Sugar-coated sensor chip and nanoparticle surfaces for the in vitro enzymatic synthesis of starch-like materials†
Ellis C.
O'Neill
a,
Abdul M.
Rashid
a,
Clare E. M.
Stevenson
a,
Anne-Claire
Hetru
a,
A. Patrick
Gunning
b,
Martin
Rejzek
a,
Sergey A.
Nepogodiev
a,
Stephen
Bornemann
a,
David M.
Lawson
a and
Robert A.
Field
*a
aDepartment of Biological Chemistry, John Innes Centre, Norwich Research Park, Norwich, NR4 7UH, UK. E-mail: rob.field@jic.ac.uk; Fax: +44 (0)1603-450018; Tel: +44 (0)1603-450720
bInstitute of Food Research, Norwich Research Park, Norwich, NR4 7UA, UK
First published on 23rd October 2013
Abstract
The insoluble glucan polymer starch is a major player in the human diet; it is also an important bulk commodity. Nonetheless, our understanding of its biochemistry remains poor, not least because of the challenge of analysing enzymes that operate across the solid–liquid interface. In the present study, the enzymatic polymerisation of glucans immobilised on gold sensor chip and nanoparticle surfaces was achieved with Arabidopsis phosphorylase AtPHS2. The basis of the action of AtPHS2 on surface glucans could be rationalised through consideration of the X-ray crystal structure of this enzyme, which identified a previously unreported enzyme surface binding site for glucans. Extension of the glucan-coated sensor chip surfaces could be monitored in real time by SPR, enabling kinetic analysis of AtPHS2-mediated glucan synthesis, which showed similar efficiency to in solution analyses. Extension of both sensor and nanoparticles surfaces coated with glucan was analysed by TEM, which confirmed glucan polymerisation. The arrangement of newly formed glucan chains into ordered helical arrangements was evident from iodine staining, as well as from enzyme response characteristics that proved typical of starch-like material. As such, the glucan-modified sensor chip and nanoparticle surfaces represent novel tools with which to analyse starch-active enzymes.
Introduction
Starch, a branched homo-polymer of glucose,1 makes the single highest calorific contribution to the human diet2 and is also a bulk commodity material, with a billions of tonnes per year global market.3 The ever-increasing demand for starch-based products cannot be met by further commitment of agricultural land to starch-based crops alone; substantial increases in crop yield are clearly needed, as are new ways to produce fit-for-purpose starch-based materials.4 The production of modified starches in planta is attractive.5 For example, the ratio of linear amylose to branched amylopectin in starch can be lowered6 or raised7 using genetic engineering, altering the gelatinisation properties of the starch. However, the generation time of plants and the rather empirical nature of gene selection would benefit from a more informed and targeted approach. To this end, a better understanding of the action of enzymes naturally involved in starch metabolism, and those employed for industrial starch modification, is called for. In the former case, we have begun to deploy chemical genomics approaches to understand the in vivo action of starch-active enzymes.8,9 There are many isoforms of the enzymes involved in starch granule synthesis and breakdown, but differences in enzyme action are often interpreted in terms of gene and protein expression profiles, rather than activity and substrate specificity. Furthermore an inherent complication of starch enzymology is often ignored – namely that starch is not water soluble, while the enzymes that act upon it are. The biochemistry of starch, and indeed the in vitro enzymatic modification of starch, therefore takes place across a solid–liquid interface, which has a profound impact on enzyme action. Reflecting this point, the activity of granule bound starch synthase, for instance, has been shown to be enhanced on crystalline amylopectin compared to soluble substrate.10 In order to better understand and exploit enzymes in starch (bio)chemistry, we were drawn to consider methods for the analysis of enzymes acting at a solid–liquid interface. In the current study, we therefore had a need for a convenient enzymatic method to polymerise glucan chains immobilised on a surface, along with a straightforward means to analyse the polymerisation process.Electron microscopy has been used to characterise the products of amylosucrase-catalysed synthesis on glycogen particles, which gives rise to semi-crystalline starch-like material.11 This technique has also been employed to demonstrate that the initial attack on a starch granule surface alters the granule.12 Direct monitoring of purified starch granules during alpha-amylase degradation, either using electron microscopy13 or synchrotron UV fluorescence microscopy,14 provides snapshots of the processes involved. Clearly such experiments provide information on structures, but not on reaction rates as such. Studies involving enzyme action on crystalline maltodextrins,15 or purified starch granules,16 have been reported, but they have measured product release from the insoluble substrate and not the breakdown of the insoluble substrate per se. In order to understand starch surface biochemistry, one really needs to monitor what is happening to the insoluble substrate, and not the released by-product, in real time. Previously, quartz crystal microbalance technology has been used to monitor phosphorylase-catalysed amylopectin extension in real time17 and to provide kinetic information.18 In addition, plant oligosaccharide microarrays are beginning to enable high through-put analysis of carbohydrates immobilised on surfaces19 a topic that has recently been reviewed.20
Amylose can be readily synthesised in solution, either by the debranching of amylopectin with isoamylase (EC 3.2.1.68)21 or by the extension of acceptor glucans with amylosucrase (EC 2.4.1.4) or phosphorylase (EC 2.4.1.1).22 Amylosucrase has been used to synthesise crystalline material,23 dendritic nanoparticles based on glycogen11 and, in combination with a branching enzyme, highly branched glucan from sucrose.24 Bacterial and mammalian glucan phosphorylases have been employed to coat glycogen with a range of sugars, including glucuronic acid,25 2-deoxy-glucose26 and deoxyfluoroglucoses.27 However, plant phosphorylases, with their lack of allosteric regulation,28 have proven much more useful in the synthesis of amylose derivatives, both by extension of covalent acceptors, such as maltoheptaose immobilised on chitosan29 or polystyrene,30 or by twining polysaccharides around a hydrophobic core to form a macromolecular complex, such as amylose-wrapped lipid.31
We have previously shown that surface plasmon resonance (SPR) can be used to measure the synthesis of soluble polysaccharides on surfaces.32,33 We have also demonstrated that gold nanoparticles provide an excellent system for displaying carbohydrates on a surface such that they are accessible to carbohydrate-binding proteins.34,35 In the current study, we report the use of Arabidopsis thaliana glucan phosphorylase PHS2 (AtPHS2) for the enzymatic extension of glucan-coated SPR biosensor and gold nanoparticle surfaces. These approaches produce insoluble glucan architectures that may be deployed for the assessment of starch-active enzymes.
Results and discussion
AtPHS2-catalysed synthesis of amylose in solution
In the current study, and following the literature,36A. thaliana PHS2 (AtPHS2) was cloned and over-expressed in E. coli as described in the ESI,† producing ca. 2.5 mg of greater than 99% pure enzyme per litre of culture (Fig. S1†). The specific activity of this enzyme in the synthetic direction was assessed by measuring phosphate released from glucose-1-phosphate (Glc-1-P) using a molybdate-based assay.37 Under optimal conditions (pH 6.0, 50 °C, Fig. S2†), the specific activity of AtPHS2 was 18 μmol min−1 mg−1 (Fig. S3†), similar to that of the rabbit muscle enzyme (25 μmol min−1 mg−1)38 and an order of magnitude faster than plant starch synthases (0.1–3 μmol min−1 mg−1).39,40 This is also 2–3 times faster than the degradative reaction of AtPHS2 (6–8 μmol min−1 mg−1).36 The minimum acceptor length for glucosylation by PHS2 was found to be maltotetraose (Table S1†), as is the case for most enzymes of this class,41 an important consideration when using this enzyme in a synthetic sense.In order to conveniently monitor AtPHS2-catalysed amylose synthesis in solution, we used carbohydrate electrophoresis with laser-induced fluorescence detection (CE-LIF) to monitor extension of glucan chains carrying an 8-aminopyrene-1,3,6-trisulfonate (APTS) label reductively-aminated to their reducing termini. We have found that this method, which is more generally used for carbohydrate structure analysis42 and glycoprofiling,43 provides both a sensitive and convenient approach for the direct monitoring of enzymatic reactions.44 Using a CE approach, the separation of glucan oligomers, released from starch by hydrolysis and having a DP of up to 80, has previously been achieved.45 Varying the Glc-1-P![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acceptor ratio is an established means to control the length of the polymer formed.46 In our hands, AtPHS2 extended APTS-labelled maltopentaose to at least 50 glucose residues, with an average DP of approximately 30 (Fig. 1 – purple), and up to 100 residues could be achieved by increasing AtPHS2 concentration (data not shown). AtPHS2-catalysed reactions can easily be driven in either direction, with glucan synthesis achieved in the presence of excess Glc-1-P, while newly synthesised glucan polymers can be shortened by phosphorolysis in the presence of excess phosphate (Fig. 1 – red). Amyloglucosidase, a specific α-1,4-glucan hydrolase, can also degrade the polymers synthesised by AtPHS2, confirming they are indeed amylose chains (Fig. 1 – blue).
:acceptor ratio is an established means to control the length of the polymer formed.46 In our hands, AtPHS2 extended APTS-labelled maltopentaose to at least 50 glucose residues, with an average DP of approximately 30 (Fig. 1 – purple), and up to 100 residues could be achieved by increasing AtPHS2 concentration (data not shown). AtPHS2-catalysed reactions can easily be driven in either direction, with glucan synthesis achieved in the presence of excess Glc-1-P, while newly synthesised glucan polymers can be shortened by phosphorolysis in the presence of excess phosphate (Fig. 1 – red). Amyloglucosidase, a specific α-1,4-glucan hydrolase, can also degrade the polymers synthesised by AtPHS2, confirming they are indeed amylose chains (Fig. 1 – blue).
 | ||
| Fig. 1 AtPHS2 catalyses the reversible transfer of glucose from Glc-1-P onto maltooligosaccharide acceptors. Black. APTS–maltopentaose (2.2 μM). Purple. AtPHS2 (5 μg ml−1) transfers glucose from Glc-1-P (10 mM) onto APTS–maltopentaose (2.2 μM) in 24 hours at 30 °C to form polymers with DP up to at least 50. Red. AtPHS2 (5 μg ml−1) phosphorolyses APTS–glucan polymers to APTS–maltotetraose in the presence of elevated phosphate levels (100 mM) at 30 °C in 24 hours. Blue. Amyloglucosidase (50 μg ml−1) hydrolyses APTS–glucan polymers to APTS–maltose and APTS–maltotriose. All reactions were performed in MES (20 mM, pH 6.0) at 30 °C for 24 h. | ||
SPR analysis of surface glucan synthesis by AtPHS2
SPR is typically employed to measure protein–ligand interactions, such as the binding of lectins to carbohydrates.47,48 It can also be used to monitor enzymatic synthesis of polymers such as DNA49 and polysaccharides.32,33 In order to act as an acceptor for AtPHS2-mediated synthesis on a surface, fractionated glucan (FG), a partially hydrolysed amylopectin fragment, was immobilised on a gold sensor chip on which a self-assembled monolayer (SAM) had been prepared.50 Initially, a low sugar density surface was generated by immobilising FG on a 2% carboxyl-modified SAM. Monitoring by SPR demonstrated that while Glc-1-P alone did not bind to the surface (Fig. 2 – pale blue), AtPHS2 was able to bind rapidly and tightly to the surface (Fig. 2 – blue), which prompted structural analysis of AtPHS2 and glucan ligands (vide infra). Upon co-injection of Glc-1-P and AtPHS2, a substantial increase in signal was noted (1200 RU over enzyme alone; Fig. 2 – purple) and the surface became uneven, as judged by AFM (Fig. 2c), with a maximum peak to trough height increasing from 4.5 to 21.2 nm (SD from mean surface height increased from 1 to 2.8 nm). We interpret these data as demonstrating the on-chip synthesis of a relatively unstructured ‘loose’ surface composed of amylose polysaccharide chains. In support of this notion, co-injection of inorganic phosphate and AtPHS2 across the newly-formed surface resulted in phosphorolysis of the surface, according to reduction of the signal (Fig. 2 – brown). Alternatively, the specific α-1,4-glucan hydrolases amyloglucosidase and β-amylase could also degrade AtPHS2-extended surfaces (Fig. 2 – yellow and orange, respectively) confirming that these newly formed surfaces are indeed amylose-based and that inter-chain association is not prevalent.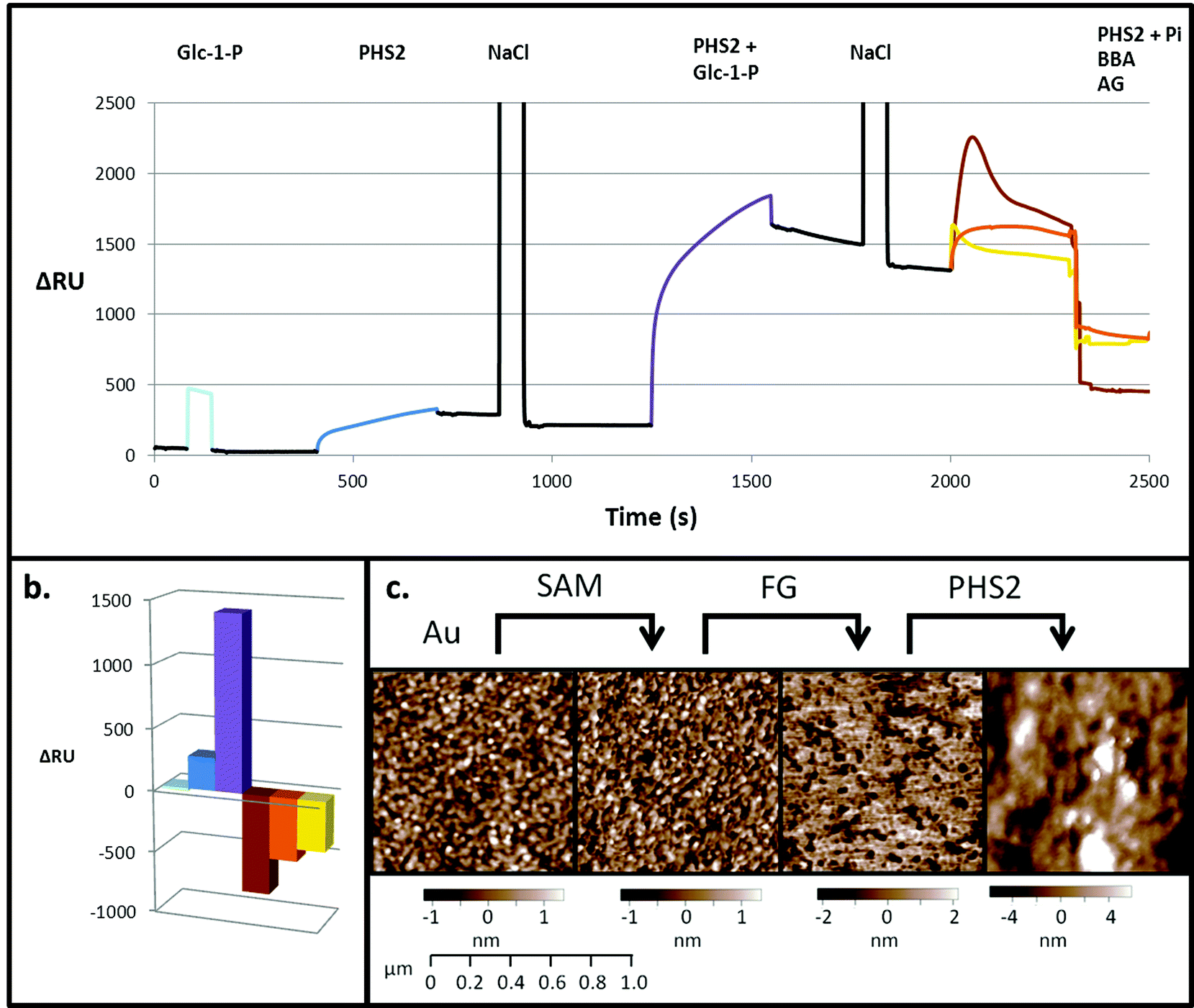 | ||
| Fig. 2 PHS2-catalysed glucan synthesis on a gold sensor surface, as analysed by SPR and AFM. (a) Composite SPR sensorgram, and (b) quantification of the final change in response, using the low sugar density surface. Pale blue. When Glc-1-P (10 mM) was injected over the surface (20 μl injection) there was a bulk change that returns to baseline when the injection ends. Blue. When AtPHS2 (5 μg ml−1) was injected alone there was around 250 RU signal increase, which did not decrease significantly when the injection ended, or with NaCl wash (100 mM, 20 μl). Purple. When AtPHS2 (5 μg ml−1) and Glc-1-P (10 mM) were co-injected, a large response (1200 RU) was seen, giving rise to an elevated and stable baseline after NaCl wash (100 mM, 20 μl). Brown. When AtPHS2 (5 μg ml−1) and Pi (10 mM) were co-injected a large decrease in signal (800 RU) was observed. Yellow. Amyloglucosidase (AG, 1 mg ml−1) caused a significant decrease in signal (500 RU) when injected over the surface. Orange. β-Amylase (BBA, 3 mg ml−1) also gave a decrease (400 RU) in signal when injected over the surface. All enzyme reactions used 100 μl injections. (c) AFM of SPR chip showing a gold sputtered surface, with a standard deviation, corresponding to surface roughness, of 536 pm. After addition of the 2% SAM layer the surface remains smooth (SD 563 pm). Coupling of FG to the surface caused an increase in roughness (SD 2.069 nm) in the region of the SPR flow cell; when AtPHS2 (5 μg ml−1) and Glc-1-P (10 mM) were co-injected, a large change in surface morphology was observed, with an increase in SD from the mean of the surface thickness to 2.768 nm. | ||
Experiments with a high sugar density surface, prepared by attachment of FG to the SAM linker before immobilising on the SPR surface without dilution, showed rapid and tight binding of AtPHS2 (Fig. 3), as was observed with the low sugar density surface. The kinetics of the AtPHS2-catalysed synthesis on the high sugar density surface was assessed using SPR (Fig. S5†). From these experiments it was possible to calculate a KMapp for Glc-1-P of 7.1 ± 2.5 mM, in reasonable agreement with that obtained for the corresponding solution phase analysis (KMapp 3.9 mM). It is also possible to calculate from the total synthesis on the surface over the course of the injection (ca. 3400 ± 1200 RU) and the total enzyme bound to the surface (ca. 240 ± 40 RU) a turnover number of ca. 30 ± 8 s−1 for AtPHS2-catalysed synthesis on the higher sugar density surface, again in reasonable agreement with the corresponding activity in solution at this temperature (7.0 ± 1.8 s−1).32
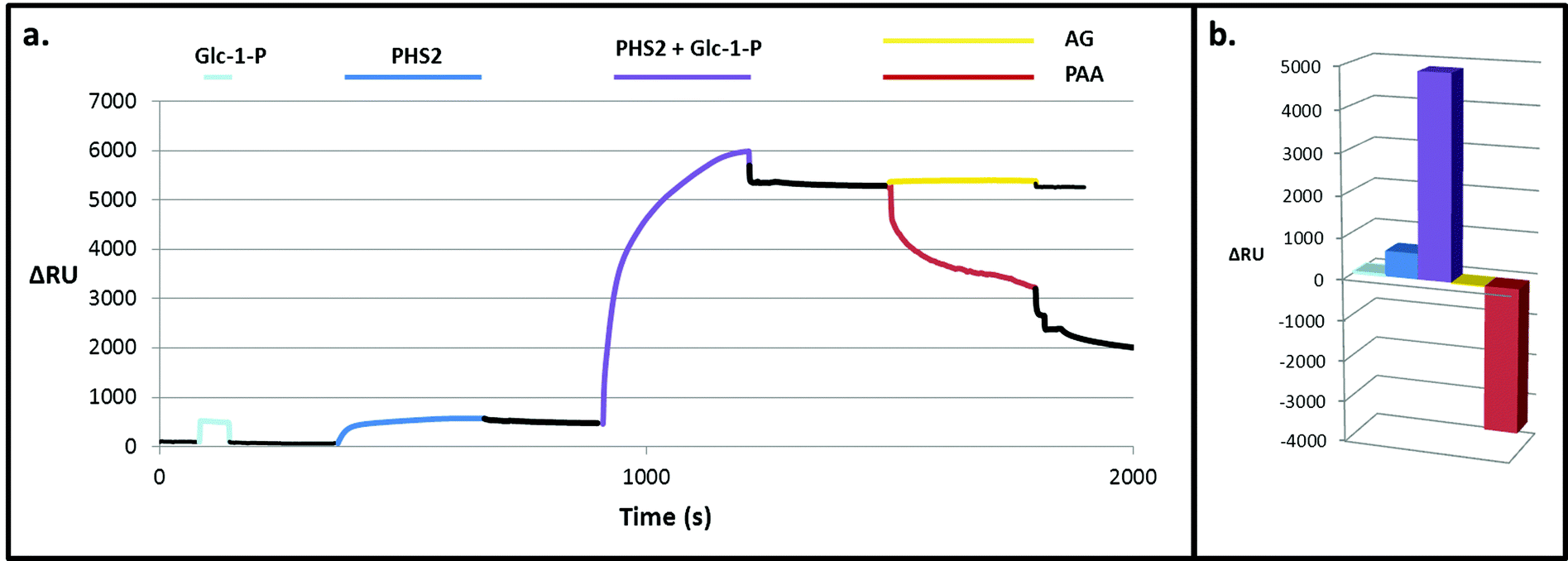 | ||
| Fig. 3 PHS2-catalysed glucan synthesis on a high density glucan surface. (a) Composite SPR sensorgram and (b) quantification of the change in response. Pale blue. When Glc-1-P (10 mM) was injected over the surface there was a bulk refractive index change, which returns to baseline when the injection ends. Blue. When AtPHS2 (5 μg ml−1) was injected alone there is around 600 RU increase in signal, which did not decrease much after the injection. Purple. When AtPHS2 (5 μg ml−1) and Glc-1-P (10 mM) were co-injected a large (5000 RU) response was seen. Yellow. AG (1 mg ml−1) caused no change when injected over the surface. Red. PPA (3 mg ml−1) gave a large decrease (3500 RU) in signal when injected over the surface. | ||
On co-injection of AtPHS2 with Glc-1-P on the high sugar density surface, a much larger response was seen (ca. 5000 RU; Fig. 3 – purple) than in the corresponding low sugar density surface experiment (ca. 1200 RU; Fig. 2 – purple), showing that synthesis was more extensive on the high sugar density surface. Amyloglucosidase, which cannot degrade insoluble starch granules, was not able to degrade the newly formed surface (Fig. 3 – yellow) but porcine pancreatic α-amylase (PPA), which can attack starch granules, hydrolysed around 2000 RU worth of the extended surface during injection and then a further 1500 RU after the injection had ended (Fig. 3 – red). PPA binds tightly to glucans51 and this continuing slow decrease in RU may be due to the further degradation during the slow dissociation. Overall, these experiments suggest that AtPHS2 action on a high sugar density surface gives rise to a structured polysaccharide material that displays some of the enzyme-responsive character of a starch-like material, which merits further assessment.
X-ray crystal structures of AtPHS2
In order to better understand the initial interaction of AtPHS2 with glucan surfaces, we undertook a structural analysis of the protein (Fig. 4). The structure of the mammalian AtPHS2 orthologue, glycogen phosphorylase (GP), has long been known, and the conformational changes associated with its allosteric and covalent control have been studied.53,54 The crystallisation of potato phosphorylase has previously been reported,55 although no structure has been deposited in the PDB. In the current study the crystal structure of native AtPHS2 was solved to 1.7 Å, revealing two copies of the protein subunit per asymmetric unit that together comprise the biologically relevant dimer. As expected, there is a pyridoxal phosphate covalently bound, about 20 Å from the surface, by formation of a Schiff base with Lys687. The phosphate group acts as a catalytic acid/base in the reaction and also has a role in coordinating the glucose-1-phosphate/phosphate in the active site.56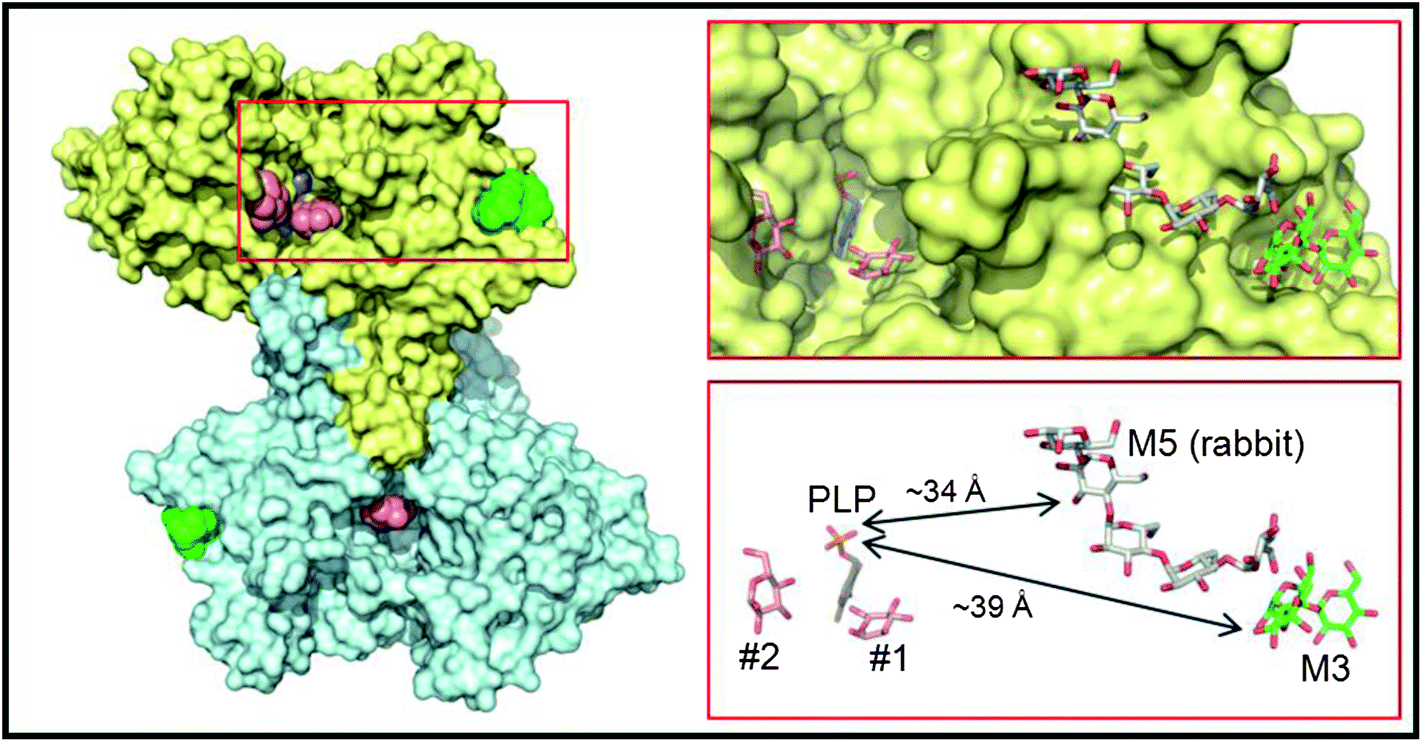 | ||
| Fig. 4 Crystal structure of PHS2. The left hand panel shows a molecular surface representation of the maltotriose-bound homodimer structure determined at 1.90 Å resolution, with one subunit in yellow and the other in cyan. Shown as van der Waals spheres are the deeply buried PLP cofactors (slate blue) and the surface maltotriose (M3; green) ligands. The salmon spheres indicate the single sugar residue binding sites identified in the active site channels of the AtPHS2 acarbose complex structure (two in the yellow subunit and one in the cyan subunit). For clarity the surface bound acarbose molecules are not shown as they are closely superposable on the maltotriose ligands (see Fig. S6†). The region bounded by the red rectangle is shown in more detail in the right hand panels where the ligands are depicted in stick representation and retain the same colour scheme for their carbon atoms. Also shown for reference with grey carbon atoms is the relative position of the surface maltopentaose (M5) site in rabbit GP (PDB code 1P29). The arrows indicate the shortest direct distances between PLP and the two surface sites. | ||
The native structure of AtPHS2 shows no binding site for the allosteric activator of the mammalian GP (AMP) and a unique conformation of the N-terminus of the protein, the site of activating covalent modification by phosphorylation. The active site of AtPHS2 is also more accessible than that of GP, with the 280s and 380s loops adopting a more open conformation and explaining the absence of allosteric regulation found in the mammalian enzyme.
Binding of carbohydrates to the surface of the catalytic subunit, as opposed to discreet carbohydrate binding modules, is an important characteristic of many enzymes,57 and can dramatically affect activity.58 Structures of AtPHS2 were obtained in complex with maltotriose and acarbose bound at the surface of the enzyme (Fig. 4). This is the first time that this surface site has been noted, although it is adjacent to, and almost contiguous with, the maltopentaose site that has previously been resolved in the rabbit muscle GP (PDB entry 1P29) (Fig. 4). However, the latter GP site borders the mouth of the active site channel, whilst the plant carbohydrate is bound 14 Å further away from the active site. Inspection of residue conservation between the two proteins and across the two surface binding sites, suggests that GP could potentially bind oligosaccharides at the AtPHS2 site, but not vice versa. Indeed, juxtaposition of the reducing end of the maltotriose in the AtPHS2 complex, and the non-reducing end of the maltopentaose in the GP structure, suggests that the latter could theoretically bind much longer oligosaccharides that span both sites. Despite repeated attempts to soak or to co-crystallise, no substrates could be seen in the active site of the enzyme, indicating the surface site binds glucans much more tightly and is likely responsible for the tight binding to glucan surface observed in SPR (vide supra). However, extra density could be resolved in the active site upon soaking the inhibitor acarbose into AtPHS2 crystals (Fig. S7†), which indicates a similar pattern of active site binding as previously reported for phosphorylase/inhibitor complexes.
Nanoparticles with a glucan surface
Whilst SPR employs a 2D surface, a more accurate mimic of the surface of a starch granule requires the third spatial dimension. The physical and chemical properties, uniform shape and size59 of gold nanoparticles make them excellent materials for the study of biological interactions.60 Uses include the rapid detection of DNA61 and the analysis of carbohydrate binding proteins62,63 based upon the interactions between surface plasmons, and hence colour, upon aggregation. When amylose is synthesised on glycogen particles using amylosucrase, diffuse material is initially observed in TEM images. After an extended period of time much smaller, denser particles are observed, corresponding to more organised and tightly packed glucan chains.64 In the present study, glycogen was extended upon incubation in the presence of AtPHS2 and Glc-1-P such that it stains strongly with iodine (Fig. S8†), presumably indicating the presence of ordered and stacked amylose helices, and was visible as much larger particles when imaged using TEM (100 nm diameter, as opposed to 20 nm).Using the same SAM chemistry as for the high sugar density SPR surface, FG was immobilised on citrate-stabilised gold nanoparticles with an average diameter of 14.7 ± 0.1 nm,59 to form FG-NPs. When imaged by TEM (Fig. 5) after negative staining with 2% uranyl acetate (UA), a narrow unstained zone was visible around the FG-NPs (0.9 ± 0.05 nm), indicating the thickness of the SAM and FG layer on the particles (Fig. S9†). When imaged immediately after a 1 hour incubation with AtPHS2 and 10 mM Glc-1-P, a much larger unstained zone (4.0 ± 0.13 nm) could be seen around the particles, which stained positively for carbohydrate using the sugar-specific periodic acid-thiocarbohydrazide-silver (PATAg) method.65 After 24 hours of aging the particles at 4 °C, this clear zone had collapsed to a much smaller size (2.3 ± 0.09 nm), suggesting that some reordering and packing of the newly synthesised glucan chains had occurred.
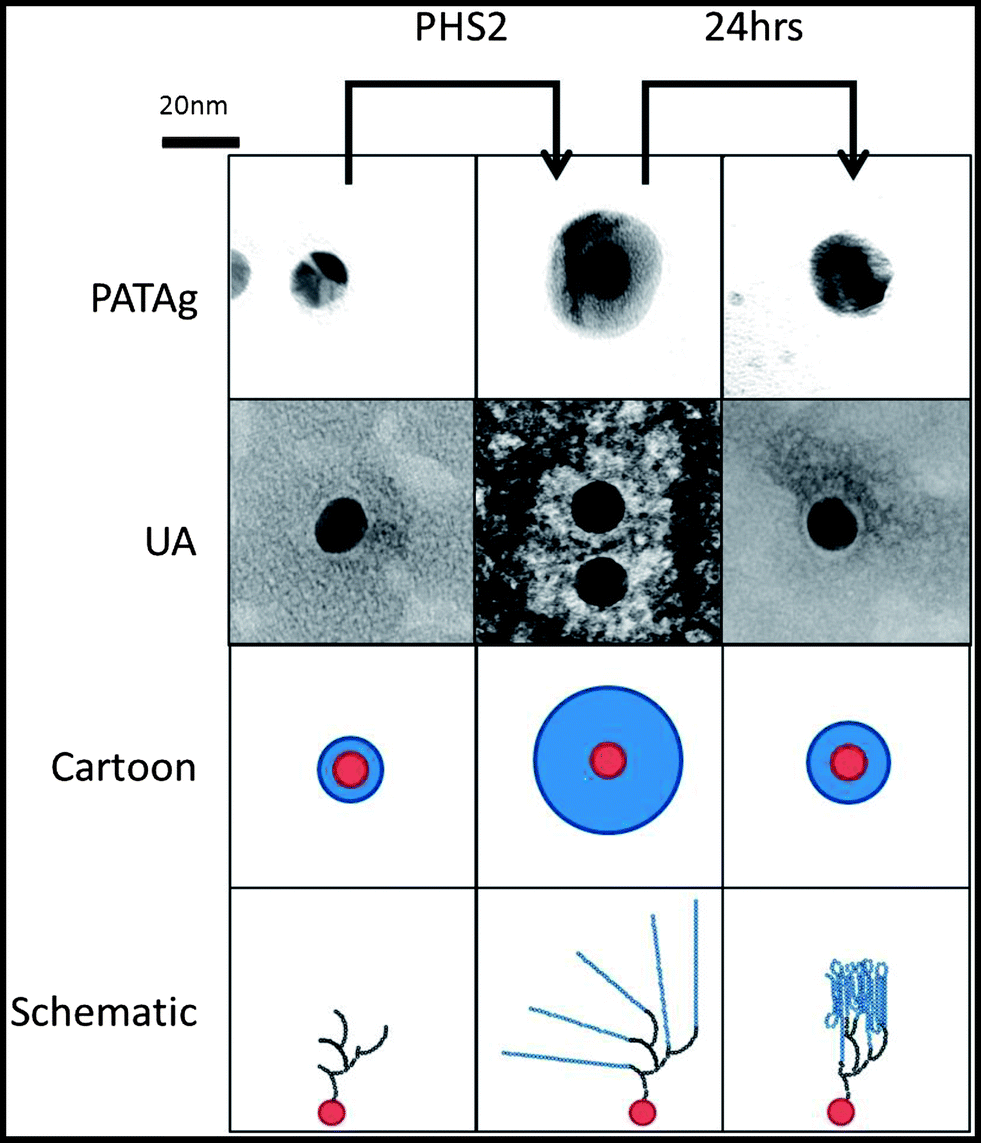 | ||
| Fig. 5 Representative images of the glucan layer on the surface of the gold nanoparticles. Gold nanoparticles with FG immobilised on the surface were imaged with TEM 20 min and 24 hours after AtPHS2 mediated extension, either stained with PATAg or 2% uranyl acetate. Both staining methods show, as illustrated in the cartoon, the carbohydrate (blue) on the nanoparticles (red), increases after treatment with AtPHS2 (10 μg ml−1) and Glc-1-P (10 mM), and rearranging to form a thinner layer after 24 hours. The schematic shows that FG (black) is extended by AtPHS2 (blue) and then rearranges by forming intra and inter-chain interactions to produce a more condensed overall structure. | ||
The characteristic optical properties of 15 nm gold nanoparticles, which result in a change from red to purple upon aggregation, has allowed their use in biomolecular detection.66 The AtPHS2-mediated extension of FG-nanoparticles had no effect on the red colour of the gold nanoparticles themselves (Fig. 6b & d), indicating that the synthesised material was not affecting the particles per se. In contrast to the starting FG-nanoparticles (Fig. 6c), which did not stain with iodine, the corresponding AtPHS2-extended FG-nanoparticles stained strongly (Fig. 6e),67 demonstrating that the material synthesised on the particles had arranged to form ordered/stacked amylose helices, rather than a random unstructured arrangement of chains.
 | ||
| Fig. 6 Images of iodine staining of glucan extension on gold nanoparticles. (a) Five μl of ethanol saturated with iodine diluted into 1 ml water has a pale yellow colour. (b) Gold nanoparticles with FG. (c) Iodine only weakly stains the FG nanoparticles. (d) The extension of the immobilised carbohydrate on the surface of the nanoparticles by AtPHS2 does not change the colour of the gold nanoparticles. (e) Iodine stains nanoparticles black after treatment with PHS2. | ||
Having aged AtPHS2-extended FG-nanoparticles for 24 hours, treatment with amyloglucosidase, caused no significant change in the unstained zone in TEM images, whilst treatment with PPA caused a modest reduction (0.7 ± 0.09 nm) in the extended surface (Fig. S9†). Extended incubation with AtPHS2 caused formation of a precipitate of nanoparticles in an entangled micron scale amylose matrix, without causing a change in the colour of the gold particles, indicating that there was no nanoparticle aggregation as such (Fig. S10†).
To summarise, PHS2 is able to synthesise polymeric α-1,4-glucans from a fractionated glucan primer immobilised on gold nanoparticles. The resultant glycopolymer stains with iodine in a manner similar to amylose and has the same resistance to amyloglucosidase, and susceptibility to α-amylase, as native starch granules.
Sequential enzymatic digestion of on-chip synthesised glucan
Building on the projections of glucan chain arrangement and 3D architecture arising from the above glyconanoparticle experiments, we revisited the SPR chip-synthesised amylo-polysaccharides with a view to assessing in more detail the nature of the material synthesised and its suitability for the analysis of starch-active enzymes. The degradation of native starch granules in vivo involves the concerted attack of amylases and debranching enzymes,52 with the action of the former being a pre-requisite for the action of the latter. Our aim was therefore to determine whether the sequential action of different starch-active enzymes could be used to assess the structure and linkage of the pseudo-starch surfaces generated by AtPHS2 (Fig. 7).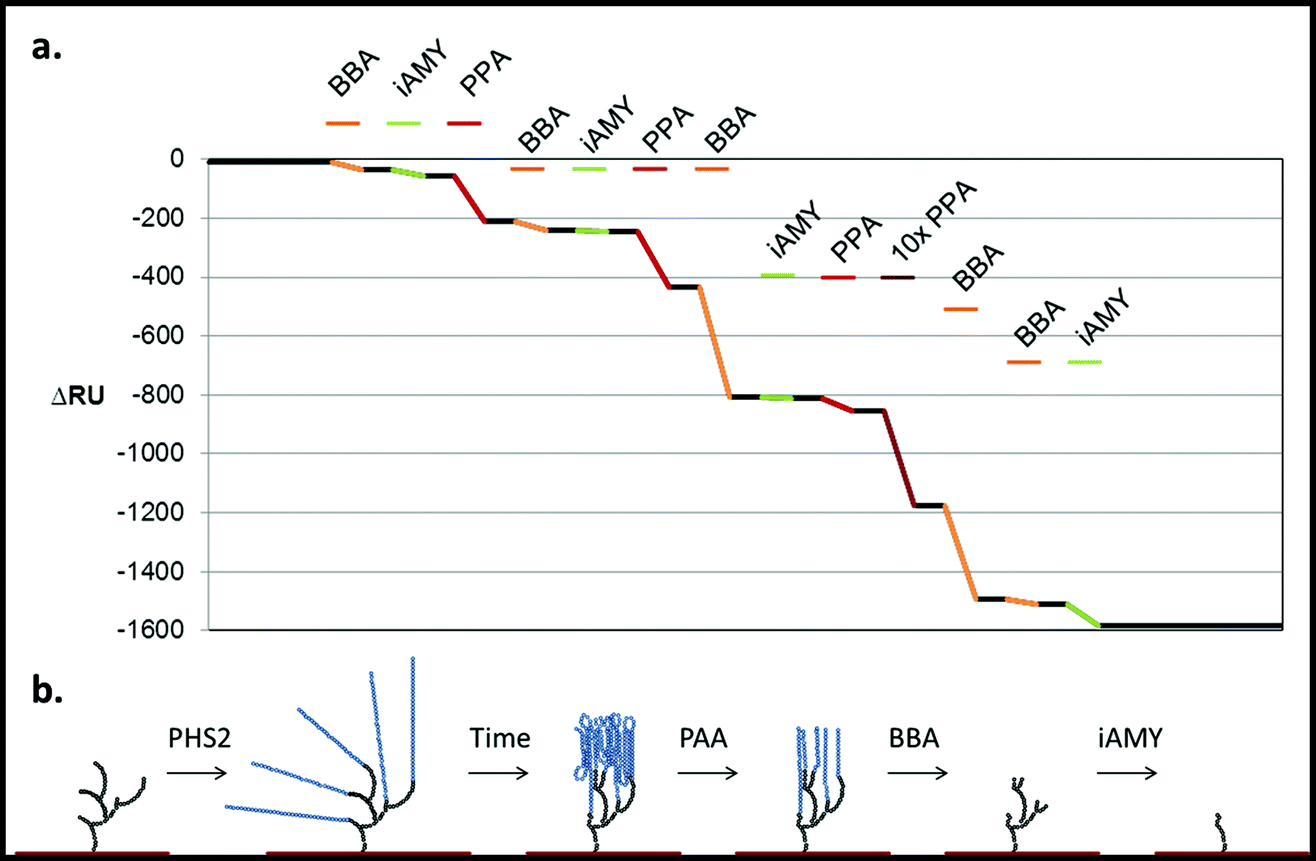 | ||
| Fig. 7 Degradation of the PHS2-synthesised glucan, as analysed by SPR. PHS2 (5 μg ml−1, Glc-1-P 10 mM) was used to extended the high sugar density surface by around 4000 RU. (a) Average profile of the injection (100 μl) of BBA (1 U ml−1, orange), iAMY (1 U ml−1, green) and PPA (1 mU ml−1, red) over this surface. Quantification in Fig. S11.† (b) Schematic of the effect of the enzymes on the glucans on the surface. Initially only PPA had any effect on the surface, but once around 500 RU had been removed BBA was able to remove around 500 RU during the injection. Once most of the material had been degraded from the surface BBA was unable to remove any further material, but iAMY was able to remove some of the material from the surface. | ||
Initially AtPHS2 was used to extend a high density FG surface to around 4000 RU-worth of material. The surface was probed sequentially with BBA (orange), isoamylase (iAMY, green) and PPA (red) (Fig. 7). Until PPA had been used to disrupt the surface, neither BBA nor iAMY had a significant effect on the surface, as judged by SPR responses (baseline change). Once the surface had been partially degraded by PPA, BBA was able to significantly degrade the surface to form a limit dextrin, which it could no longer affect. iAMY was able to degrade this material, removing the branch points of the limit dextrin.
By tightly packing the carbohydrate surface, the AtPHS2-extended material takes on sensitivity and resistance to enzymatic digestion that is reminiscent of native starch granules. These observations are in keeping with the schematics of surface glycan architecture presented in Fig. 5 and 7, where the bulk of the material generated is highly organised and likely semi-crystalline and where the 1,6-linked branch points are buried towards the base of the polysaccharide structure, close to the gold sensor/nanoparticle surfaces.
Conclusions and outlook
The surface enzymology of the starch granule led us to explore new ways to investigate enzymatic reactions at solid–liquid interfaces. Due to its catalytic efficiency, we have explored the plant phosphorylase AtPHS2 for the rapid synthesis of linear amylose chains. In solution assays, linear amylose-like structures of up to 100 sugar units could be assembled. Similar syntheses could also be achieved on gold sensor chip surfaces: on its own, the phosphorylase enzyme bound tightly to the glucan coated sensor surface, presumably using the surface binding site elucidated in the crystal structure of AtPHS2. In contrast, co-injection of AtPHS2 and the donor substrate, glucose-1-phosphate, gave rise to glucan synthesis, which could be monitored conveniently in real time by SPR. The resulting extended surfaces were responsive to starch-active enzymes in a manner reminiscent of native starch granules, suggesting the potential for this approach to generate physiologically relevant starch-like surfaces.Continuing with gold surfaces but moving to 15 nm diameter gold nanoparticles, we were able to demonstrate AtPHS2-catalysed glucan polymerisation, as judged by TEM. Significantly, it became apparent that these nanoparticle-based glucan surfaces were subject to aging, with the glucan layer appearing to reorganise and assemble into a much more ordered, tightly packed structure over time. We interpret this observation as glucan chains wrapping back on themselves to produce inter- or intra-chain hydrogen bonds, as proposed for crystalline material generated by the enzymatic debranching of starch68 or the amylosucrase-mediated extension of glycogen nanoparticles.64 These data were recapitulated on SPR sensor chips, where high density glucan surfaces could be demonstrated to posses the susceptibility to essential enzymatic digestion that one would expect of a starch granule. With high enzyme concentrations and at extended time periods, glucan polymer hundreds of nm in length could be accessed by AtPHS2-mediated extension of glyconanoparticles. While some way short of the 0.5–100 μm length scale of the typical starch granules,69 these data show potential for the elaboration of micron scale carbohydrate architectures by AtPHS2-meditated synthesis.
To conclude, the 2D and 3D surfaces developed in this study represent novel tools for the study of surface enzymology, which will be explored further to investigate enzymes associated with starch granule metabolism and the industrial processing of this bulk commodity.
Experimental
Fractionated glucan (FG)
Glucidex IT12 (Roquette) with 3% branching (as judged by 1H NMR spectroscopy) and with an average molecular weight of 19 kDa (DP ∼ 110) is known to have low DP contaminants including 1% Glc, 3% maltose and 5% maltotriose.70 Fractionated Glucan (FG) was therefore prepared from Glucidex IT12 by spin filtration with a 30 kDa MWCO spin filter (Amicon), washing three times with 3 ml MilliQ H2O (MQ, Millipore), with 0.5 ml retained each time, and lyophilising the material retained by the filter.Preparation of ADH-tethered fractionated glucan (ADH–FG)
FG (10 mg) was reacted with adipic acid dihydrazide (ADH, 17.4 mg, 0.1 mmol) in sodium acetate (500 μl, 1 M, pH 4.5), at 37 °C for 4 hours. The product was purified by gel filtration chromatography (on a TSK HW40S – 90 × 0.75 cm column using 20 mM ammonium bicarbonate, pH 7.0, 0.5 ml min−1) and characterised by 1H NMR spectroscopy at 25 °C (400 MHz, Bruker). The characteristic reducing terminal anomeric signals [5.14 ppm (d, J = 3.7 Hz, H1α) and 4.57 ppm (d, J = 8.0 Hz, H1β)] disappeared completely and a new signal appeared, corresponding to the glycoside anomeric proton in the FG–ADH adduct [4.03 ppm (d, J = 9.3 Hz, H1β)].Preparation of SPR sensor chips
The sensor surface was prepared by cleaning a commercial gold chip (Sensor Chip Au, BIAcore BR-1005-42) using 5 μl Piranha solution (3:1 sulphuric acid (conc.):30% H2O2) and washing exhaustively with MQ water to remove the Piranha solution, before drying with compressed air and immobilising a self-assembled monolayer (SAM) on the surface.48
Components for the preparation of self-assembled monolayers (SAMs)
SAMs were prepared using: SAM spacer {1-mercapto-11-undecyl tri(ethylene glycol) [HSC11-(EG)3OH]; (Assemblon)} and SAM linker {1-mercapto-11-undecyl carboxymethyl-hexa(ethylene glycol) [HSC11-(EG)6OCH2COOH]; (Prochimia)}.Generation of low sugar density gold sensor surfaces
SAM spacer (98 mM) and SAM linker (2 mM) in DMF (10 μl) were placed onto a gold sensor chip surface and left overnight at 4 °C to assemble into a homogenous SAM. EDC (1-ethyl-3-(3-dimethylaminopropyl) carbodiimide; 10 μl, 0.2 M) and N-hydroxysuccinimide (NHS) (10 μl, 0.05 M) in water were added to the surface and incubation was continued at room temperature for 30 min. FG–ADH adduct (10 μl, 10 mg ml−1) in water was added to the surface and incubation was continued for a further 30 min at room temperature. The excess liquid was removed with a pipette and the surface was washed with MQ water before inserting the surface into the sensor chip holder. This sensor chip assembly was inserted into the spectrometer and interrogated using SPR.Enzyme reactions on gold sensor surfaces
SPR experiments were performed using a BiacoreX instrument (Biacore) at 25 °C using a continuous flow rate of 20 μl min−1 MES (40 mM, pH 6.0, previously degassed under vacuum) and injections were performed after the baseline had stabilised. Injections were of 100 μl and washes with NaCl (20 μl, 100 mM) were made after each injection of protein. Experiments with the high sugar density surface showed the PHS2-catalysed surface extension was proportional to the amount of substrate added (Fig. S5†).Accession numbers
Coordinates and structure factors for the three AtPHS2 structures and complexes described herein have been deposited in the Protein Data Bank with accession numbers 4BQE, 4BQF and 4BQI.Acknowledgements
These studies were supported by the UK BBSRC Institute Strategic Programme Grant on Understanding and Exploiting Metabolism (MET) [BB/J004561/1], the John Innes Foundation and European Union Framework Programme 6 Marie Curie Early Stage Training grant [MEST-CT-2005-019727]. Structural studies were carried out with the support of the Diamond Light Source, and we acknowledge the assistance of the beam line scientists on I02 and I03. We thank Alison Smith for advice on starch biochemistry, Kim Findlay for assistance with TEM and Christian Ruzanski for providing Arabidopsis cDNA.References
- A. M. Smith, Plant J., 2008, 54, 546–558 CrossRef CAS PubMed.
- A. R. S. U.S. Department of Agriculture, in What We Eat in America, US Department of Agriculture, Agricultural Research Service, http://www.ars.usda.gov/ba/bhnrc/fsrg, 2012, vol. NHANES 2009–2010 Search PubMed.
- R. P. Ellis, M. P. Cochrane, M. F. B. Dale, C. M. Duffus, A. Lynn, I. M. Morrison, R. D. M. Prentice, J. S. Swanston and S. A. Tiller, J. Sci. Food Agric., 1998, 77, 289–311 CrossRef CAS.
- J. Diouf, Popul. Dev. Rev., 2009, 35, 837–839 CrossRef.
- S. Jobling, Curr. Opin. Plant Biol., 2004, 7, 210–218 CrossRef CAS PubMed.
- S. A. Jobling, R. J. Westcott, A. Tayal, R. Jeffcoat and G. P. Schwall, Nat. Biotechnol., 2002, 20, 295–299 CrossRef CAS PubMed.
- R. G. F. Visser, I. Somhorst, G. J. Kuipers, N. J. Ruys, W. J. Feenstra and E. Jacobsen, Mol. Gen. Genet., 1991, 225, 289–296 CrossRef CAS.
- D. Stanley, M. Rejzek, H. Naested, M. Smedley, S. Otero, B. Fahy, F. Thorpe, R. J. Nash, W. Harwood, B. Svensson, K. Denyer, R. A. Field and A. M. Smith, Plant Physiol., 2011, 155, 932–943 CrossRef CAS PubMed.
- M. Rejzek, C. E. Stevenson, A. M. Southard, D. Stanley, K. Denyer, A. M. Smith, M. J. Naldrett, D. M. Lawson and R. A. Field, Mol. BioSyst., 2011, 7, 718–730 RSC.
- A. Edwards, A. Borthakur, S. Bornemann, J. Venail, K. Denyer, D. Waite, D. Fulton, A. Smith and C. Martin, Eur. J. Biochem., 1999, 266, 724–736 CrossRef CAS.
- J. L. Putaux, G. Potocki-Veronese, M. Remaud-Simeon and A. Buleon, Biomacromolecules, 2006, 7, 1720–1728 CrossRef CAS PubMed.
- Z. Sun and C. A. Henson, Plant Physiol., 1990, 94, 320–327 CrossRef CAS PubMed.
- V. Planchot, P. Colonna, D. J. Gallant and B. Bouchet, J. Cereal Sci., 1995, 21, 163–171 CrossRef CAS.
- G. Tawil, F. Jamme, M. Refregiers, A. Vikso-Nielsen, P. Colonna and A. Buleon, Anal. Chem., 2011, 83, 989–993 CrossRef CAS PubMed.
- M. Hejazi, J. Fettke, O. Paris and M. Steup, Plant Physiol., 2009, 150, 962–976 CrossRef CAS PubMed.
- C. Edner, J. Li, T. Albrecht, S. Mahlow, M. Hejazi, H. Hussain, F. Kaplan, C. Guy, S. M. Smith, M. Steup and G. Ritte, Plant Physiol., 2007, 145, 17–28 CrossRef CAS PubMed.
- A. Murakawa, T. Mori and Y. Okahata, Chem. Lett., 2007, 36, 312–313 CrossRef CAS.
- H. Nishino, A. Murakawa, T. Mori and Y. Okahata, J. Am. Chem. Soc., 2004, 126, 14752–14757 CrossRef CAS PubMed.
- H. L. Pedersen, J. U. Fangel, B. McCleary, C. Ruzanski, M. G. Rydahl, M.-C. Ralet, V. Farkas, L. von Schantz, S. E. Marcos, M. C. F. Andersen, R. Field, M. Ohlin, J. P. Knox, M. H. Clausen and W. G. T. Willats, J. Biol. Chem., 2012, 287, 39429–39438 CrossRef CAS PubMed.
- C. J. Gray, M. J. Weissenborn, C. E. Eyers and S. L. Flitsch, Chem. Soc. Rev., 2013, 42, 6378–6405 RSC.
- T. Harada, A. Misaki, H. Akai, K. Yokobayashi and K. Sugimoto, Biochim. Biophys. Acta, Enzymol., 1972, 268, 497–505 CrossRef CAS.
- M. Yanase, T. Takaha and T. Kuriki, J. Appl. Glycosci., 2007, 54, 125–131 CrossRef CAS.
- G. Potocki-Veronese, J.-L. Putaux, D. Dupeyre, C. Albenne, M. Remaud-Siméon, P. Monsan and A. Buleon, Biomacromolecules, 2005, 6, 1000–1011 CrossRef CAS PubMed.
- F. Grimaud, C. Lancelon-Pin, A. Rolland-Sabaté, X. Roussel, S. Laguerre, A. Viksø-Nielsen, J.-L. Putaux, S. Guilois, A. Buléon, C. D'Hulst and G. Potocki-Véronèse, Biomacromolecules, 2013, 14, 438–447 CrossRef CAS PubMed.
- Y. Umegatani, H. Izawa, M. Nawaji, K. Yamamoto, A. Kubo, M. Yanase, T. Takaha and J.-i. Kadokawa, Carbohydr. Res., 2012, 350, 81–85 CrossRef CAS PubMed.
- H. W. Klein, D. Palm and E. J. M. Helmreich, Biochemistry, 1982, 21, 6675–6684 CrossRef CAS.
- S. G. Withers, Carbohydr. Res., 1990, 197, 61–73 CrossRef CAS.
- T. Fukui, S. Shimomura and K. Nakano, Mol. Cell. Biochem., 1982, 42, 129–144 CrossRef CAS.
- Y. Kaneko, S. Matsuda and J. Kadokawa, Biomacromolecules, 2007, 8, 3959–3964 CrossRef CAS PubMed.
- K. Loos and A. H. E. Müller, Biomacromolecules, 2002, 3, 368–373 CrossRef CAS PubMed.
- G. G. Gelders, H. Goesaert and J. A. Delcour, Biomacromolecules, 2005, 6, 2622–2629 CrossRef CAS PubMed.
- C. Cle, A. P. Gunning, K. Syson, L. Bowater, R. A. Field and S. Bornemann, J. Am. Chem. Soc., 2008, 130, 15234–15235 CrossRef CAS PubMed.
- C. Cle, C. Martin, R. A. Field, P. Kuzmic and S. Bornemann, Biocatal. Biotransform., 2010, 28, 64–71 CrossRef CAS.
- C. L. Schofield, R. A. Field and D. A. Russell, Anal. Chem., 2007, 79, 1356–1361 CrossRef CAS PubMed.
- C. L. Schofield, B. Mukhopadhyay, S. M. Hardy, M. B. McDonnell, R. A. Field and D. A. Russell, Analyst, 2008, 133, 626–634 RSC.
- Y. Lu, J. M. Steichen, J. Yao and T. D. Sharkey, Plant Physiol., 2006, 142, 878–889 CrossRef CAS PubMed.
- M. R. M. De Groeve, G. H. Tran, A. Van Hoorebeke, J. Stout, T. Desmet, S. N. Savvides and W. Soetaert, Anal. Biochem., 2010, 401, 162–167 CrossRef CAS PubMed.
- G. Vereb, A. Fodor and G. Bot, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1987, 915, 19–27 CrossRef CAS.
- J. L. Ozbun, J. S. Hawker and J. Preiss, Biochem. J., 1972, 126, 953–963 CAS.
- C. Pollock and J. Preiss, Arch. Biochem. Biophys., 1980, 204, 578–588 CrossRef CAS.
- T. Suganuma, J. I. Kitazono, K. Yoshinaga, S. Fujimoto and T. Nagahama, Carbohydr. Res., 1991, 217, 213–220 CrossRef CAS.
- F. Goubet, P. Jackson, M. J. Deery and P. Dupree, Anal. Biochem., 2002, 300, 53–68 CrossRef CAS PubMed.
- W. Laroy, R. Contreras and N. Callewaert, Nat. Protoc., 2006, 1, 397–405 CrossRef CAS PubMed.
- E. Prifti, S. Goetz, S. A. Nepogodiev and R. A. Field, Carbohydr. Res., 2011, 346, 1617–1621 CrossRef CAS PubMed.
- M. K. Morell, M. S. Samuel and M. G. O'Shea, Electrophoresis, 1998, 19, 2603–2611 CrossRef CAS PubMed.
- J. van der Vlist and K. Loos, Macromol. Symp., 2007, 254, 54–61 CAS.
- M. Fais, R. Karamanska, S. Allman, S. A. Fairhurst, P. Innocenti, A. J. Fairbanks, T. J. Donohoe, B. G. Davis, D. A. Russell and R. A. Field, Chem. Sci., 2011, 2, 1952–1959 RSC.
- Z. L. Zhi, N. Laurent, A. K. Powell, R. Karamanska, M. Fais, J. Voglmeir, A. Wright, J. M. Blackburn, P. R. Crocker, D. A. Russell, S. Flitsch, R. A. Field and J. E. Turnbull, ChemBioChem, 2008, 9, 1568–1575 CrossRef CAS PubMed.
- M. Buckle, R. M. Williams, M. Negroni and H. Buc, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 889–894 CrossRef CAS.
- J. Lahiri, L. Isaacs, J. Tien and G. M. Whitesides, Anal. Chem., 1999, 71, 777–790 CrossRef CAS.
- F. J. Warren, P. G. Royall, S. Gaisford, P. J. Butterworth and P. R. Ellis, Carbohydr. Polym., 2011, 86, 1038–1047 CrossRef CAS PubMed.
- A. M. Smith, S. C. Zeeman and S. M. Smith, Annu. Rev. Plant Biol., 2005, 56, 73–98 CrossRef CAS PubMed.
- D. Barford, S. H. Hu and L. N. Johnson, J. Mol. Biol., 1991, 218, 233–260 CrossRef CAS.
- J. L. Buchbinder, V. L. Rath and R. J. Fletterick, Annu. Rev. Biophys. Biomol. Struct., 2001, 30, 191–209 CrossRef CAS PubMed.
- A. Kamogawa, T. Fukui and Z. Nikuni, J. Biochem., 1968, 63, 361–369 CAS.
- D. Palm, H. W. Klein, R. Schinzel, M. Buehner and E. J. M. Helmreich, Biochemistry, 1990, 29, 1099–1107 CrossRef CAS.
- S. Cuyvers, E. Dornez, J. A. Delcour and C. M. Courtin, Crit. Rev. Biotechnol., 2012, 32, 93–107 CrossRef CAS PubMed.
- C. Ragunath, S. G. A. Manuel, V. Venkataraman, H. B. R. Sait, C. Kasinathan and N. Ramasubbu, J. Mol. Biol., 2008, 384, 1232–1248 CrossRef CAS PubMed.
- B. V. Enustun and J. Turkevich, J. Am. Chem. Soc., 1963, 85, 3317–3328 CrossRef CAS.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- J. M. Nam, S. I. Stoeva and C. A. Mirkin, J. Am. Chem. Soc., 2004, 126, 5932–5933 CrossRef CAS PubMed.
- H. Otsuka, Y. Akiyama, Y. Nagasaki and K. Kataoka, J. Am. Chem. Soc., 2001, 123, 8226–8230 CrossRef CAS PubMed.
- D. C. Hone, A. H. Haines and D. A. Russell, Langmuir, 2003, 19, 7141–7144 CrossRef CAS.
- A. Buleon, G. Veronese and J. L. Putaux, Aust. J. Chem., 2007, 60, 706–718 CrossRef CAS.
- J. P. Thiery, J. Microsc., 1967, 6, 987–1018 CAS.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078–1081 CrossRef CAS.
- F. L. Bates, D. French and R. E. Rundle, J. Am. Chem. Soc., 1943, 65, 142–148 CrossRef CAS.
- A. Pohu, V. Planchot, J. L. Putaux, P. Colonna and A. Buléon, Biomacromolecules, 2004, 5, 1792–1798 CrossRef CAS PubMed.
- J.-L. Jane, T. Kasemsuwan, S. Leas, H. Zobel and J. F. Robyt, Starch/Staerke, 1994, 46, 121–129 CrossRef CAS.
- D. Kilburn, J. Claude, T. Schweizer, A. Alam and J. Ubbink, Biomacromolecules, 2005, 6, 864–879 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details; cloning, expression and purification of AtPHS2; enzyme activity assays and CE protocols; preparation of SAMs; details of surface plasmon resonance experiments; crystallisation and structure determination of AtPHS2; preparation of glycan-modified gold nanoparticles and the enzymatic modification thereof. See DOI: 10.1039/c3sc51829a |
| This journal is © The Royal Society of Chemistry 2014 |