh and B. Chu, Macromolecules, 1998, 31, 8157–8163 CrossRef CAS.
- C. K. Liu and G. G. Warr, Langmuir, 2012, 28, 11007–11016 CrossRef CAS PubMed.
- F. B. Rosevear, J. Am. Oil Chem. Soc., 1954, 31, 628–639 CrossRef CAS.
- E. S. Blackmore and G. J. T. Tiddy, J. Chem. Soc., Faraday Trans. 2, 1988, 84, 1115–1127 RSC.
- F. Ricoul, M. Dubois, T. Zemb, M. P. Heck, A. Vandais, D. Plusquellec, I. Rico-Lattes and O. Diat, J. Phys. Chem. B, 1998, 102, 2769–2775 CrossRef CAS.
- A. Gulik, H. Delacroix, G. Kirschner and V. Luzzati, J. Phys. II, 1995, 5, 445–464 CrossRef CAS.
- J. McAlpine, Honours, The University of Sydney, 2005 Search PubMed.
- E. S. Blackmore and G. J. T. Tiddy, Liq. Cryst., 1990, 8, 131–151 CrossRef CAS.
- T. Wärnheim and A. Jönsson, J. Colloid Interface Sci., 1988, 125, 627–633 CrossRef.
- D. Maciejewska, A. Khan and B. Lindman, in New Trends in Colloid Science, ed. H. Hoffmann, SpringerBerlin/Heidelberg, 1987, vol. 73, pp. 174–179 Search PubMed.
- X. Auvray, C. Petipas, R. Anthore, I. Rico and A. Lattes, J. Phys. Chem., 1989, 93, 7458–7464 CrossRef CAS.
- K. M. McGrath, Langmuir, 1995, 11, 1835–1839 CrossRef CAS.
- J. D. Morgan, D. H. Napper, G. G. Warr and S. K. Nicol, Langmuir, 1994, 10, 797–801 CrossRef CAS.
- J. D. Morgan, D. H. Napper and G. G. Warr, J. Phys. Chem., 1995, 99, 9458–9465 CrossRef CAS.
- H. N. Patrick and G. G. Warr, J. Phys. Chem., 1996, 100, 16268–16274 CrossRef CAS.
- K. D. Collins, Biophys. Chem., 2006, 119, 271–281 CrossRef CAS PubMed.
- M. Y. Kiriukhin and K. D. Collins, Biophys. Chem., 2002, 99, 155–168 CrossRef CAS.
- R. Buchner, F. Samani, P. M. May, P. Sturm and G. Hefter, ChemPhysChem, 2003, 4, 373–378 CrossRef CAS PubMed.
- B. Gao and Z. F. Liu, J. Phys. Chem. A, 2005, 109, 9104–9111 CrossRef CAS PubMed.
- G. Para and P. Warszynski, Colloids Surf., A, 2007, 300, 346–352 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sm52339j |
| This journal is © The Royal Society of Chemistry 2014 |