Dynamic reactions of liposomes†
Sebastian A.
Berg
and
Bart Jan
Ravoo
*
Organic Chemistry Institute, Westfälische Wilhelms-Universität Münster, Corrensstrasse 40, 48149 Münster, Germany. E-mail: b.j.ravoo@uni-muenster.de
First published on 1st October 2013
Abstract
Dynamic chemical reactions at the surface of liposomes are of fundamental interest to the understanding of physiological processes at biological membranes and can be exploited to prepare responsive soft materials. In this paper we describe a covalent yet reversible reaction between liposomes. To this end we explored the reversible thioester exchange reaction of membrane embedded amphiphilic thioesters with dithiols from solution. It was found that thioester exchange leads to the formation of covalent inter-liposomal cross-links and aggregation of liposomes. Liposome aggregation has a characteristic lag phase and the rate of aggregation depends on the concentration of dithiols as well as on the concentration of liposomes in solution. The reversibility of the aggregation could be demonstrated by dissociation of the liposome clusters by the addition of a monothiol. Furthermore we developed a fluorescence anisotropy assay to monitor the thioester exchange reaction on the membrane surface. To the best of our knowledge, the formation of a dynamic covalent network of liposomes is unprecedented.
Introduction
Biological membranes are fundamental to life since they are responsible for the segregation and compartmentalization of cells and organisms. Phospholipid membranes are the fundamental scaffold for biological functions and cellular structures. Amongst others, membranes sustain chemical and potential gradients, are vital for the signal transduction between cells, are instrumental in biosynthesis and are a crucial line of defense against pathogens.1–3 Despite the eminent role of phospholipids in biology, the investigation of these biomolecules lags behind compared to others such as oligonucleotides, peptides, proteins, or carbohydrates. This can be attributed to the complexity of their structure and function in biological membranes in combination with their pleiotropic nature and the problems posed by the investigation of dynamic biological systems in vivo.1–3 Therefore, model systems that enable the detailed investigation of phospholipid membranes are most useful and ever since the preparation of the first liposomes by Bangham4 numerous simple as well as more sophisticated models have been developed to study the correlation of the chemistry of bilayer membranes and their biological function.1,5,6Chemical reactions such as phosphorylations, glycosylations, etc. on or near biological membrane surfaces are of great physiological relevance but have received relatively little attention in model systems such as liposomes. Various reactions have been meanwhile utilized to modify the surface of liposomes: amongst others, the Michael addition of thiols and maleimides,7–10 the substitution of bromoacetyl functions with thiols11 and the addition of aldehydes to hydrazones12 have been used for this purpose. The 1,3-dipolar cycloaddition of azides and alkynes catalyzed by Cu(I) (CuAAC or “click chemistry”) has also been very versatile. The great potential of this reaction inspired Kros et al.13 to develop a FRET based colorimetric assay demonstrating that the cycloaddition occurs at the liposome surface. A CuAAC induced lateral polymerization of the membrane of liposomes (“synthetic cytoskeleton”) could be achieved by incorporating lipids functionalized with three propagyl groups into the membrane and linking them together by a diazide.14 The encapsulation efficiency of these liposomes was strongly increased and the incorporation of a disulfide bridge into the diazide allowed for a cleavage of the polymer shell under reducing conditions thus forming a switchable drug release system.
Pioneering work on reactions at the phospholipid bilayer was done by Menger et al. who explored the reaction of an electrophile and a nucleophile incorporated into different liposomes which reacted upon collision.15,16 Menger et al. also described the covalent cross-linking of maleimide-functionalized liposomes in the presence of dithiothreitol.17 They also showed that the covalent cross-links could be eliminated by oxidation.17 Zumbuehl et al. used CuAAC to covalently link liposomes together and ultimately fuse them in the process.18 To this end they incorporated alkyne and azide modified lipids into liposomes and “clicked” them together in the presence of Cu(I). A comparable click reaction was reported for dendrisomes (vesicles composed of amphiphilic dendrimers).19
The covalent reactions on liposome surfaces described above are irreversible reactions under physiological conditions. This is quite remarkable considering that in nature many transformations are inherently reversible and hence highly dynamic. This observation is especially true for recognition and adhesion of cellular membranes. To the best of our knowledge, there are only very few reports describing reversible, dynamic covalent reactions20,21 at or on the interface of liposomes (or synthetic vesicles) and water. The first example is the work of Regen et al. on nearest neighbor recognition in phospholipid membranes, which elegantly exploits reversible disulfide formation.22 A more recent example is the work of van Esch et al.,23 which describes the dynamic formation of amphiphiles in water and the subsequent self-assembly of these amphiphiles into vesicles. In that study, the authors exploit the condensation of water soluble amines and aldehydes to form amphiphilic diimines. It was shown that the vesicles dissociate upon dilution or a change in pH, which leads to hydrolysis of the imines. In a follow-up paper, it was also shown that a combination of dialdehydes and diimines can lead to the formation of a pH responsive vesicle gel.24 The third example is the work of Otto et al.,25 which involves dynamic combinatorial chemistry at the surface of liposomes. In that study, the thioester exchange reactions in bulk solution and at the surface of liposomes are compared. It was shown that the reaction of thiols from solution with aromatic thioesters bound to the membrane or free in solution reaches the equilibrium state in about 24 h. This is fast in comparison to other dynamic reactions such as disulfide or hydrazine exchange and within the lifetime range of liposomes making these reactions suitable for dynamic reactions at the phospholipid bilayer. It was also shown that at a pH of 8.0 and a temperature of 40 °C, thioester hydrolysis is not competitive with the thioester exchange in bulk or at liposomes.
In this article we demonstrate that the thioester exchange reaction at the surface of liposomes can be used to form reversible covalent cross-links between different liposomes. To this end, liposomes containing an amphiphilic thioester were reacted with a dithiol. The covalent cross-linking of the liposomes was monitored in real time by using optical density (OD400) measurements and dynamic light scattering (DLS). Additionally we introduce an alternative method to follow the thioester exchange reactions at the phospholipid bilayer in situ by straightforward fluorescence anisotropy measurements. To the best of our knowledge, the formation of a dynamic covalent network of liposomes is unprecedented.
Results and discussion
The synthesis of aromatic thioesters (1) and (2) is described in the ESI.† The analytical data for (1) and (2) are consistent with the structure shown in Fig. 1. Both thioesters contain a hydrophobic oleic acid anchor to incorporate them into the membrane of liposomes. The thioester is connected to its hydrophobic anchor via a triethylene glycol spacer to enhance the accessibility of the thioester function at the liposome surface to molecules from solution. In (1) the thioester is composed of a benzoic acid and thioglycerol, whereas in (2) the thioester is composed of a benzoic acid and a fluorescein dye (excitation at 497 nm, emission at 527 nm). It is known that aromatic thioesters are more reactive than aliphatic thioesters.25 As a consequence of the hydrophilic character of the triethylene glycol spacer, the thioglycerol and the fluorescein, the thioesters should be exposed to the water surrounding the phospholipid bilayer.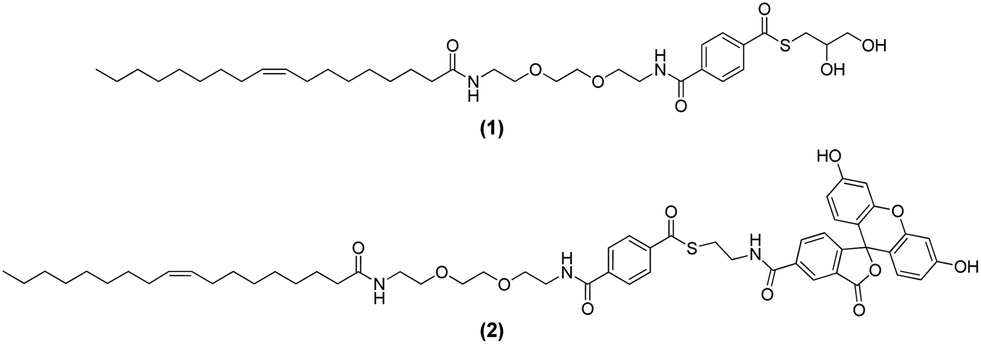 | ||
| Fig. 1 Amphiphilic thioesters (1) and (2) incorporated into liposomes. | ||
The design of the amphiphilic thioester (1) should enable the formation of liposomes with thioesters on their surface simply by adding thioester (1) to any desired mixture of lipids. A schematic representation of liposomes decorated with (1) is provided in the left part of Fig. 2. Upon addition of dithiol 2,2′-(ethylenedioxy)diethanethiol (3) to the liposome solution a thioester exchange reaction should take place that leads to the formation of a network of liposomes that are covalently linked together by multiple dithioesters. A schematic representation of the liposome network is shown in the right part of Fig. 2 and the thioester exchange reaction is shown in the lower part of Fig. 2.
 | ||
| Fig. 2 Schematic description of the cross-linking of liposomes induced by reversible thioester exchange reactions between membrane-bound thioester (1) and a dithiol (3) from solution. | ||
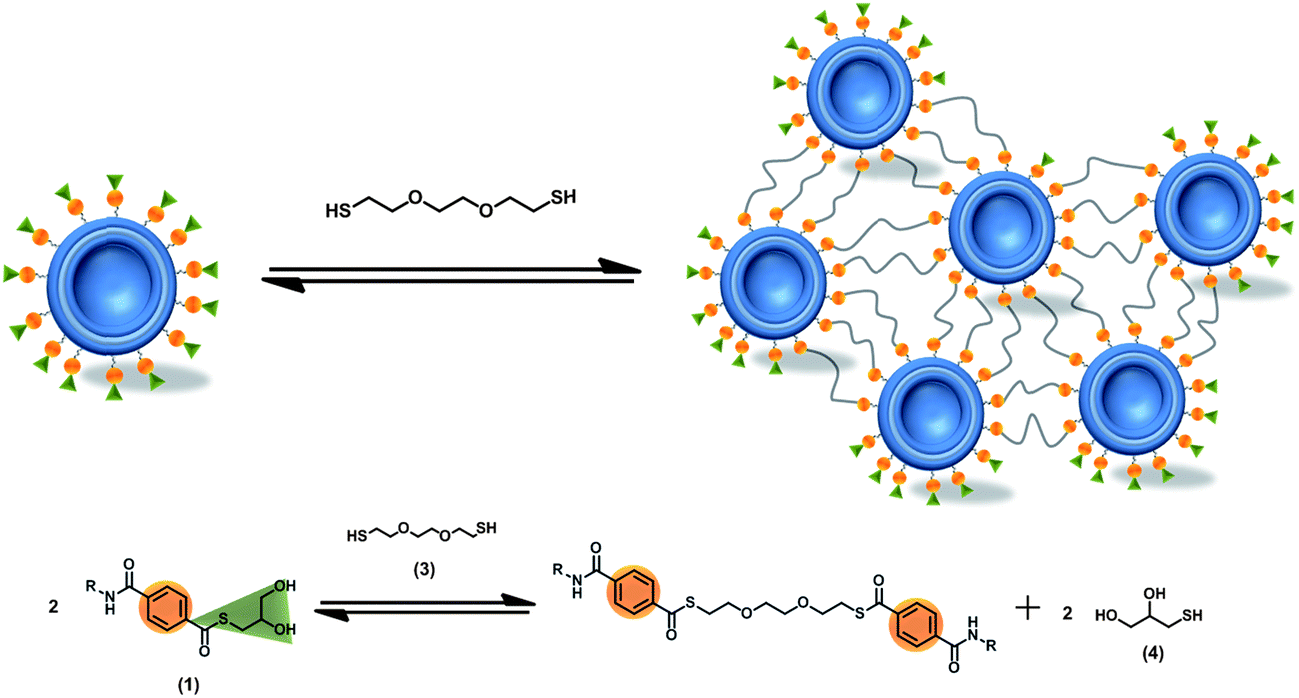
Based on this hypothesis we prepared liposomes of soy bean lecithin and amphiphilic thioester (1) in 100 mM phosphate buffer at pH 8.00 (see ESI† for details of the preparation of liposomes). The liposome solution consisted of 0.1 mM lecithin and 0.025 mM thioester (1) (i.e. 20 mol% of thioester) and had an average size of 100 nm and a polydispersity index of 0.178 according to DLS measurements. All thioester exchange experiments were carried out at a temperature of 50 °C under stirring at 200 rpm in completely filled closed PMMA cuvettes. OD400 and DLS measurements were used to monitor the aggregation of the liposomes in the presence of dithiol (3). The results of a set of OD400 measurement are shown in Fig. 3.
 | ||
| Fig. 3 OD400 measurement of liposomes containing 20 mol% of thioester (1) in the presence of dithiol (3). Conditions: 100 nm liposomes; 0.1 mM Lecithin; 0.025 mM (1); 100 mM phosphate buffer at pH 8.00; T = 50 °C; stirred at 200 rpm. After approx. 10 min dithiol (3) was added to the liposomes resulting in the following concentrations: red: 10 mM, blue: 15 mM, green: 20 mM dithiol (3). | ||
It can be seen from Fig. 3 that all samples of the liposome solution initially have an OD400 of around 0.01, consistent with a transparent solution of unilamellar liposomes. After 10 min dithiol (3) was added to each sample resulting in a fast but very modest increase of the OD400 to values of 0.05 to 0.2 depending on the concentration of dithiol (3) added. The OD400 stayed nearly constant for 6–7 h. Only after this period a much larger increase in the OD400 was observed. The OD400 continued to increase until it leveled off at a value of around 3 after 30–50 h depending on the concentration of dithiol. The most straightforward explanation for the increase in OD400 is an aggregation of the liposomes as a result of a covalent cross-linking by dithiol (3). Indeed, the corresponding DLS measurements (see Fig. S1 in the ESI†) conducted before addition of the dithiol and after 16 h also show a significant increase in the average particle size. It should be noted that the average particle size obtained after 16 h varies from sample to sample and that it is difficult to obtain quantitatively reliable data by DLS when the OD400 is much higher than 1 due to the low transparency of the sample and the broad size distribution of the liposome aggregates.
In contrast to the addition of dithiol (3), the addition of a monothiol such as thioglycerol (4) to the liposome solution did not induce any aggregation of liposomes over a period of 24 h (see Fig. S2 in the ESI†). This important control experiment clearly demonstrates that the aggregation of liposomes containing (1) requires a dithiol, not a thiol. We also note that aggregation of liposomes could be observed at temperatures below 50 °C and at lower concentrations of dithiol (3), but that in that case the rate of aggregation became too slow to obtain meaningful results within a time window suitable for liposome solutions. The long term aggregation of liposomes in the presence of dithiol also underscores the stability of thioesters towards hydrolysis under the conditions of the experiment. We emphasize that the aggregation of liposomes induced by the addition of dithiol (3) also occurred in the presence of the reducing agent tris(2-carboxyethyl)phosphine (TCEP) which indicates that the formation of disulfides is not involved in the aggregation process.
From these data it can be deduced that the aggregation process proceeds in two separate phases. In the initial phase (up to 6 h under the experimental conditions investigated here) no liposome aggregation occurs whereas in the second phase (after about 6 h), the aggregation starts and leads to the formation of large liposome aggregates observed by OD400 and DLS. Considering the observation of Otto et al.25 that a substantial conversion of membrane bound thioesters to thiols from solution occurs within a few hours and considering the large excess of dithiol (3) present in solution (400, 600 or 800 fold excess compared to thioester (1)) we hypothesize that this initial phase of the aggregation process can be attributed to the solution to liposome exchange of thioester (1) with dithiol (3) resulting in liposomes coated with a thiol-terminated ethylene glycol layer. Continuing from this point three different reaction pathways are conceivable. One pathway is a repeated solution to liposome exchange reaction of the formed thiol-terminated ethylene glycol thioester with another dithiol molecule from solution. Even though this exchange reaction probably proceeds rapidly due to the high concentration of dithiol in solution it does not in any way change the surface composition of the liposomes and it cannot lead to an aggregation of the liposomes. The second pathway is an intravesicular thioester exchange between two of the formed thioesters that results in a looped thioester where a dithiol bridges two thioesters incorporated into the same liposome. This reaction may be promoted by the relatively high local concentration of the thioester (20 mol%) on the membrane surface but it cannot be responsible for an aggregation of the liposomes since it does not result in cross-links between liposomes and it is unlikely to have a significant effect on the surface properties of the liposomes. The third pathway is an intervesicular or liposome to liposome reaction between two thioesters on two different liposomes that react to form a dithioester that is embedded in the membranes of two different liposomes. This reaction gives rise to the formation of covalent cross-links between liposomes. In view of the relatively short ethylene glycol dithiol (3) such a reaction can only occur when two liposomes come very close together or even collide. It should be emphasized that this third pathway is in competition with its back reaction and the other two described possibilities, but the formation of only a few covalent cross-links – perhaps even a single cross-link – would result in liposome aggregation. It is reasonable to assume that aggregation of the liposomes is ultimately a consequence of the different environments and different local concentrations of reacting partners in the bulk solution and at the phospholipid bilayer. As soon as the first covalent (yet reversible) link between two liposomes is formed, the two liposomes are held close together and the local environment between them changes drastically due to hydrophobic effects of the membrane and crowding of the reaction partners at the interface.26–33 This effectively results in a steep decrease of the local concentration of dithiol (3) and a substantial increase of the local concentration of the thiol-terminated thioesters in the contact area of the liposomes. As a consequence, the reaction equilibrium shifts towards thioesters linking the liposomes resulting in a stable multivalent network of liposomes. It is reasonable to assume that aggregation thus requires a critical density of thiol-terminated thioester on the liposome surface, which may explain the lag phase of the aggregation. This mechanism also explains why the formed aggregates are stable despite the enormous excess of free dithiol in solution: even if any given individual cross-link may be broken by exchange with dithiol from solution, the two resulting non-bridging thioesters are likely to exchange back to the bridging thioesters due to the high local concentration of thioester present at the contact area of two liposomes.
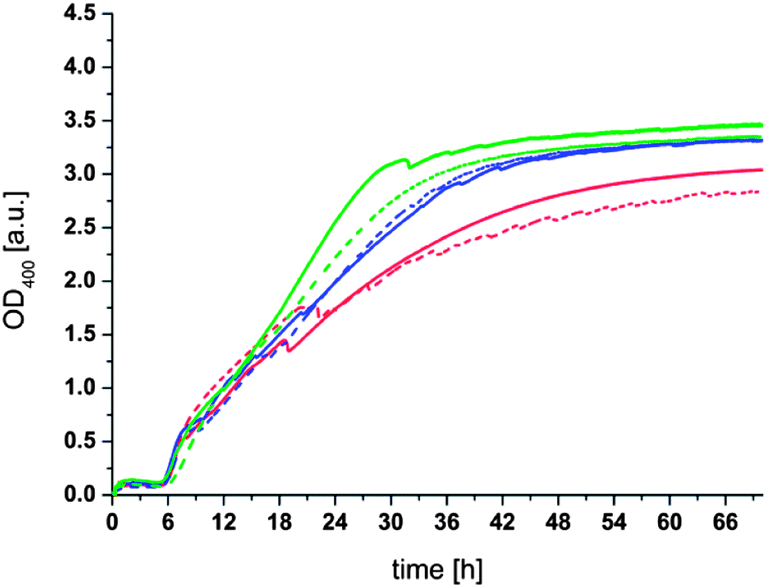
If the proposed exchange and cross-link mechanism is correct it should be possible to shift the equilibrium of the thioester formation to a compound that is not able to link the liposomes together. To verify this we added a large excess of monothiol (thioglycerol (4)) to the aggregated liposomes after 16 h. To minimize the complexity of the dynamic system we selected thioglycerol (4) for this purpose since an exchange reaction of this compound with the thioesters on the liposome surface should lead to the formation of the initial thioester (1). As can be seen in Fig. 4 the OD400 gradually decreases after addition of thioglycerol (4) to a value of about 0.25 after 30 h. An initial instantaneous decrease can be attributed to the dissociation of the largest aggregates by the shear stress applied during mixing with a pipette. The decrease of the OD400 during 30 h can be attributed to a dissociation of the liposome clusters to very small aggregates or individual liposomes due to the disruption of the intervesicular cross-links and (re)formation of the thioester (1). Based on the large excess of thioglycerol (4) to thioester (1) (∼5000×) it can be assumed that once reformed these thioesters are not, or only to a very small extent, reacting again with a bridging thioester. We assume that the disaggregation process takes about 30 h despite the large amount of thioglycerol in solution due to the molecular crowding and high local concentration of bridging thioesters at the contact areas of the liposomes which hinders the diffusion of thioglycerol into proximity of the thioesters. As a consequence, the exchange reaction can only take place at the “rim” of the cross-linking areas, slowing down the overall process considerably. We note that this interpretation is supported by the fact that no decrease in the OD400 due to hydrolysis can be observed during the entire duration of the experiment (see Fig. 3). We also note that the observation that the aggregation of liposomes can be completely reversed rules out that significant fusion of cross-linked liposomes occurs, since it is highly unlikely that fusion would be reversible.
 | ||
| Fig. 4 OD400 measurement of agglutinated liposomes upon addition of thioglycerol (4). Conditions: 100 nm liposomes; 0.1 mM lecithin; 0.025 mM (1); 100 mM phosphate buffer at pH 8.00; T = 50 °C; stirred at 200 rpm; dithiol (3) concentrations: red: 10 mM, blue: 15 mM, green: 20 mM. After approx. 10 min, 120 mM thioglycerol (4) were added to the samples. | ||
Another prediction resulting from the proposed mechanism of the aggregation process is that the concentration of liposomes in solution should influence the rate of aggregation since it determines the statistical probability of two liposomes coming near enough to react with each other. To verify this assumption, liposome solutions with the same concentration of phospholipid and thioester (1) but different average sizes of 125 nm (“large”) and 74 nm (“small”) were prepared. From these size differences the ratio of small to large liposomes can be calculated to be 2.8 (see Formula S1 in the ESI†). A comparison of the aggregation behavior of these two liposome populations showed the expected increase in the aggregation rate for the higher concentration of liposomes present in solution (Fig. 5). Moreover, it was observed that the aggregation had a shorter lag time (4.5 h) for small liposomes compared to large liposomes (6–7 h). The increase in the aggregation rate and the shorter lag time for smaller liposomes can be attributed to a higher statistical chance for the initial cross-linking step of two liposomes due to the higher number of liposomes in solution. Additionally the faster diffusion of the smaller liposomes probably accelerates the aggregation process. The differences between the additions of 10 or 20 mM dithiol (3) are, in the case of the small liposomes, comparable to the observations described above. In the case of the larger liposomes the aggregation process has not proceeded far enough to show a significant difference between the two samples but from the slope of the OD400 curves a difference between the additions of 10 or 20 mM dithiol (3) comparable to results presented above can be expected.
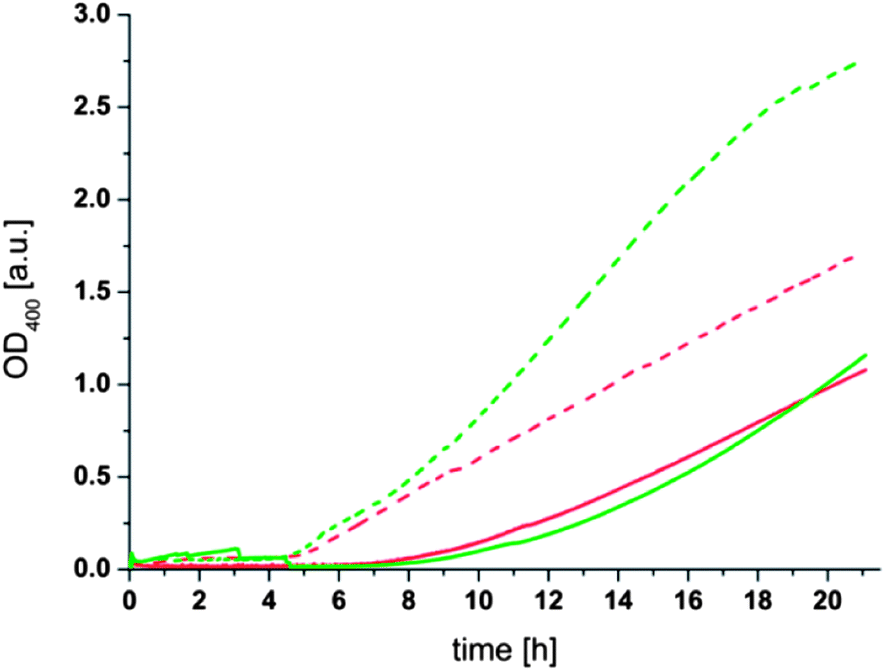 | ||
| Fig. 5 OD400 measurement of differently sized liposomes with thioester (1) and dithiol (3). Conditions: 0.1 mM lecithin, 0.025 mM (1), 100 mM phosphate buffer at pH 8.00, T = 50 °C, stirred at 200 rpm; dotted lines: small liposomes (74 nm); solid lines: large liposomes (125 nm.) After approx. 10 min, dithiol (3) was added to the solutions resulting in the following concentrations: red: 10 mM, green: 20 mM. | ||
To further investigate the thioester exchange reaction at the surface of liposomes we synthesized the fluorescently labeled thioester (2) and incorporated it into the membrane of liposomes. Since the fluorescent dye is anchored to the liposome, its fluorescence exhibits a pronounced fluorescence anisotropy.34 Upon addition of a thiol to the solution a solution to liposome thioester exchange reaction should occur that liberates the fluorescent dye from the liposome surface so that it can diffuse freely in the solution. The increase in molecular mobility enables us to follow the exchange reaction by using fluorescence anisotropy measurements. To this end, fluorescent thioester (2) was incorporated into the membrane of preformed liposomes (0.1 mM lecithin and 0.0025 mM (2), i.e. 2.5 mol% of thioester) with a size of 125 nm in 100 mM phosphate buffer at pH 8.0. The fluorescent thioester (2) was added after preparation of the liposomes so that the majority of the fluorophore would reside at the outer surface of the liposomes. The fluorescence anisotropy of thioester (2) embedded in liposomes was around 0.125 for each sample investigated. The change in anisotropy upon addition of thioglycerol (4) at 50 °C was measured over time (Fig. 6).
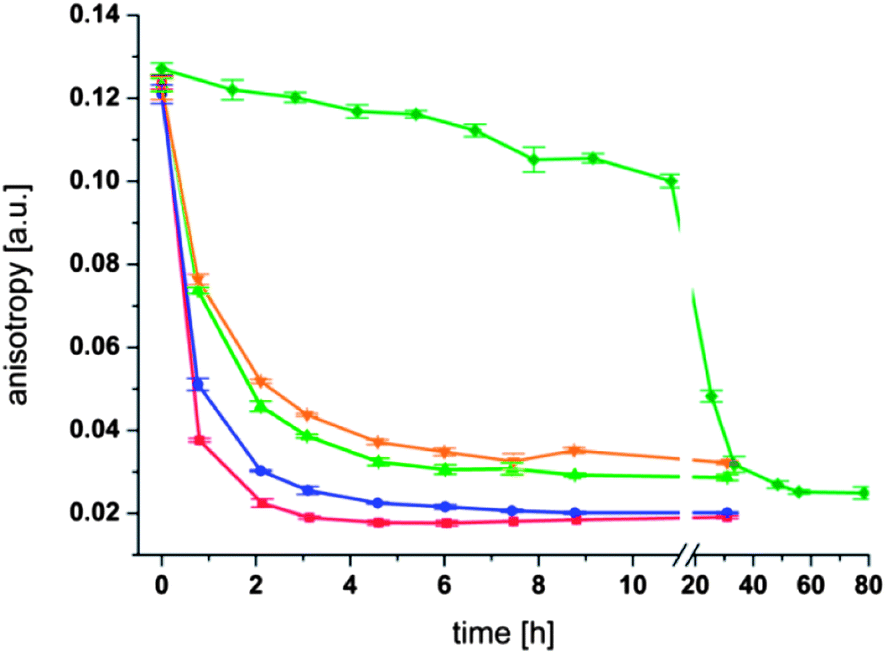 | ||
| Fig. 6 Fluorescence anisotropy measurements of liposomes with fluorescent thioester (2). Conditions: 0.1 mM Lecithin; 0.0025 mM thioester (2); 125 nm liposomes; 100 mM phosphate buffer at pH 8.00; T = 50 °C, stirred at 200 rpm; red: 1 mM thioglycerol (4); blue: 0.5 mM thioglycerol (4); green: 0.2 mM thioglycerol (4); orange: 0.15 mM thioglycerol (4); dark green: no thioglycerol (4). | ||
It can be seen from Fig. 6 that in each experiment the fluorescence anisotropy decreases with time. However, the rate of decrease depends strongly on the concentration of thioglycerol. A clear trend is visible where the addition of 1 mM thioglycerol (400 fold excess compared to the thioester (2)) leads to the fastest decrease of the anisotropy as well as the lowest residual value (∼0.018 after 3 h). The addition of less thioglycerol leads to a slower decrease in anisotropy and to a higher base value at the end of the measurement. Thus an addition of 0.15 mM thioglycerol (4) (60 fold excess compared to the thioester (2)) leads to the slowest decrease of anisotropy and the highest residual value (∼0.076 after 6 h) of all four measurements. Importantly, the fluorescence anisotropy for liposomes in the absence of thioglycerol (4) decreases only very slowly over the first 10 h confirming that the observed effect is due to a thioester exchange reaction at the membrane surface and that the extent of hydrolysis of the thioester is very modest under these conditions.
The different residual anisotropy values of the liposomes reacted with thioglycerol indicate that the reactions reach an equilibrium state after a few hours (3–6 h) depending on the amount of thiol added to the solution. Due to the fact that only two states of the fluorophore are existent, namely the bound and the unbound state, the fraction of free fluorophore in solution can be estimated according to eqn (1):
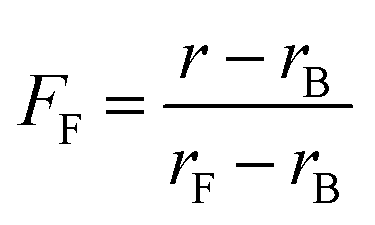 | (1) |
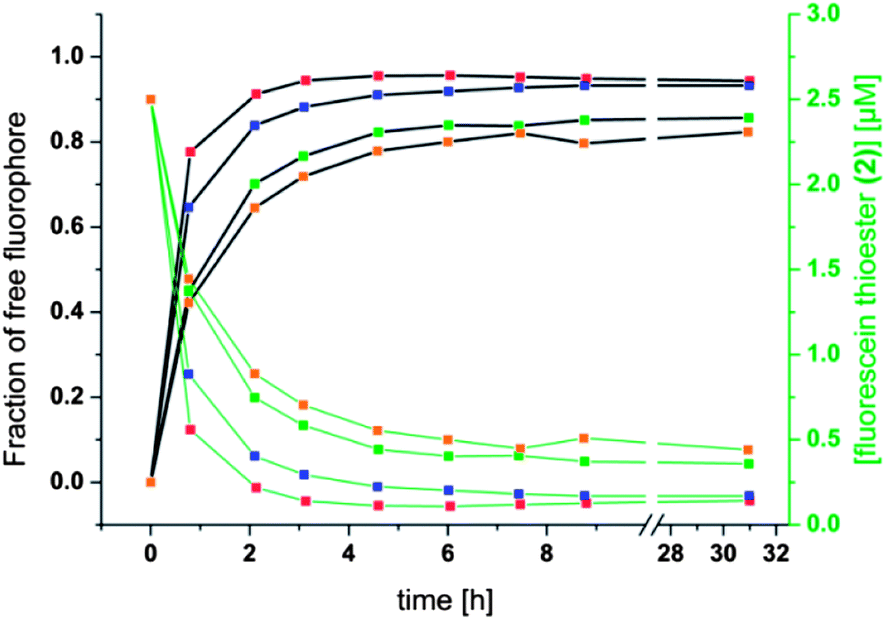 | ||
| Fig. 7 Fraction of free fluorophore against time (left, black scale and black curves) and concentration of fluorescein thioester (2) against time (right, green scale and green curves). Conditions and color coding as in Fig. 6 with red = 1 mM thioglycerol, blue = 0.5 mM thioglycerol, green = 0.2 mM thioglycerol, orange = 0.15 mM thioglycerol. | ||
Conclusions
In this study we have demonstrated the formation of liposome aggregates by a dynamic covalent reaction at the liposome surface. Thioester exchange reactions are suitable for this purpose since the kinetics of these exchange reactions are clearly within the lifetime of liposomes under the experimental conditions used. The incorporation of aromatic thioesters into the membranes of liposomes provided the required reaction site at the liposome that selectively reacts with free thiols from solution. The addition of a dithiol to the solution results in a two-step aggregation process of the liposomes. The rate of aggregation is dependent on the concentration of dithiol and on the concentration of liposomes. Moreover, the aggregation process can be entirely reversed by addition of a monothiol. Finally, the thioester exchange reaction was monitored in real time by fluorescence anisotropy measurements. It was found that the thioester exchange at liposomes is a dynamic reaction under thermodynamic equilibrium where the final composition depends on the concentrations of the reagents participating in the reaction. Thus, anisotropy measurements are a powerful approach for the investigation of covalent exchange reactions at the phospholipid bilayer. We anticipate that the results presented here will further the understanding the chemical transformations at the interface of biological membranes and their surrounding aqueous environment.Acknowledgements
This research was funded by the Deutsche Forschungsgemeinschaft DFG (SFB 858).References
- D. E. Vance and J. E. Vance, Biochemistry of Lipids, Lipoproteins and Membranes, Elsevier, Amsterdam, Netherlands, 5th edn, 2008 Search PubMed.
- M. Luckey, Membrane structural biology: with biochemical and biophysical foundations, Cambridge University Press, New York, USA, 2008 Search PubMed.
- P. L. Yeagle, The Structure of Biological Membranes, CRC Press, Taylor & Francis Group, Boca Raton, Fl, USA, 3rd edn, 2012 Search PubMed.
- A. D. Bangham and R. W. Horne, J. Mol. Biol., 1964, 8, 660 CrossRef CAS.
- J. Voskuhl and B. J. Ravoo, Chem. Soc. Rev., 2009, 38, 495–505 RSC.
- H. Robsen Marsden, I. Tomatsu and A. Kros, Chem. Soc. Rev., 2011, 40, 1572–1585 RSC.
- P. Benzinger, G. Martiny-Baron, P. Reusch, G. Siemeister, J. T. Kley, D. Marmé, C. Unger and U. Massing, Biochim. Biophys. Acta, 2000, 1466, 71–78 CrossRef CAS.
- M. Fleiner, P. Benzinger, T. Fichert and U. Massing, Bioconjugate Chem., 2001, 12, 470–475 CrossRef CAS PubMed.
- J. T. Kley, T. Fichert and U. Massing, Monatsh. Chem., 1998, 129, 319–327 CAS.
- F. J. Martin and D. Papahadjopoulos, J. Biol. Chem., 1982, 257, 286–288 CAS.
- P. Schelte, C. Boeckler, B. Frisch and F. Schuber, Bioconjugate Chem., 2000, 11, 118–123 CrossRef CAS PubMed.
- L. Bourel-Bonnet, E. I. Pecheur, C. Grandjean, A. Blanpain, T. Baust, O. Melnyk, B. Hoflack and H. Gras-Masse, Bioconjugate Chem., 2005, 16, 450–457 CrossRef CAS PubMed.
- S. Cavalli, A. R. Tipton, M. Overhand and A. Kros, Chem. Commun., 2006, 3193–3195 RSC.
- S. Zhang and Y. Zhao, Bioconjugate Chem., 2011, 22, 523–528 CrossRef CAS PubMed.
- F. M. Menger and V. A. Azov, J. Am. Chem. Soc., 2000, 122, 6492–6493 CrossRef CAS.
- F. M. Menger, K. L. Caran and V. A. Seredyuk, Angew. Chem., Int. Ed., 2001, 40, 3905–3907 CrossRef CAS.
- F. M. Menger, J. Bian and V. A. Seredyuk, Angew. Chem., Int. Ed., 2004, 43, 1265–1267 CrossRef CAS PubMed.
- F. Loosli, D. A. Doval, D. Grassi, P. L. Zaffalon, F. Favarger and A. Zumbuehl, Chem. Commun., 2012, 48, 1604–1606 RSC.
- H. Jin, W. Huang, Y. Zheng, Y. Zhou and D. Yan, Chem.–Eur. J., 2012, 18, 8641–8646 CrossRef CAS PubMed.
- J. M. Lehn, Chem.–Eur. J., 1999, 5, 2455–2463 CrossRef CAS.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- S. M. K. Davidson and S. L. Regen, Chem. Rev., 1997, 97, 1269–1280 CrossRef CAS PubMed.
- C. B. Minkenberg, F. Li, P. van Rijn, L. Florusse, J. Boekhoven, M. C. A. Stuart, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2011, 50, 3421–3424 CrossRef CAS PubMed.
- C. B. Minkenberg, W. E. Hendriksen, F. Li, E. Mendes, R. Eelkema and J. H. van Esch, Chem. Commun., 2012, 48, 9837–9839 RSC.
- F. Mansfeld, H. Au-Yeung, J. K. M. Sanders and S. Otto, J. Syst. Chem., 2010, 1, 12 CrossRef CAS.
- H. X. Zhou, G. N. Rivas and A. P. Minton, Annu. Rev. Biophys., 2008, 37, 375–397 CrossRef CAS PubMed.
- S. B. Zimmerman and A. P. Minton, Annu. Rev. Biophys., 1993, 22, 27–65 CrossRef CAS PubMed.
- A. P. Minton, J. Biol. Chem., 2001, 276, 10577–10580 CrossRef CAS PubMed.
- S. B. Zimmerman and S. O. Trach, J. Mol. Biol., 1991, 222, 599–620 CrossRef CAS.
- J. Spitzer, Microbiol. Mol. Biol. Rev., 2011, 75, 491–506 CrossRef CAS PubMed.
- R. S. Fuller, J. M. Kaguni and A. Kornberg, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 7370–7374 CrossRef CAS.
- R. J. Ellis, Trends Biochem. Sci., 2001, 26, 597–604 CrossRef CAS.
- D. S. Goodsell, Trends Biochem. Sci., 1991, 16, 203–206 CrossRef CAS.
- V. Borenstain and Y. Barenholz, Chem. Phys. Lipids, 1993, 64, 117–127 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3sm51515j |
| This journal is © The Royal Society of Chemistry 2014 |