Multiwalled functional colloidosomes made small and in large quantities via bulk emulsification†
Jonathan S.
Sander
and
André R.
Studart
*
Complex Materials, Department of Materials, ETH Zurich, 8093 Zurich, Switzerland. E-mail: andre.studart@mat.ethz.ch
First published on 9th October 2013
Abstract
Colloidosomes are attractive microcarriers for encapsulation and controlled release. If their shell is composed of multiple layers of particles they are very robust against defects, which ensures a well-defined permeability. So far most capsules with multilayer particle shells could only be assembled using microfluidic emulsification, a technique that offers unparalleled encapsulation control but that is difficult to scale up for high material throughput. Here we propose a method that allows for high throughput assembly of multiwalled colloidosomes using double emulsion templates made in two simple bulk emulsification steps. Stabilization of the emulsion with interfacially adsorbed particles is key to enable control over the colloidosome architecture, which can display single or multiple internal compartments depending on the magnitude of the applied shear. In addition to the high throughput, our method also allows for the assembly of capsules with magnetically responsive shells and controllable release properties in a size range that has previously not been possible for multiwalled colloidosomes. The potential of such systems is illustrated by producing large quantities of multiwalled colloidosomes that are small enough to display Brownian motion in a fluid and that can be externally triggered to release molecular encapsulants on demand.
Introduction
Colloidosomes are hollow capsules containing nanoparticles in the shell to provide size-selective permeability through controlled particle interstitials.1,2 Deliberate selection of the nanoparticles that comprise the shell enables functionalization of the colloidosomes for specific functions, including for instance the encapsulation of living organisms,3,4 guided transport5–7 or triggered release.8–10 The semi-permeable wall, the rigid shell structure and the versatility in terms of capsule size and shell materials make colloidosomes promising candidates for microreactors in bioanalytics or delivery vehicles for drugs, flavors, food additives or rheology modifiers.11–15Most current examples of colloidosomes consist of microcapsules with a single particle layer forming the shell. The particle layer is typically formed by the interfacial adsorption of colloidal particles at the liquid interface of single emulsions or on the surface of particle templates.1,2,5,16,17 Although such a monolayer offers an interesting platform for fundamental studies on packing and topological defects on non-planar surfaces,18 the size-selective permeability of single layer colloidosomes is sensitive to such packing defects19 making the release of encapsulants difficult to control.20 More recent developments have shown that double emulsions can be successfully used as templates for the assembly of colloidosomes with multiple particle layers in the shell that are more robust against topological flaws. Except for the approach recently described by San Miguel et al. to form double-layered colloidosomes through bulk emulsification,10 microfluidics is the method most commonly used to prepare double emulsions utilized as templates for the creation of multiwalled colloidosomes.7,21 Microfluidics provides very high encapsulation efficiencies and control over the geometry of the final colloidosomes at the expense of a lower material throughput compared to bulk emulsification approaches. Here, we define bulk emulsification approaches as those processes in which droplets are concomitantly created throughout the bulk mixture of immiscible fluids, as opposed to microfluidic and membrane techniques that generate emulsions through single dripping events. While its continuous nature and the increasing efforts to scale up microfluidic emulsification will likely circumvent its currently low material throughput,22,23 offering bulk emulsification as a possible means to prepare multiwalled colloidosomes can be an attractive strategy in applications where the higher cost and low productivity of standard microfluidics remains a major obstacle.
In addition to achieving high material throughputs, another challenge in the field is to produce multiwalled colloidosomes that are small enough to remain suspended by Brownian motion in fluids and that can be applied for example in the blood stream. Colloidosomes of a few microns in size or smaller can only be produced with a monolayer of particles in the shell,15,16,24 since the required double emulsion template dimensions are currently difficult to achieve using microfluidic techniques. Therefore, the development of methods that allow for the large-scale assembly of multiwalled colloidosomes in the micron range would open new opportunities for the use of such capsules as robust delivery microsystems.
Here, we report a method to produce multiwalled colloidosomes from double emulsion templates using a truly scalable, high throughput bulk emulsification process. The adsorption of particles at the oil–water interface and the resulting viscoelastic interfacial film are found to be crucial for the large-scale assembly of colloidosomes with a tunable number of internal compartments. Because the method is compatible with high-energy emulsification techniques, Brownian colloidosomes with a few microns in size can be easily produced at high throughput rates. To illustrate the potential of this approach in generating functional microcapsules, we use the proposed bulk emulsification route to create complex colloidosomes with functional particles in the shell7 or physically entrapped in the capsule to allow for triggered release on demand.25 The preparation of small multiwalled colloidosomes with high material throughput should facilitate the exploitation of such semi-permeable capsules in many industrial applications.
Experimental
Modification of particles
Double emulsification
Double emulsions were prepared in a two-step process. First, single emulsions were made by mixing a 2 mM phosphate buffer aqueous solution at pH 5 with a 12–30 wt% silica-loaded toluene suspension. The silica suspension was sonicated for 5 minutes prior to use. The buffer solution was prepared by adding 0.01 wt% monosodium phosphate and 0.04 wt% disodium phosphate into distilled water and diluting the resulting solution to achieve the phosphate concentration of 2 mM (both 99.0% pure, Sigma Aldrich, Switzerland). The fraction of the aqueous phase in the single emulsion was varied within the range of 20–45 vol%. The water-in-oil (w/o) emulsions were prepared by mixing the aqueous and oil phases either with a simple pipette or using a high-shear rotor–stator mixer (Ultra-Turrax, T10 Basic, IKA process, Germany). The formation of w/o emulsions was confirmed by conductivity measurements (Knick Portamess Cond 913, Knick, Berlin, Germany), which indicated the presence of oil with very low conductivity as a continuous phase. The size of the emulsion droplets was adjusted by the time of mixing, which typically varied from 30 to 60 seconds for the manual pipette mixing and from 10 to 30 seconds using the rotor–stator mixer. Emulsification in the rotor–stator was performed using typical mixing speeds in the range of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–24000 rpm, depending on the required size of the droplets.
000–24000 rpm, depending on the required size of the droplets.
To obtain double emulsions, the single w/o emulsion was added to water containing 5 mM NaOH–borax buffer and 1 wt% poly(vinyl alcohol) (PVA, 87–89% hydrolysed, Mw = 31000–51000 g mol−1, Sigma Aldrich, Switzerland) to reach a volume fraction of a single w/o emulsion in water of up to 25 vol%. The mixture was emulsified immediately by mixing with either a pipette or using the rotor–stator. The mixing time was similar to that employed in the first emulsification step. In this second step, the droplet size was adjusted by varying the mixing speed in the rotor–stator mixer within the range of 8000–12000 rpm. The NaOH–borax buffer was prepared by adding NaOH to a solution of 0.1 wt% Na2B4O7·10H2O (Sigma-Aldrich, Switzerland, 98% and 99.5% pure, respectively) until a pH of 10 was reached. This typically required a NaOH concentration of 0.000144 wt% with respect to the total water content. The solution was diluted to the right molarity with double distilled water. Small double emulsions were prepared by using an ultrasonic horn for emulsification. For each emulsification step a two second ultrasound pulse was applied (Vibra cell VCX 130, Sonics, USA).
Colloidosomes
Colloidosomes were created by diluting the double emulsions at least tenfold with double distilled water and leaving the double emulsion in an open container for at least 2 days to remove the toluene from the dispersed phase. We observed that the toluene is completely removed when the colloidosomes remain intact and stable after the addition of ethanol to the aqueous continuous phase. For the setup used in this study, the removal of toluene occurs mostly within the first day after formation of the double emulsions. Although the removal process was not optimized in this work, the time period for removal can be significantly reduced by decreasing the amount of toluene in the middle phase, creating smaller colloidosomes with an inherently higher surface-to-volume ratio, optimizing the design of the container and promoting air circulation.:Al2O3 and Fe3O4–SiO2-aggregate:SiO2 volume ratio of about 1:1 during the first emulsification process. The second emulsification was performed as described above.
:1 volume ratio mixture of ferrofluid (7.5 vol%, EMG-605, Ferrotec, Germany) and water containing 4 mM phosphate buffer at pH 5. Before complete removal of the toluene, the individual Fe3O4-loaded double emulsions were prone to slow coalescence of the intermediate oil phase likely induced by the surfactant present in the ferrofluid. This was prevented by storing them in a large beaker to minimize permanent contact between several emulsions at the early stage of the toluene removal.
Interfacial rheology
Interfacial rheology was measured in a Physica MCR 300 rheometer (Anton Paar, Ostfildern, Germany) using a biconal disc geometry.26,27 After addition of approximately 50 ml of 2 mM phosphate aqueous buffer at pH 5 into the sample container, the bicone was accurately positioned at the air–water interface. About 18 ml of a toluene suspension containing 5 wt% SiO2 particles was then carefully pipetted on top. The particle suspension was sonicated for 10 minutes prior to the measurements. The experiments were started immediately after the addition of the particle suspension by applying an oscillatory strain with a constant maximum strain amplitude of 0.1% and a frequency of 1 rad s−1 for a period of 1 hour. Directly after this first time-sweep measurement, an amplitude sweep with increasing maximum strain from 0.01% to 30% was performed at an angular frequency of 1 rad s−1.Microscopy
Triggered cargo release
000 g mol−1, Sigma Aldrich, Switzerland) and 2 mM phosphate buffer at pH 5. 1.3 ml of the innermost phase was emulsified by manual mixing with a pipette in 2 ml of a suspension containing 20 wt% SiO2 particles in toluene. The resulting w/o emulsion was re-emulsified in 5 ml water with 1% PVA. The double emulsions were diluted 100-fold with 0.1 mM phosphate buffer. After removal of the toluene, the supernatant of the resulting colloidosome suspension was replaced at least 3 times before measuring the release properties of the microcapsules.
Results and discussion
The proposed method to assemble colloidosomes from double emulsion templates is based on a two-step bulk process that is scalable with existing emulsification techniques (Fig. 1, ESI Movie S1†). In a typical assembly procedure, we first mix a particle loaded toluene suspension with the inner aqueous phase containing the encapsulant (Fig. 1a and b). Because the particles are partially surface hydrophobized, emulsification using bulk techniques such as rotor–stator mixing leads to stable water-in-oil (w/o) emulsions with the particles adsorbed at the interface (Fig. 1c and 2a). This emulsion is mixed with the continuous aqueous phase containing the amphiphilic polymer poly(vinyl alcohol) (PVA, 87–89% hydrolyzed) and is eventually emulsified again (Fig. 1d and e). The inner w/o droplets of the resulting double emulsions are stabilized by particles while the outer oil-in-water (o/w) emulsion is stabilized by PVA macromolecules (Fig. 1e and 2b). As the toluene of the intermediate phase is slightly soluble in water, it can be continuously removed from the double emulsion until the hydrophobic SiO2 particles eventually jam together to form the colloidosome shell (Fig. 1f and 2c). In the example shown in Fig. 1, we use lab-scale equipment to process within 1 minute the same amount of material that would require 30 minutes to be obtained using a state-of-the-art single-dripping microfluidic device. This corresponds to a throughput rate of 0.6 g min−1 as compared to the value of 0.02 g min−1 obtained by microfluidics using a single drop maker. Much higher production speeds can potentially be reached by replacing the small laboratory mixers and beakers used in this study by larger scale emulsification equipment. The high throughput rate of the bulk emulsification process is achieved at the expense of the yield of encapsulant-tight colloidosomes, which was observed to be in the range of 50–70% as compared to the nearly 100% obtained with microfluidics. | ||
| Fig. 1 Illustration of the two-step bulk emulsification process for the preparation of large quantities of multiwalled colloidosomes from double emulsion templates. (a) The aqueous phase with the encapsulant is (b) first mixed with the oil phase containing the modified silica particles and (c) then emulsified using for instance a rotor–stator mixer. The stable water-in-oil single emulsion is (d) mixed with a second aqueous phase containing surface active polymers and (e) is emulsified again. (f) After removal of the oil phase by dissolution in water the modified particles give rise to a semi-permeable shell, generating a large quantity of colloidosomes with physically entrapped encapsulants. | ||
 | ||
| Fig. 2 (a) Single emulsion stabilized with SiO2 particles, and (b) double emulsion stabilized with particles and PVA. (c) Silica colloidosome containing 1 μm hydrophilic silica particles as a model encapsulant. Scale bars: 25 μm. | ||
The formation of double emulsions using such a high throughput process requires efficient stabilization of the inner w/o and the outer o/w droplets during both emulsification steps.28–31 In our approach, we rely solely on particles to stabilize the inner w/o interface during the first emulsification step. Following our earlier work,26 we modified the surface of the silica particles to promote their adsorption at the interface. Surface modification using an alkane-terminated silane alone (trimethoxy(octadecyl)-silane) was found to be insufficient for the stabilization of the inner droplets, probably because of the low wettability of the modified particles in water. Instead, a combination of alkane- and amine-terminated silanes as modifiers increased the wettability of the particles in water, making them sufficiently surface active to adsorb at the w/o interface and thus form more stable Pickering emulsions.
Interfacial rheology measurements show that the modified silica particles adsorbed on the w/o droplets are able to form a strong viscoelastic film at the interface (Fig. 3). Although the interfacial rheology experiments do not replicate the timescales and the complex shear conditions encountered during bulk emulsification, they provide insightful information about the role of the nanoparticles in the stabilization of such double emulsions. The long timescales required in the rheological experiment to form a strong interfacial film are likely shorter during the bulk emulsification process due to the expected increase in the frequency of collisions between particles and interfaces under high shear conditions. Due to its high strength, the interfacial film withstands prolonged shearing preventing further rupture of the inner w/o droplets during the second emulsification step. This allows us to obtain outer o/w droplets that are close in size to the inner w/o droplets, leading to a high fraction of double emulsions with a 1:1 number ratio of inner-to-outer water droplets when intensive shearing is applied (Fig. 4a and c). Such proportion is usually difficult to achieve when the inner w/o surface is only stabilized by surfactants, since the absence of a viscoelastic film leads to continuous break-down of the inner droplets as shearing is intensified.28–30 The low number ratio of inner-to-outer droplets increases the encapsulation efficiency and minimizes shape heterogeneities that can lead to defects in the final colloidosome. Removal of the oil phase from the multiple emulsions results in colloidosomes with one, two, three or more compartments with effectively entrapped encapsulants (Fig. 4d). For each emulsification batch, a mixture of colloidosomes with different number of compartments is produced. The relative fraction of colloidosomes with one particular number of compartments in the mixture can be adjusted by tuning the energy provided in the second emulsification step.
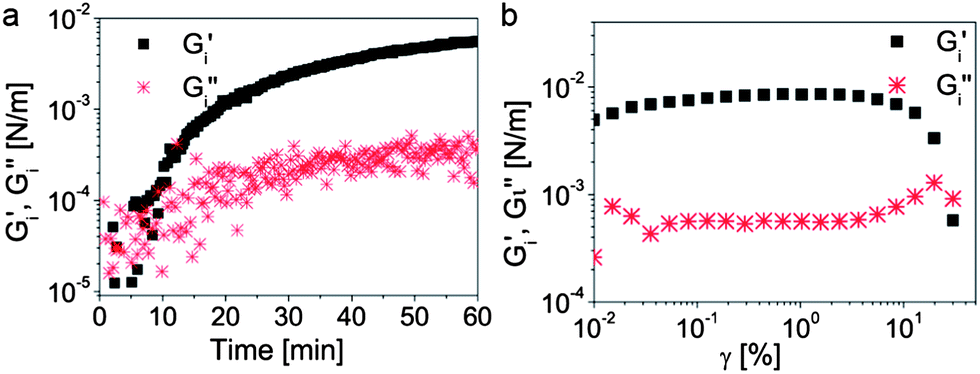 | ||
| Fig. 3 (a) Rheological properties of the toluene–water interface as a function of time, indicating the gradual interfacial adsorption of silica particles initially added to the toluene phase. (b) Viscoelastic response of the resulting film as a function of the applied strain. The crossover of the storage (G′) and the loss (G′′) moduli at high strains clearly indicates the formation of a strong film at the toluene–water interface. | ||
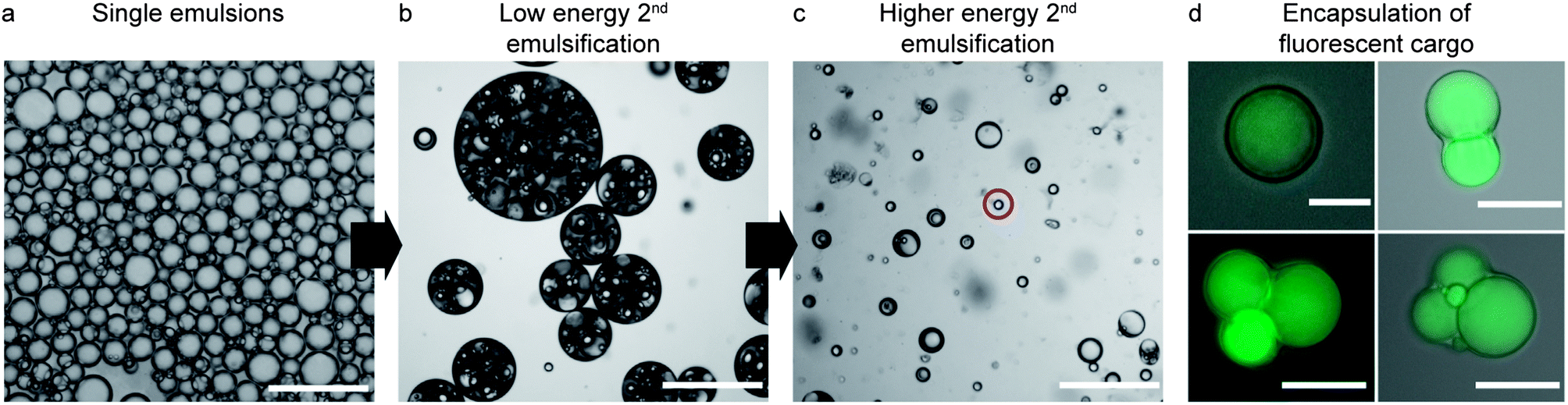 | ||
| Fig. 4 Tuning of the number of compartments in each colloidosome by controlling the input energy during the second emulsification step. (a) Single water-in-toluene emulsion stabilized by interfacially adsorbed silica particles. (b and c) Double emulsions stabilized by particles added to the intermediate toluene phase and PVA present in the continuous phase after (b) short/low energy shearing or (c) long/high energy shearing during the second emulsification step. In images (b) and (c), the energy input was increased by simply mixing the emulsions longer by manual pipetting. The red circle indicates a single o/w emulsion. Scale bars in (a–c): 250 μm. (d) Colloidosomes obtained after removal of toluene exhibiting different number of compartments loaded with an encapsulated fluorescent dye. Scale bars: 10 μm in the top left image and 75 μm in the others. | ||
The shearing forces imposed during the second emulsification step to reduce the size of the outer droplets also generate smaller oil-in-water single droplets, resulting from the removal of the oil from the intermediate phase. Thus, reducing the average size of the outer droplets inevitably leads to a mixture of single and double emulsions (Fig. 4c). We show that simple, scalable techniques based on sieving, creaming/sedimentation or magnetic manipulation can be employed to remove the undesired single oil droplets from the double emulsions. For instance, complete removal of the oil phase from the system through dissolution and evaporation (Fig. 1f) makes single emulsion droplets considerably smaller than double emulsion droplets. This enables their removal from the mixture by simple sieving. The smaller size of single droplets results from the strong shrinkage observed during dissolution of the oil in the continuous aqueous phase. In contrast, the shrinkage of double emulsion droplets during oil removal is constrained by the inner w/o droplet. The collapsed single emulsions obtained after oil dissolution can easily be removed using standard polymeric filter papers to generate a large quantity of colloidosomes alone (Fig. 5a).
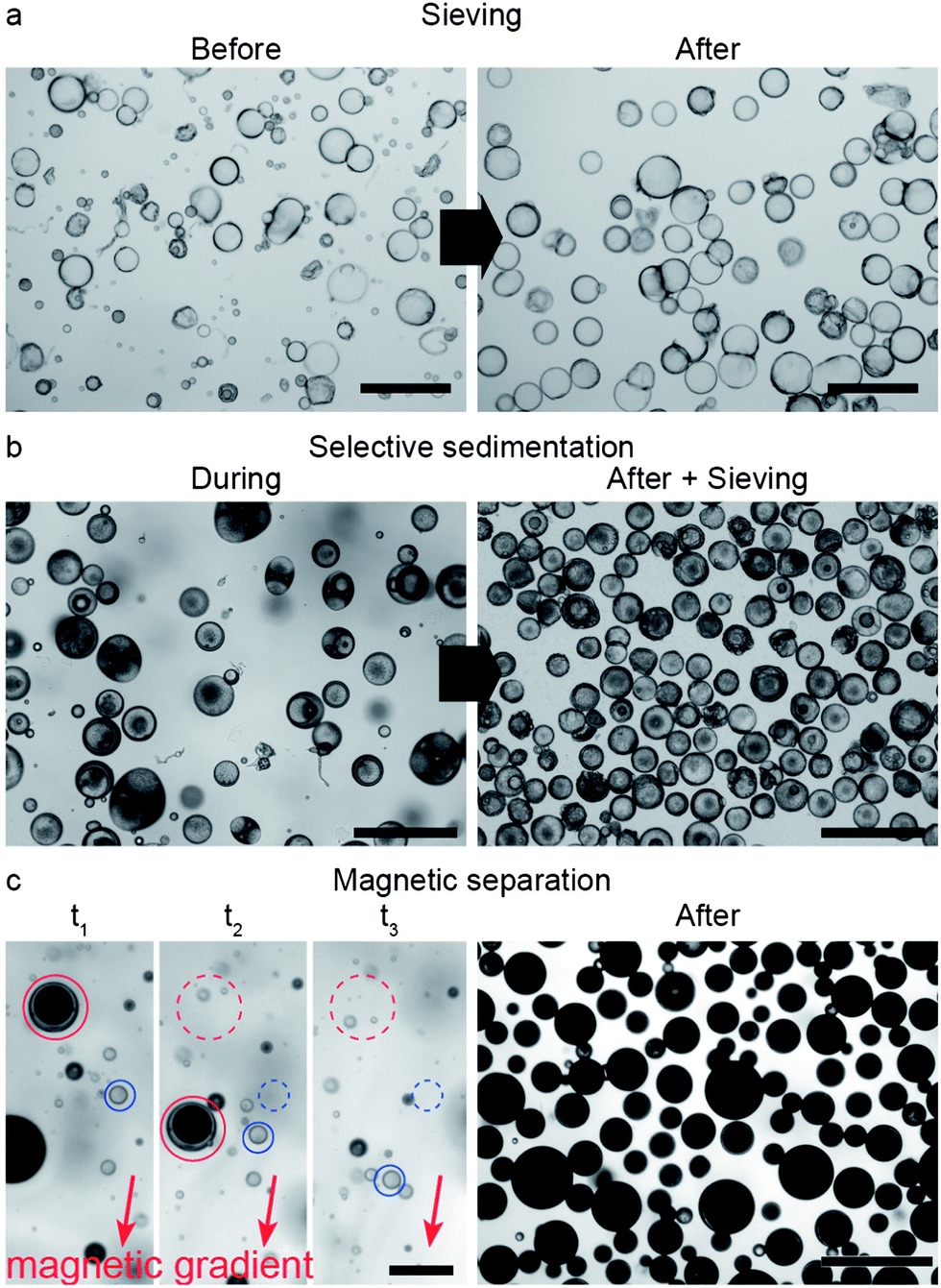 | ||
| Fig. 5 Separation of colloidosomes from single o/w droplets via (a) sieving, (b) selective sedimentation followed by sieving, and (c) magnetic manipulation. (a) Colloidosomes obtained from double emulsion templates before and after sieving with a 60 μm polymer mesh (scale bars: 250 μm). (b) Mixture of single oil droplets and double emulsion templates filled with 1 μm silica particles during selective sedimentation (left). The final colloidosomes obtained after sedimentation followed by an additional sieving step are depicted on the right image (scale bars: 250 μm). (c) Snapshots showing double emulsion templates loaded with superparamagnetic particles being separated from nonmagnetic single oil droplets using a magnetic field gradient (left, scale bar: 250 μm). The image on the right displays the large quantity of magnetically responsive colloidosomes obtained after the separation process (scale bar: 500 μm). As a guide to the eye, the initial and present positions of a double emulsion (red) and of a single emulsion (blue) are marked with dashed and full lines, respectively. At the elapsed time t3 the marked double emulsion has already left the area of the image. | ||
In another approach, even higher yields are achieved by the selective sedimentation of double emulsion droplets from the initial mixture. In this case, effective separation is accomplished by purposefully increasing the specific gravity of the double emulsions as compared to the single o/w droplets. At particle contents in the middle oil phase lower than around 18 wt%, the toluene suspension is lighter than water and thus both double and single emulsions cream. However, the double emulsions can be made heavier than water to selectively sediment by simply adding high-density colloidal particles into their innermost phase (Fig. 5b and ESI Movie S2†). By combining it with a final sieving step, we were able to achieve a fraction of double emulsions as high as 93.5% using such a selective sedimentation approach. Alternatively, selective separation can be accomplished by adding other responsive particles that are for instance superparamagnetic. These enable facile separation of the magnetic double emulsion from the nonmagnetic single o/w droplets under mild external magnetic fields (Fig. 5c and ESI Movie S3†). Using this approach, it is possible to completely separate the double emulsions from the initial mixture (100% yield), even for initial mixtures containing number fractions of double emulsions as low as 30%.
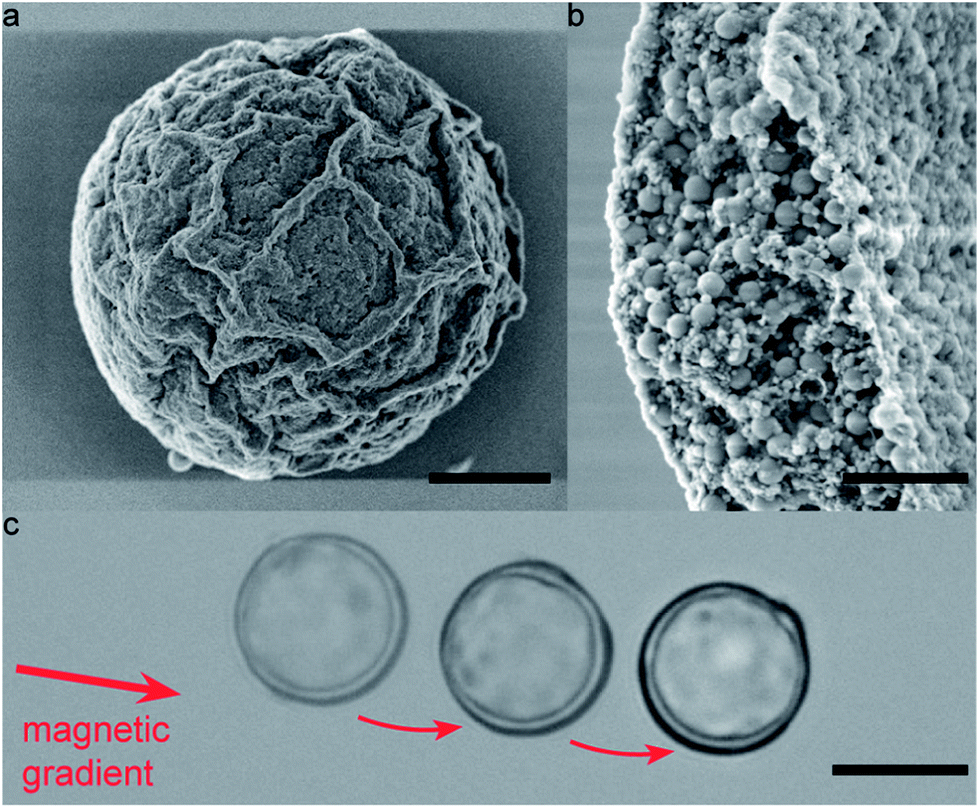
Superparamagnetic particles can also be used to sort colloidosomes according to their size, r. Since the magnetic force scales with the volume (∼r3) and the drag force at low velocities scales with the size (∼r), larger colloidosomes are attracted more strongly to an imposed magnetic gradient (Fig. 6 and ESI Movie S4†).
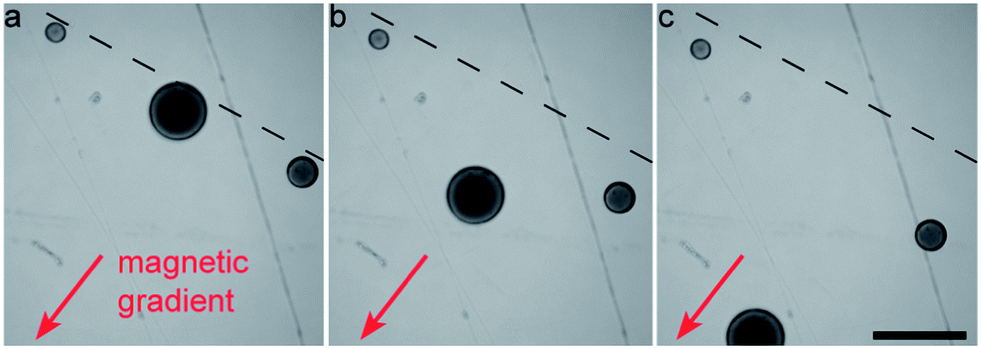 | ||
| Fig. 6 Snapshots showing the separation of Fe3O4-filled colloidosomes by size using a magnetic field gradient. The initial positions of three colloidosomes of different sizes are marked with a dashed line. Images were taken with elapsed time differences of 1.5 s. Scale bar: 250 μm. | ||
The two-step technique employed for the fabrication of multiwalled colloidosomes is also flexible regarding the shell material. By combining different types of nanoparticles in the intermediate oil phase we can adapt the shell to obtain colloidosomes with specific functional properties. For instance, particles of different sizes can be used to make the interstitial openings smaller and thus change the shell permeability, as illustrated in Fig. 7 for a composite colloidosome containing 250 nm SiO2 and 40 nm Al2O3 particles. In another example, Fe3O4 particles can be added to the shell to make the colloidosomes magnetically responsive, enabling guided transport of the capsule using an external field gradient (Fig. 7c and ESI Movie S5†).
 | ||
| Fig. 7 Functional multiwalled colloidosomes made by bulk emulsification. (a and b) Example of an Al2O3–SiO2 composite colloidosome with a multiwalled shell. The 40 nm Al2O3 particles are located within the 250 nm SiO2 particles and also inhabit the outer and inner oil–water interface. Scale bars: 4 μm in (a); 1 μm in (b). (c) Overlay of several snapshots taken from a SiO2–Fe3O4 colloidosome transported by a magnetic field gradient. Scale bar: 15 μm. | ||
The proposed two-step bulk emulsification technique also enables the creation of multiwalled colloidosomes that are only a few microns in diameter, which are one to two orders of magnitude smaller than similar capsules obtained by state-of-the-art single-dripping microfluidic devices.7,21 As an example, Fig. 8a and b display small silica colloidosomes made from double emulsion templates prepared using high-energy ultrasonication. This technique is usually considered inappropriate for two-step emulsification due to the high energy imposed during the second step.29 However, the high interfacial strength provided by the silica particles adsorbed on the w/o droplets makes our approach also applicable in high-energy emulsification processes. Remarkably, the multiwalled semi-permeable capsules obtained via this simple method are sufficiently small to even undergo Brownian motion when dispersed in water. The random walk diffusion that characterizes such systems was evidenced by tracking the position of a 2.5 μm large colloidosome over a time period of 112 s (Fig. 8c and ESI Movie S6†). Brownian motion was also clear from the erratic rotation of an anisotropic dumbbell colloidosome in water, as shown by the series of snapshots displayed in Fig. 8d–i (see also ESI Movie S7†). Despite their small dimensions, the Brownian colloidosomes were sufficiently tight to effectively encapsulate a fluorescent dye (see Fig. S1 in the ESI†). Given the topological defects expected on such highly curved surfaces,2,18 the tightness of such capsules clearly shows the advantage of the multilayered wall in providing a capsule shell that is less sensitive to particle packing defects.
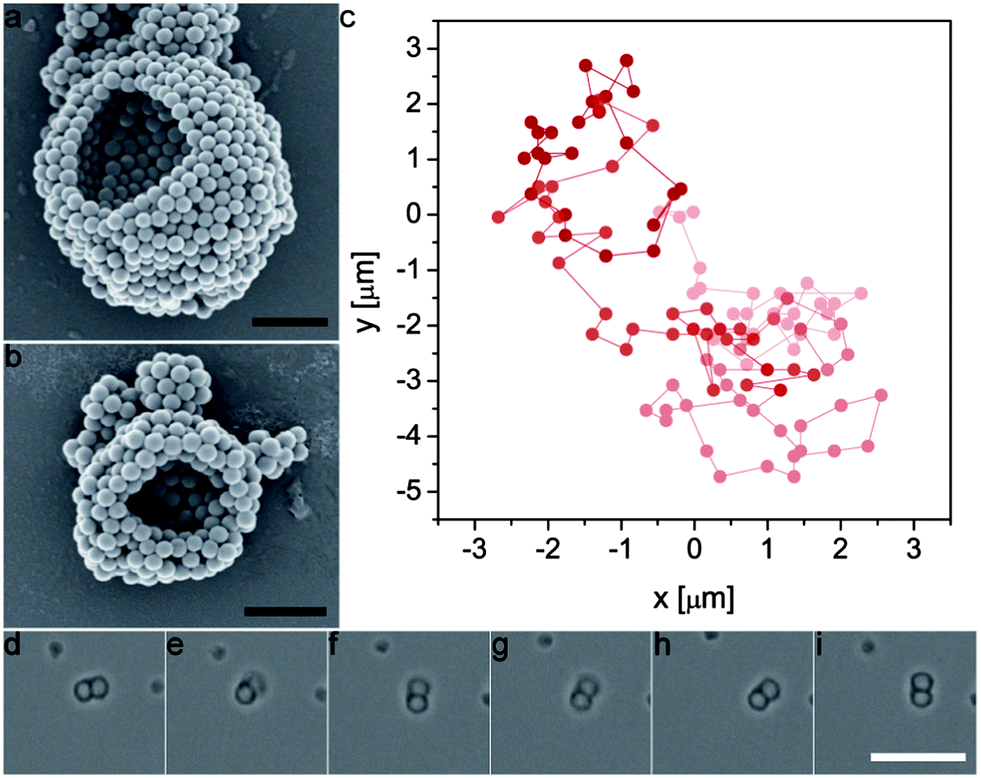 | ||
| Fig. 8 (a and b) SEM images of colloidosomes made from ultrasonicated double emulsions. Scale bars: 1 μm. (c) Position of a colloidosome initially located at (x, y) = (0, 0) after many 1 second time steps. For clarity, earlier positions are displayed in lighter colours. (d–i) Snapshots taken with 2 s time intervals showing Brownian rotation of a dimer colloidosome. Scale bar: 10 μm. | ||
The high flexibility of the proposed method allows us to apply previously developed techniques to trigger the release of encapsulants from nanoparticle-filled colloidosomes.25 For example, by encapsulating pH-swellable particles inside the colloidosome, it is possible to rupture the capsule shell and thus release its content upon addition of a base, as shown in Fig. 9 (see also ESI Movie S8†). In this example, colloidosomes were loaded with a fluorescently labeled dextran dye as a model cargo (Mw = 500000 g mol−1, Sigma Aldrich, Switzerland) and alkali swellable microgel particles as on-demand chemical triggers. Due to their large size, the dye and the microgel particles are initially trapped in the colloidosomes. Addition of a few drops of 0.1 M KOH leads to a rapid increase in the pH, resulting in swelling of the microgel particles and rupture of the shell to release the cargo (Fig. 9a–d). Such pH triggered rupture and release is also possible with small Brownian colloidosomes (Movie S9 in the ESI†).
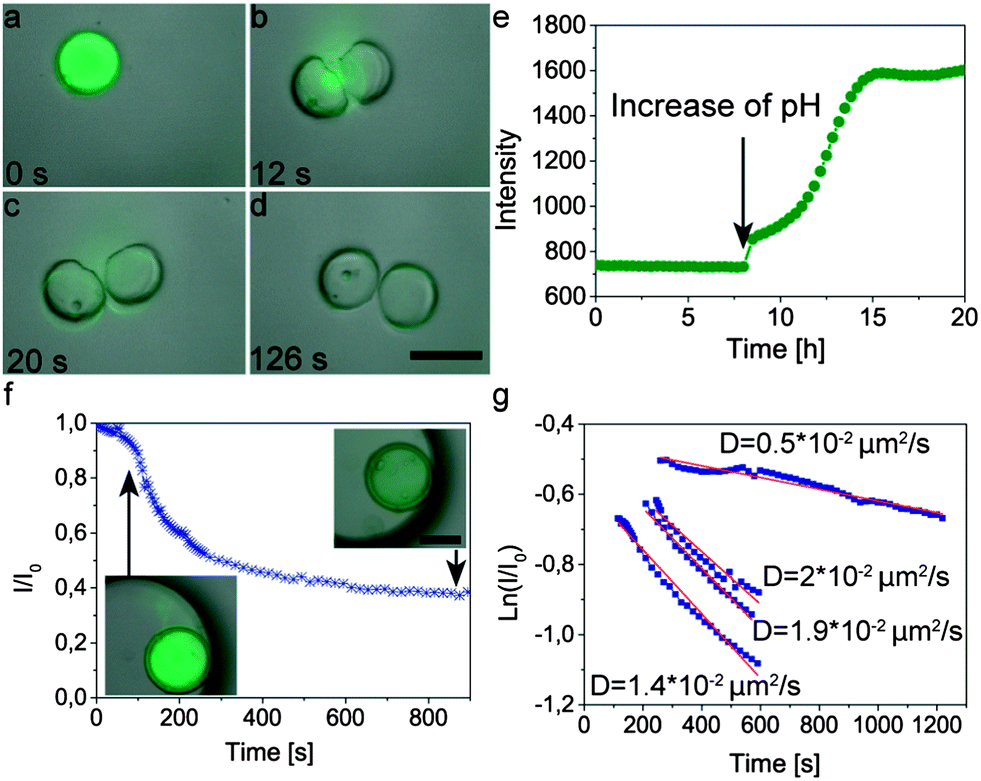 | ||
| Fig. 9 Release kinetics of fluorescently labeled dextran (Mw = 500000 g mol−1) upon (a–d) abrupt rupture or (e–g) cracking of multiwalled colloidosomes due to swelling of internal microgel particles. Images (a–d) show overlays of fluorescence and brightfield microscopy snapshots of a ruptured colloidosome after increase of pH by addition of 0.1 M KOH. Scale bar: 75 μm. Graph (e) shows the fluorescence intensity as a function of time measured in the supernatant of a solution containing sedimented colloidosomes. (f) Fluorescence intensity inside a colloidosome that was gently cracked through the addition of 0.2 mM buffer at pH 10. After an initial burst (inset on left), the intensity gradually decreases over time (inset on right). Scale bar in inset: 100 μm (g) Exponential fittings to the intensity data during slow release from four different capsules. The calculated diffusion coefficients of the dye through the cracked shells are indicated in the graph. | ||
To study the triggered release mechanism, we monitored the concentration of the dye released by measuring the fluorescence intensity of the supernatant of a 1 ml suspension containing 1.3 mg colloidosomes sedimented at the bottom of a measuring cuvette (Fig. 9e). During the first 8 h of the measurement the colloidosomes are immersed at pH 5 and consequently the fluorescence intensity in the supernatant remains constant. Replacement of the supernatant by a 0.2 mM buffer solution at pH 10 leads to a continuous increase of the fluorescence intensity of the supernatant solution, confirming the release of the dye and its diffusion from the bottom to the measurable window at a position higher up in the cuvette. As opposed to the abrupt rupture of the shell achieved by addition of the KOH solution (Fig. 9a–d), the use of a buffer solution (pH 10) leads to a more gentle change in pH and thus the formation of smaller cracks on the shell that are not visible with the optical microscope (insets in Fig. 9f and ESI Movie S8†).
To better quantify the triggered release of fluorescently labeled cargo we also measured the average intensity of the fluorescent dye throughout a cross-section inside the colloidosomes with small cracks (Fig. 9f). In this case, the change in fluorescence can be directly linked to the release event and is not affected by diffusion of the dye from the bottom to the measuring area in the cuvette. The rapid drop of intensity and dye concentration observed in the first 200 seconds after addition of the buffer solution (pH 10, Fig. 9f) is caused by cracking of the colloidosome wall as a result of the large pressure exerted by the hydrogel particles upon swelling. The initial burst is followed by a slower release of the dye through the cracks generated within the colloidosome wall. The release kinetics at this slow phase can be fitted reasonably using a simple exponential equation of the form:
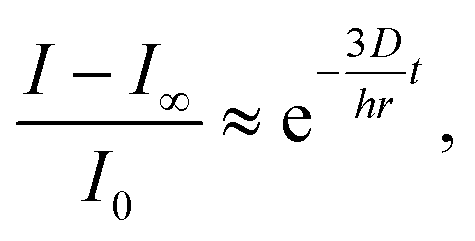 | (1) |
Conclusions
Thickwalled colloidosomes ranging from hundreds of microns to a few microns in size can be created at high throughput rates if particle-stabilized single emulsions are used as templates in a bulk, two-step double emulsification process. In the first emulsification step, silica nanoparticles adsorbed on the surface of single w/o emulsion templates were found to form a strong viscoelastic film at the interface that effectively stabilizes the droplets against rupture in the second emulsification process. This allows us to apply sufficiently high shear rates in the second emulsification to create colloidosome mixtures with a tunable average number of internal compartments, including capsules with one single compartment alike those obtained by microfluidic emulsification. While the yield of tight colloidosomes is lower than that obtained via microfluidics, the method proposed offers a simple high-throughput route for the preparation of colloidosomes with tunable permeability, shell material and sizes. We exploited the high flexibility of this approach to create various types of functional colloidosomes, including composite capsules that can be manipulated with magnetic fields and multiwalled colloidosomes that are sufficiently small to undergo Brownian motion in fluids and that can release their encapsulant upon pH-triggered rupture of the shell. The ability to trigger the release from semi-permeable multiwalled capsules of such small sizes offers unique opportunities for efficient on-demand delivery of minor amounts of encapsulants in several potential applications.Acknowledgements
We thank Patrick Rühs and Dr Peter Fischer from the Laboratory of Food Process Engineering at the Institute of Food, Nutrition and Health from ETH Zurich for kindly providing the tools and expertise for the interfacial rheology experiments. We also greatly acknowledge the Swiss National Science Foundation (grant number 200021_126646) and ETH Zurich for the financial support.Notes and references
- O. D. Velev, K. Furusawa and K. Nagayama, Langmuir, 1996, 12, 2374–2384 CrossRef CAS.
- A. D. Dinsmore, M. F. Hsu, M. G. Nikolaides, M. Marquez, A. R. Bausch and D. A. Weitz, Science, 2002, 298, 1006–1009 CrossRef CAS PubMed.
- P. H. R. Keen, N. K. H. Slater and A. F. Routh, Langmuir, 2011, 28, 1169–1174 CrossRef PubMed.
- P. H. R. Keen, N. K. H. Slater and A. F. Routh, Langmuir, 2012, 28, 16007–16014 CrossRef CAS PubMed.
- I. Akartuna, E. Tervoort, A. R. Studart and L. J. Gauckler, Langmuir, 2009, 25, 12419–12424 CrossRef CAS PubMed.
- H. W. Duan, D. Y. Wang, N. S. Sobal, M. Giersig, D. G. Kurth and H. Mohwald, Nano Lett., 2005, 5, 949–952 CrossRef CAS PubMed.
- J. S. Sander and A. R. Studart, Langmuir, 2011, 27, 3301–3307 CrossRef CAS PubMed.
- O. J. Cayre, J. Hitchcock, M. S. Manga, S. Fincham, A. Simoes, R. A. Williams and S. Biggs, Soft Matter, 2012, 8, 4717–4724 RSC.
- A. S. Miguel and S. H. Behrens, Soft Matter, 2011, 7, 1948–1956 RSC.
- A. S. Miguel, J. Scrimgeour, J. E. Curtis and S. H. Behrens, Soft Matter, 2010, 6, 3163–3166 RSC.
- J.-M. Rabanel, X. Banquy, H. Zouaoui, M. Mokhtar and P. Hildgen, Biotechnol. Prog., 2009, 25, 946–963 CrossRef CAS PubMed.
- V. Taly, B. T. Kelly and A. D. Griffiths, ChemBioChem, 2007, 8, 263–272 CrossRef CAS PubMed.
- M. A. Augustin and Y. Hemar, Chem. Soc. Rev., 2009, 38, 902–912 RSC.
- Y. Pan, J. Gao, B. Zhang, X. Zhang and B. Xu, Langmuir, 2010, 26, 4184–4187 CrossRef CAS PubMed.
- T. Bollhorst, T. Grieb, A. Rosenauer, G. Fuller, M. Maas and K. Rezwan, Chem. Mater., 2013, 25, 3464–3471 CrossRef CAS.
- C. S. Wagner, A. Fortini, E. Hofmann, T. Lunkenbein, M. Schmidt and A. Wittemann, Soft Matter, 2012, 8, 1928–1933 RSC.
- R. K. Shah, J.-W. Kim and D. A. Weitz, Langmuir, 2010, 26, 1561–1565 CrossRef CAS PubMed.
- A. R. Bausch, M. J. Bowick, A. Cacciuto, A. D. Dinsmore, M. F. Hsu, D. R. Nelson, M. G. Nikolaides, A. Travesset and D. A. Weitz, Science, 2003, 299, 1716–1718 CrossRef CAS PubMed.
- H. N. Yow and A. F. Routh, Soft Matter, 2006, 2, 940–949 RSC.
- R. T. Rosenberg and N. R. Dan, J. Colloid Interface Sci., 2011, 354, 478–482 CrossRef CAS PubMed.
- D. Lee and D. A. Weitz, Adv. Mater., 2008, 20, 3498–3503 CrossRef CAS.
- T. Nisisako, T. Ando and T. Hatsuzawa, Lab Chip, 2012, 12, 3426–3435 RSC.
- M. B. Romanowsky, A. R. Abate, A. Rotem, C. Holtze and D. A. Weitz, Lab Chip, 2012, 12, 802–807 RSC.
- C. Yuan, Y. Xu, N. Jiang, G. Chen, B. Xu, N. He and L. Dai, Soft Matter, 2011, 7, 3366–3372 RSC.
- J. S. Sander and A. R. Studart, 2013, submitted.
- J. S. Sander, L. Isa, P. A. Ruhs, P. Fischer and A. R. Studart, Soft Matter, 2012, 8, 11471–11477 RSC.
- P. Erni, P. Fischer, E. J. Windhab, V. Kusnezov, H. Stettin and J. Lauger, Rev. Sci. Instrum., 2003, 74, 4916–4924 CrossRef CAS.
- N. Garti, Colloids Surf., A, 1997, 123, 233–246 CrossRef.
- A. T. Florence and D. Whitehill, Int. J. Pharm., 1982, 11, 277–308 CrossRef CAS.
- J. Bibette, F. L. Calderon and P. Poulin, Rep. Prog. Phys., 1999, 62, 969–1033 CrossRef CAS.
- B. P. Binks, A. K. F. Dyab and P. D. I. Fletcher, Chem. Commun., 2003, 2540–2541 RSC.
- T. C. Laurent, L. O. Sundelof, K. O. Wik and B. Warmegard, Eur. J. Biochem., 1976, 68, 95–102 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Movies S1–S9. See DOI: 10.1039/c3sm51900g |
| This journal is © The Royal Society of Chemistry 2014 |