New evidence for 5f covalency in actinocenes determined from carbon K-edge XAS and electronic structure theory†
Stefan G.
Minasian
ab,
Jason M.
Keith‡
b,
Enrique R.
Batista
*b,
Kevin S.
Boland
b,
David L.
Clark
b,
Stosh A.
Kozimor
*b,
Richard L.
Martin
*b,
David K.
Shuh
*a and
Tolek
Tyliszczak
a
aLawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. E-mail: dkshuh@lbl.gov
bLos Alamos National Laboratory, Los Alamos, NM 87545 LA-UR-13-25346, USA. E-mail: erb@lanl.gov; stosh@lanl.gov; rlmartin@lanl.gov
First published on 5th November 2013
Abstract
Evidence for metal–carbon orbital mixing in thorocene and uranocene was determined from DFT calculations and carbon K-edge X-ray absorption spectra (XAS) collected with a scanning transmission X-ray microscope (STXM). Both the experimental and computational results showed that the 5f orbitals engaged in significant δ-type mixing with the C8H82− ligands, which increased as the 5f orbitals dropped in energy on moving from Th4+ to U4+. The first experimental evidence for extensive ϕ-orbital interactions has been provided by the C K-edge XAS analysis of thorocene; however, ϕ-type covalency in uranocene was negligible. The results highlighted two contrasting trends in orbital mixing from one pair of highly symmetric molecules, and showed that covalency does not increase uniformly for different molecular orbital interactions with later actinides.
Introduction
Actinide sandwich complexes – “actinocenes”, (C8H8)2An (An = Th, Pa, U, Np, Pu) – have played a central role in the development of electronic structure models in organoactinide chemistry.1–4 Prior experimental and theoretical studies of (C8H8)2An have provided considerable evidence of covalent mixing between the ligands and metal-based 5f and 6d orbitals.5–18 Much of our current understanding of actinocene bonding and orbital mixing is based on earlier gas phase photoelectron spectroscopy (PES) studies, which probed the occupied molecular orbitals.9,19–21 Covalency in the empty or partially occupied 5f and 6d orbitals has not been studied extensively, despite the foundational role of actinocenes in the development of ligand field theory for organoactinide complexes.Recent advances in soft X-ray synchrotron radiation have shown that studies of empty or partially occupied 5f and 6d orbitals are within reach using ligand K-edge X-ray absorption spectroscopy (XAS). The technique measures ligand centered 1s → np (n = principle quantum number) transitions, whose intensities are proportional to coefficients of orbital mixing between metals and ligands.22 Applications of ligand K-edge XAS to quantify orbital mixing in M–Cl and M–S bonds is well-established for transition metals22 and actinides,23,24 and recent developments in C and O K-edge XAS has enabled quantitative examinations of a variety of metal interactions with light atoms in molecular systems.25–29
In this study, C K-edge XAS and time-dependent density functional theory (TDDFT) was used to evaluate the electronic structures of thorocene, (C8H8)2Th, and uranocene, (C8H8)2U. These molecules were ideal for evaluating the relative role of 5f versus 6d covalency in actinide bonding because of their variable electron count and because hybridization of the 5f and 6d orbitals is symmetry-forbidden in the D8h point group. The results were consistent with earlier synthetic,8,30,31 spectroscopic, and theoretical analyses of actinocenes, and when taken together provide a comprehensive bonding picture for both the occupied and unoccupied actinide orbitals.5,8,9,12–17 Experimental evidence for an unusual ϕ-bonding interaction in (C8H8)2Th is also discussed.
Results and discussion
Ground state electronic structure and molecular orbital description
Before discussing the C K-edge XAS spectra in detail it is instructive to provide a framework for evaluating the molecular orbital interactions in each complex. Because the electronic structures for actinocenes are well established, this discussion will focus on the metal-based unoccupied orbitals that are relevant to the C K-edge XAS experiment. For the purposes of this study, we define covalency in bonding as mixing between two orbitals, in accordance with recently published studies.17 The currently accepted actinocene bonding model5,8,9,12–18 is consistent with results of our DFT calculations on (C8H8)2Th and (C8H8)2U as shown in Fig. 1 and 2 and Table 1. In general, the DFT calculations showed that bonding in the (C8H8)2An compounds involved orbital mixing between the sixteen (C8H8)24− orbitals that are C–C π-bonding (perpendicular to the individual ring planes) and the metal 5f and 6d orbitals. The actinide 6d valence orbitals interacted to form molecular antibonding orbitals of 2a1g (6d–σ), 2e1g (6d–π), and 2e2g (6d–δ) symmetries, while the actinide 5f valence orbitals were split into the 2a2u (5f–σ), 2e1u (5f–π), 2e2u (5f–δ), and 1e3u + 2e3u (5f–ϕ) molecular orbitals. In accordance with previous theoretical studies,12,33,34 our DFT calculations confirmed a dominant role in bonding for the gerade and ungerade e2 (δ) orbitals and showed that the a (σ) and e1 (π) orbitals were engaged in significantly less mixing. It has been proposed that the origin of this increased mixing is related to the directionality of the lobes of the 2e2u (5f–δ) orbitals, which pointed more towards the carbon atoms of the C8H82− rings.5 The DFT calculations also revealed an unusual case of ϕ-bonding in the bonding 1e3u and antibonding 2e3u (5f–ϕ) orbitals, which has been discussed in previous theoretical reports.35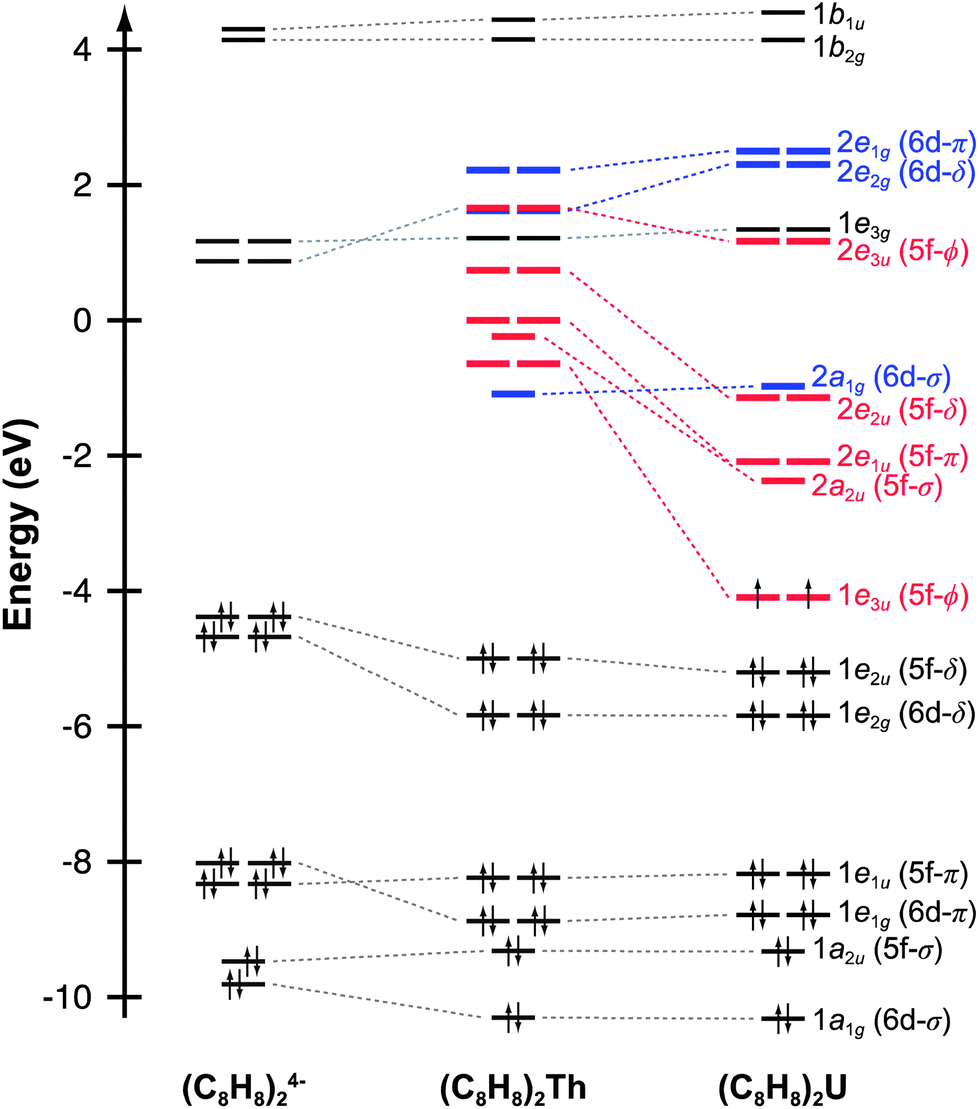 | ||
| Fig. 1 Quantitative molecular orbital diagram showing density functional theory (DFT) calculated energies for an idealized (C8H8)24− fragment in D4h symmetry (left) in comparison to those for (C8H8)2Th (middle) and (C8H8)2U (right). For (C8H8)2U, alpha spin–orbital energies are used. | ||
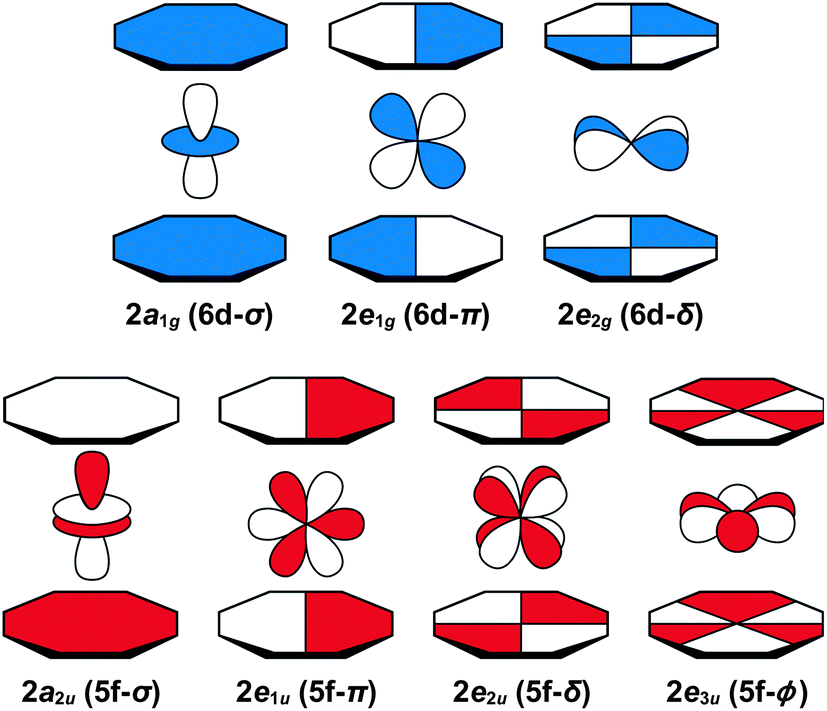 | ||
| Fig. 2 Representations of the 5f- and 6d-antibonding interactions in actinocenes. For a complete table of all the virtual Kohn–Sham orbitals for (C8H8)2Th and (C8H8)2U, refer to the ESI.† | ||
| Orbital | Energy (eV) | MO (DFT, %) | |||||
|---|---|---|---|---|---|---|---|
| C 2p | C 2s | M 6d | M 5f | M 7s | M 7p | ||
| a Alpha spin–orbital energies are reported for (C8H8)2U. b The use of a non-orthogonal basis set can cause Mulliken analysis to have nonphysical results such as compositions >100% or <0.92 | |||||||
| (C8H8)2Th | |||||||
| 1b1u (An-C nb) | 4.40 | 0.98 | 0 | 0 | 0 | 0 | 0 |
| 1b2g (An-C nb) | 4.10 | 0.97 | 0 | 0 | 0 | 0 | 0 |
| 2e1g (6d–π) | 2.18 | 0.10 | −0.18 | 0.71 | 0 | 0 | 0 |
| 2e3u (5f–ϕ) | 1.62 | 0.49 | 0 | 0 | 0.47 | 0 | 0 |
| 2e2g (6d–δ) | 1.57 | 0.23 | −0.02 | 0.75 | 0 | 0 | 0 |
| 1e3g (An-C nb) | 1.17 | 0.96 | −0.01 | 0 | 0 | 0 | 0 |
| 2e2u (5f–δ) | 0.69 | 0.13 | 0 | 0 | 0.86 | 0 | 0 |
| 2e1u (5f–π) | −0.05 | 0.01 | 0.01 | 0 | 0.91 | 0 | 0.05 |
| 2a2u (5f–σ) | −0.29 | 0.03 | 0 | 0 | 0.88 | 0 | 0 |
| 1e3u (5f–ϕ) | −0.69 | 0.33 | 0 | 0 | 0.65 | 0 | 0 |
| 2a1g (6d–σ) | −1.13 | 0.03 | 0.02 | 0.73 | 0 | 0.14 | 0 |
| (C8H8)2U | |||||||
| 1b1u (An-C nb) | 4.54 | 0.98 | 0 | 0 | 0 | 0 | 0 |
| 1b2g (An-C nb) | 4.14 | 0.94 | 0 | 0 | 0 | 0 | 0 |
| 2e1g (6d–π) | 2.50 | 0.13 | −0.13 | 0.44 | 0 | 0 | 0 |
| 2e2g (6d–δ) | 2.30 | 0.22 | −0.02 | 0.74 | 0 | 0 | 0 |
| 1e3g (An-C nb) | 1.35 | 0.96 | −0.01 | 0 | 0 | 0 | 0 |
| 2e3u (5f–ϕ) | 1.17 | 0.89 | 0 | 0 | 0.07 | 0 | 0 |
| 2a1g (6d–σ) | −0.97 | 0.02 | 0 | 0.77 | 0 | 0.03 | 0 |
| 2e2u (5f–δ) | −1.14 | 0.28 | 0 | 0 | 0.71 | 0 | 0 |
| 2e1u (5f–π) | −2.09 | 0.02 | 0 | 0 | 0.97 | 0 | 0 |
| 2a2u (5f–σ) | −2.37 | 0.03 | 0 | 0 | 0.96 | 0 | 0 |
| 1e3u (5f–ϕ) | −4.10 | 0.06 | 0 | 0 | 0.94 | 0 | 0 |
C K-edge and N5,4-edge measurements
Obstacles associated with measuring the C K-edge XAS spectra of nonconducting molecular compounds were recently addressed in the study of M–C bonding for Group 4 bent metallocene dichlorides, (C5H5)2MCl2 (M = Ti, Zr, Hf).29 Using that methodology, small droplets of (C8H8)2Th and (C8H8)2U were allowed to evaporate on Si3N4 windows, forming thin crystallites that enabled transmission XAS measurements that were within the linear regime of the Beer–Lambert law. A scanning transmission X-ray microscope (STXM) was utilized to record images, elemental maps, C K-edge XAS, and actinide N5/4-edge spectra.36 Normal contrast images and elemental maps of representative crystals of (C8H8)2Th and (C8H8)2U used in the XAS experiments have been provided in Fig. 3. | ||
| Fig. 3 Three images each of the crystals of (C8H8)2Th (top row) and (C8H8)2U (bottom row) from which C K-edge and N5/4-edge XAS data were obtained: normal contrast images obtained with a photon energy of 300 eV (left column); elemental distribution maps obtained by subtraction using photon energies of 286–276 eV (middle top), 292–282 eV (middle bottom), 675–660 eV (right top) and 738–718 eV (right bottom) with regions containing carbon, thorium or uranium shown as white using a standard grayscale. | ||
Spectra at the N5,4-edges were collected because they probed valence 5f orbitals directly with electronic excitations from core 4d orbitals,32,37–39 and because previous studies have shown that the branching ratio of N5 and N4 peaks can be sensitive to changes in valence electronic structure.40–43 The Th and U N5/4-edge XAS spectra and elemental maps also provided confidence that pure (C8H8)2An particles had been selected for subsequent C K-edge XAS analyses. Background-subtracted actinide N5,4-edge XAS spectra for (C8H8)2Th and (C8H8)2U have been included in Fig. 4. For each (C8H8)2An complex, the major contribution to the N5,4-edge absorption intensities likely arose from dipole allowed 4d105fn → 4d95fn+1 transitions (Th, n = 0; U, n = 2). These transitions were split by 36.7 (Th) and 41.3 eV (U) into two primary actinide N5 (4d5/2) and N4 (4d3/2) edges due to spin–orbit coupling of the core–hole. The actinide N5 and N4-edge transitions are found at 675.3 and 712.0 eV for (C8H8)2Th and 737.3 and 778.6 eV for (C8H8)2U. On moving from (C8H8)2Th to (C8H8)2U the N5 peak widths at half-maxima broadened from 4.0 eV to 5.2 eV while the N4 peaks broadened from 4.4 to 5.1 eV. As shown by a graphical analysis of the 2nd derivative of the raw spectra (see ESI†),42 the branching ratio A5/(A5 + A4), where A5 and A4 are the areas under the N5 and N4 peaks, increased from 0.62 for (C8H8)2Th (5f0) to 0.67 for (C8H8)2U (5f2). This increase is consistent with previous spectroscopic and theoretical studies that showed larger branching ratios with greater 5f-orbital occupancies for the early actinide metals Th, U, Np, Pu, and Am.40,44 Because the N5,4-edges were primarily reflective of multiplet structure, additional insights into ligand field strength and effects from covalency were sought using C K-edge XAS measurements.
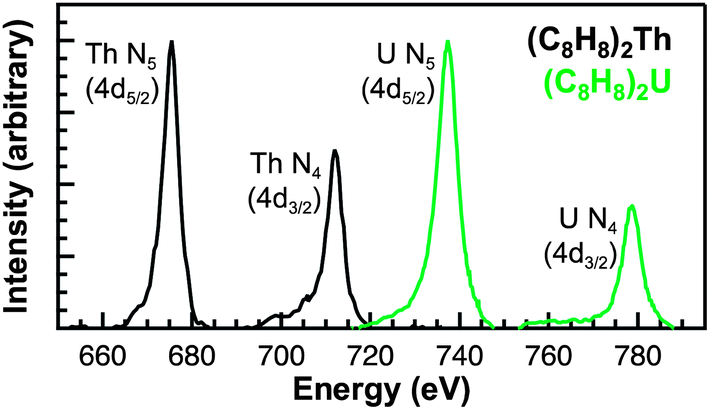 | ||
| Fig. 4 Thorium and uranium N5,4-edge (4d5/2,3/2) XAS data obtained in transmission for (C8H8)2Th (black trace) and (C8H8)2U (green trace). | ||
The background subtracted and normalized C K-edge XAS spectra for the (C8H8)2An are provided in Fig. 5. The second derivative of the spectra suggested the presence of several pre-edge peaks, which were indicative of actinide 5f and 6d orbital mixing with the cyclooctatetraene dianion C–C π-orbitals (Fig. 5 and ESI†). The peaks fell into two distinct groupings centered at roughly 285.5 eV and 287 eV. They also moved as a pair to lower energy and decreased in intensity upon moving from Th to U. Though both groupings were asymmetric and comprised of multiple unique transitions, the groupings observed for (C8H8)2U were broader as a possible consequence of increased f–f electron repulsion and/or spin–orbit coupling. At this stage, it was not possible to deconvolute the spectra and assign intensity to unique transitions because multiple curve fitting models could be derived. In the absence of a well-defined model of the experimental spectra, general aspects of the spectral profiles and transition energies will be discussed.
 | ||
| Fig. 5 C K-edge XAS data obtained in transmission for (C8H8)2Th (black trace) and (C8H8)2U (green trace). | ||
TDDFT spectral simulations
Because of the complexity of both experimental spectra, TDDFT calculations were necessary to guide spectral interpretations (Fig. 6 and Table 2). The TDDFT calculations for the (C8H8)2Th and (C8H8)2U compounds suggested that transitions involving the actinide 6d orbitals were unlikely to be accurately resolved in the experimental spectra. The calculated transitions to the 2a1g (6d–σ), 2e1g (6d–π), and 2e2g (6d–δ) orbitals (285 to 289 eV) exhibited little oscillator strength. In addition, transitions involving 2e1g (6d–π), and 2e2g (6d–δ) orbitals were calculated at high energy, above the 2nd main pre-edge peak, and in a low intensity valley that likely coincided with the onset of the edge.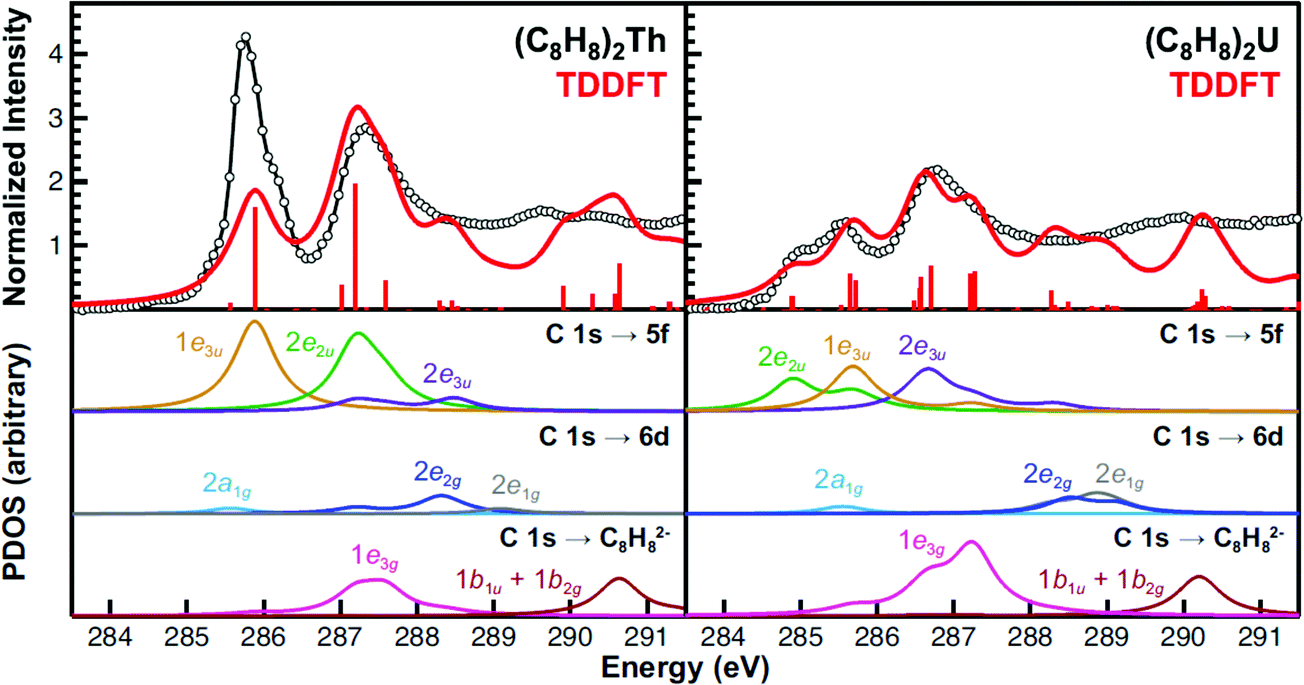 | ||
| Fig. 6 The experimental C K-edge XAS data obtained for (C8H8)2Th and (C8H8)2U (black circles and traces) are compared with the TDDFT calculations in the top panes. The red bars represent the energies and oscillator strengths for the individual transitions. Moving down, the subsequent panes show the partial density of states (PDOS) derived from the TDDFT for final states associated with the 5f, 6d, or ligand-based orbitals. Excitations to the antibonding 2a2u (5f–σ), 2e1u (5f–π) orbitals (effectively zero transition intensity), and higher lying metal 7s, 7p or other Rydberg-type orbitals are provided in the ESI.† Each simulated spectrum has been shifted by approximately +10 eV based on assignment of the C 1s → 2e2u (5f–δ) transitions to account for omission of atomic relaxation and other effects (see Experimental).93 | ||
| Final state orbital | Transition energies (eV) | |
|---|---|---|
| XAS | TDDFT | |
| a Experimental values were determined from a plot of the 2nd derivative of the spectrum (see ESI). Calculated values were taken from TDDFT simulated spectra and shifted by approximately +10 eV (see Experimental). | ||
| (C8H8)2Th | ||
| 1b1u + 1b2g (An-C nb) | 290.2 | 290.6 |
| 2e1g (6d–π) | — | 289.1 |
| 2e3u (5f–ϕ) | 288.7 | 288.5 |
| 2e2g (6d–δ) | 288.7 | 288.3 |
| 1e3g (An-C nb) | 287.4 | 287.5 |
| 2e2u (5f–δ) | 287.2 | 287.2 |
| 2e1u (5f–π) | — | 286.6 |
| 2a2u (5f–σ) | — | 286.4 |
| 1e3u (5f–ϕ) | 285.7 | 285.9 |
| 2a1g (6d–σ) | 285.3 | 285.6 |
| (C8H8)2U | ||
| 1b1u + 1b2g (An-C nb) | 289.8 | 290.2 |
| 2e1g (6d–π) | — | 288.9 |
| 2e2g (6d–δ) | 287.9 | 288.6 |
| 1e3g (An-C nb) | 287.0 | 287.3 |
| 2e3u (5f–ϕ) | 286.7 | 286.7 |
| 1e3u (5f–ϕ) | 285.5 | 285.7 |
| 2a1g (6d–σ) | 285.5 | 285.6 |
| 2e2u (5f–δ) | 284.9 | 284.9 |
| 2e1u (5f–π) | — | 284.1 |
| 2a2u (5f–σ) | — | 283.8 |
For (C8H8)2Th, the pattern of C 1s → 5f transition energies was closely related to the relative orbital ordering described from the ground-state DFT calculation (Fig. 1 and Table 2). For example, the TDDFT for (C8H8)2Th predicted strong low energy electronic excitations centered at 285.9 and 287.2 eV from C 1s orbitals to the bonding 1e3u (5f–ϕ) and antibonding 2e2u (5f–δ) orbitals, which correlated well with the first two pre-edge features at 285.7 and 287.2 eV. For comparison, the 1.5 eV experimental splitting for transitions involving 1e3u and 2e2u orbitals was close to the 1.4 eV splitting shown in the ground-state DFT calculations for the same 1e3u and 2e2u orbitals. These values were also similar to calculations reported in earlier work with (C8H8)2Pa.34 The TDDFT calculations indicated that these C 1s → 2e2u (5f–δ) transitions overlapped with those involving the exclusively ligand-based 1e3g (Th–C nonbonding) orbitals. Additionally, the energy for the C 1s → 2e2u (5f–δ) transitions coincided with transitions to the Th–C antibonding 2e3u (5f–ϕ) orbitals (288.5 eV), which were the antibonding counterparts of the bonding 1e3u (5f–ϕ) orbitals, as shown in Fig. 1. This close correspondence between the spectrum and the one-electron orbital model was likely associated with the Th4+ ground state electronic configuration being (1s)2⋯(5f)0(6d)0, hence the excited states reflected the simple (1s)1⋯(5f)1(6d)0 and (1s)1⋯(5f)0(6d)1 electronic configurations.45
Overall, the computational approach described above supported an interpretation of the C K-edge XAS spectrum of (C8H8)2U that was similar to (C8H8)2Th, but with some added complexity due to the 5f2 ground state of U4+.12,46 The ground state DFT calculation (Fig. 1) suggested that the first excited state could result from excitation into the doubly occupied 1e3u2 (5f–ϕ) orbitals and provide a 1e3u3 final state on uranium. However, there are two factors that push transitions to the 1e3u (5f–ϕ) orbitals to higher energy. The first is associated with double occupancy of the 5f orbitals that leads to a significant Coulomb repulsion (the Hubbard U). This raises the energy of final states that involve a 1e3u3 electronic configuration and favors nearby configurations that avoid double occupancy. The second is associated with Hund's rule spin coupling, which can provide energy stabilization to these nearby excited state configurations. For example, TDDFT results demonstrated that the quartet 5f3 excited states, which resulted from excitation into the antibonding 2e1u (5f–π) and 2a2u (5f–σ) orbitals were the lowest energy excited states. However, because there was very little mixing in the 2e1u (5f–π) and 2a2u (5f–σ) orbitals, transitions to them have negligible oscillator strength and were likely not resolved in the experimental spectra (Fig. 6). Hence, the TDDFT predicts that the first low energy grouping of features at about 285 eV was best attributed to closely spaced transitions from the C 1s orbitals to both the antibonding 2e2u (5f–δ) and bonding 1e3u (5f–ϕ) orbitals. The C 1s → 1e3u (5f–ϕ) transition was primarily centered at the 285.5 eV peak maximum, while the C 1s → 2e2u (5f–δ) transition was largely responsible for the emergence of the low-energy shoulder at 284.9 eV. For comparison, relative to (C8H8)2Th there was little change in the energies of the C 1s → 1e3u (5f–ϕ) transitions, but the C 1s → 2e2u (δ) transition in (C8H8)2U was 2.3 eV lower in energy. For the high energy grouping of features at 286.8 eV, the calculations showed contributions from C 1s electron excitations to the 2e3u (5f–ϕ) and the 1e3g (An–C nonbonding) molecular orbitals. Although moving from (C8H8)2Th to (C8H8)2U had little effect on the energy of C 1s → 1e3g (An–C nonbonding) transitions – which involved exclusively ligand-based orbitals – the energy for the C 1s → 2e3u (5f–ϕ) transitions was appreciably lower (288.5 eV for Th and 286.7 eV for U).
Despite the generally good agreement in peak position between the TDDFT and the experimental data, the distribution of the TDDFT intensities did not always faithfully reproduce the experiment. For example, the first feature in the spectrum for (C8H8)2Th (ca. 285 eV) was more intense in the experimental spectrum than in the calculation. The discrepancy was less obtuse once the 10% error associated with the experimental peak intensities was considered. Additionally, there existed several sources of potential error in the calculated intensities, which are discussed briefly. Our previous studies suggested that spin–orbit effects were relatively minor in the context of the experimental resolution available from the ligand K-edge XAS experiment.24 However, spin–orbit effects will certainly alter the distribution of oscillator strength (and to some extent the energies) in transitions to the 5f-manifold. In general, it is possible to perform calculations in either Russell–Saunders coupling (LS) or by an iterative solution including the spin–orbit coupling (the jj limit). Our current approach – thus far – has interpreted XAS spectra in the LS limit, recognizing that the associated TDDFT spectrum will represent a weighted average over the appropriate spin–orbit coupled states. We prefer this approach because it has provided relative ease for comparisons with transition metal spectra and because it is not clear that interpretation in the jj coupling basis will be superior. The real answer lies somewhere between these two limits, and is not easily addressed with one-electron approximations such as TDDFT. An additional and important issue is associated with systems that contain partially filled 5f-orbitals, like the 5f2 (C8H8)2U molecule. In these cases single determinant approximations are incapable of determining all the appropriate multiplet states. Once again, the TDDFT results must be viewed as providing a spectrum that is an average over certain spin and angular momentum multiplets.
A more pressing concern with the TDDFT calculation (in our opinion) lies with the approximate nature of the functional. The hybrid functionals used here have been quite successful for determining the properties of actinide materials. However, almost all functionals (LDA, GGA, hybrids) suffer from that fact that the associated long-range potential does not decay asymptotically with distance as 1/r, which it should. This inadequacy has dramatic effects on the energies of charge-transfer like states associated with the valence electrons.47 In the present context, this shortfall may cause an incorrect description of the potential felt at the C 1s core by the valence electrons. For example, the C 1s eigenvalue is, essentially, proportional to the electrostatic potential at the core generated by the valence electrons. The inappropriate potential at the core generated by DFT is in part responsible for our need to shift the predicted C 1s energy transitions to align theory with the experimental peaks.48 In so far as it is a constant error, it need not concern us. However, the oscillator strength associated with transitions from the C 1s orbitals is a different issue, and one that to our knowledge has not been studied in depth. We have therefore conducted preliminary calculations that compared the computed oscillator strengths between the B3LYP functional and a calculation using the Hartree–Fock/single-excitation configuration–interaction approach (HF–CIS). Because the HF potential does decay properly as 1/r, and the CIS approach is quite similar to the TDDFT approximation, the comparison should principally reflect any difficulties associated with the asymptotic form of the potential. In the HF–CIS approximation, we find the intensity of the first peak in (C8H8)2Th is now significantly larger than the second, in qualitative agreement with experiment. It should be possible to retain the advantages of DFT while more faithfully reproducing the intensities with “range-corrected” density functionals.49–52 This tentative conclusion merits additional research.
Evaluating An–(C8H8) bonding
Taken together, the C K-edge XAS experiment, TDDFT, and ground-state DFT calculations were used to evaluate the relative roles of the 5f–δ and 5f–ϕ bonding interactions for (C8H8)2Th and (C8H8)2U. Moving from Th4+ to U4+ has been proposed to impart a decrease in energy for the 5f atomic orbitals,13 bringing them closer in energy to the ligand-based orbitals of e2u symmetry (Fig. 1). The resulting enhancement in orbital mixing was reflected in the DFT calculations by more C 2p character in the 2e2u (5f–δ) orbitals for (C8H8)2U (28%) relative to (C8H8)2Th (13%). Additional support for this interpretation was found in complete active space natural orbital calculations published by Kerridge and Kaltsoyannis, which indicated that occupation of the 2e2u orbitals is greater for (C8H8)2U relative to (C8H8)2Th.53 In spite of the increase in 5f–δ orbital mixing for (C8H8)2U, which was anticipated based on the ground-state DFT calculations (Table 1) and earlier PES studies,19 it was clear that the pre-edge features in the C K-edge XAS spectra and TDDFT calculations between ca. 284 and 288.5 eV were more intense for (C8H8)2Th than (C8H8)2U. According to the assignment of transition energies described above, it was likely that the intense pre-edge for (C8H8)2Th arose due to strong ϕ-type mixing between the C 2p and Th 5f orbitals of e3u symmetry. The productive mixing was reflected by intense C 1s → 1e3u (5f–ϕ) transitions for (C8H8)2Th, and by a large splitting between transitions to the unoccupied bonding 1e3u and antibonding 2e3u orbitals (XAS: 3.0 eV; TDDFT: 2.6 eV) relative to (C8H8)2U (XAS: 1.2 eV; TDDFT: 1.0 eV). As shown in Fig. 7 and Table 1, the calculated C 2p and Th 5f orbital mixing coefficients for (C8H8)2Th also showed significant mixing in the bonding 1e3u (33% C 2p, 65% Th 5f) and antibonding 2e3u orbitals (49% C 2p, 47% Th 5f). Conversely, for (C8H8)2U the bonding 1e3u orbitals were almost entirely metal based (6% C 2p, 94% U 5f), while the antibonding 2e3u orbitals were comprised mostly of ligand character (89% C 2p, 7% U 5f). Future efforts are focused on confirming these experimental and computational values using additional spectroscopic and theoretical approaches.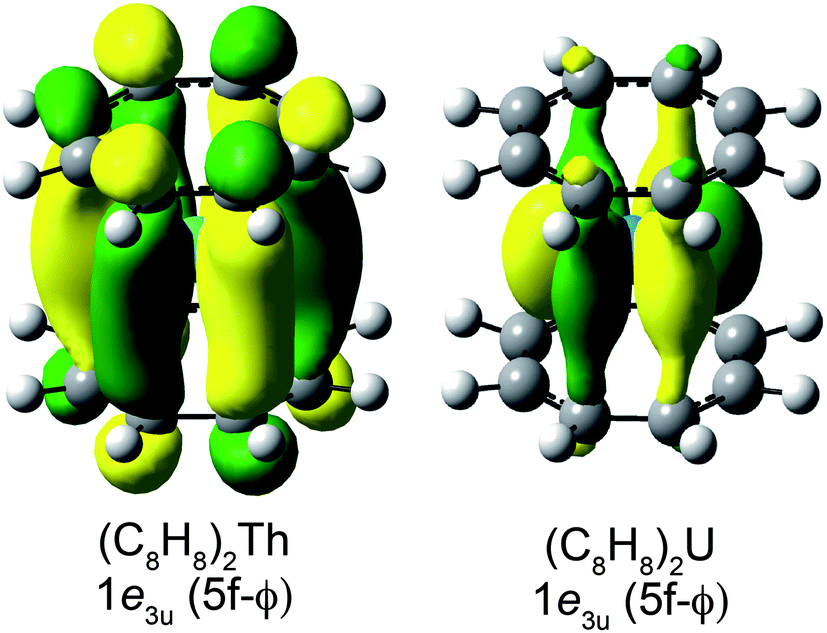 | ||
| Fig. 7 Representations of the bonding 1e3u Kohn–Sham molecular orbitals for (C8H8)2Th (left) and (C8H8)2U (right). | ||
The spectroscopic and computational results reported here demonstrated how subtle differences in actinide 5f orbital energies could influence orbital mixing. As shown above, the high-lying Th 5f orbitals were capable of forming quite covalent 1e3u (5f–ϕ) interactions. However, as the effective nuclear charge (Zeff) increased when moving further to the right in the actinide series (Th to U), the 5f orbitals dropped in energy. This likely led to an increasing energy mismatch with the (C8H8)24− orbitals of e3u symmetry, such that for (C8H8)2U the 5f–ϕ mixing was diminished and the 1e3u orbitals were best regarded as non-bonding 5f-electron reservoirs.1,34,54 Concomitantly, with the decrease in energy of the 5f orbitals, they also became better matched in energy with the filled (C8H8)24− orbitals of e2u symmetry and 5f–δ mixing was enhanced in (C8H8)2U.20
Concluding remarks
The presence of δ-type bonding interactions – where two nodal planes are coincident with the bond axis – has been invoked in both metal–metal bonded complexes55–60 and inverted arene sandwich complexes61–75 of the d- and f-block elements. To the best of our knowledge, the C K-edge XAS spectrum of thorocene represents the first experimental evidence of measurable ϕ-type orbital mixing, and supports many theoretical analyses that predict the f-elements are uniquely capable of participating in ϕ-bonding interactions.13,33–35,57,76 In this case, mixing resulted from a near perfect energy match between the Th 5f and (C8H8)24− orbitals of e3u symmetry, which has been discussed in earlier theoretical reports.5,53,77 Orbital mixing has been proposed to increase with later actinides;17,78,79 however, the e3u (5f–ϕ) orbitals for (C8H8)2Th provided an unusual case where mixing is greatest for the early actinides. As shown previously by Cloke and coworkers,80 the unoccupied 1e3u and 2e3u (5f–ϕ) orbitals did not appear to govern the chemical reactivity of (C8H8)2Th. Nevertheless, this type of ϕ-orbital mixing may be important to the chemical and physical properties of other actinide and lanthanide molecules and materials. Future work is focused on evaluating how the decrease in 5f orbital energy anticipated for (C8H8)2Np and (C8H8)2Pu affects bonding in these transuranic organometallics.Experimental
STXM sample preparation
All manipulations were performed with rigorous exclusion of air and moisture using Schlenk and glovebox techniques under an atmosphere of argon. Toluene (Fisher) was distilled from sodium metal and benzophenone prior to use. Samples of (C8H8)2Th2 and (C8H8)2U81 were prepared using the literature procedures. A small amount of each sample (∼1 mg) was dissolved in toluene (1 mL), and an aliquot of this solution (0.5 μL) was transferred to a Si3N4 window (100 nm, Silson) using a micropipette. The toluene was allowed to evaporate over a few seconds, which deposited thin crystallites of the sample on the Si3N4 membrane. After drying for several more minutes, a second window was placed over the sample, sandwiching the crystallites, and the windows were sealed together using Hardman Double/Bubble® epoxy.STXM measurements and data analysis
The STXM methodology was similar to that discussed previously.27–29,32,75 Single-energy images and carbon K-edge XAS spectra were acquired using the STXM instrument at the Advanced Light Source-Molecular Environmental Science (ALS-MES) beamline 11.0.2, which is operated in top-off mode at 500 mA, in a ∼0.5 atm He filled chamber.36 An energy calibration was performed at the C K-edge for CO2 gas (294.95 eV) and at the Ne K-edge for Ne gas (867.30 eV). For these measurements, the X-ray beam was focused with a zone plate onto the sample, and the transmitted light was detected. The spot size and spectral resolution were determined from characteristics of the 25 nm zone plate. The energy resolution was determined to be 0.04 eV at the C K-edge and 0.08 eV at the U/Th N-edges, and spectra were collected using circularly polarized radiation. During the STXM experiment, samples showed no sign of radiation damage and each spectrum was reproduced from multiple independent crystallites. Salient features of the spectra were reproducible using samples prepared from non-oriented polycrystalline particles. The C K-edge data were normalized in MATLAB using the MBACK algorithm,82 and by setting the edge jump at 295 eV to an intensity of 1.0. Second-derivative spectra were used as guides to determine the number and position of peaks (see ESI†).Electronic structure calculations
Ground state electronic structure calculations were performed on the (C8H8)2An complexes using B3LYP hybrid density functional theory (DFT)83,84 in the Gaussian 09 code.85 Th and U were modeled with the Stuttgart relativistic effective core potential (ecp) and basis set86–88 while C and H were modeled using a Pople style double-ζ 6-31 G(d′,p′) basis set with polarization functions optimized for heavy atoms.89,90 These functionals and basis sets have demonstrated good agreement between experimental and simulated ligand K-edge XAS spectra for organometallic and inorganic systems.23,27,29,91 The molecular orbital compositions of each compound were obtained by Mulliken population analysis of the individual molecular orbitals.Simulated C K-edge spectra
For (C8H8)2An, the C K-edge XAS spectra were simulated using time dependent density functional theory (TDDFT). These calculations were conducted in analogy to earlier reports.29,46 As discussed previously, an energy shift must be established to account for the omission of the atomic relaxation associated with the core excitation, relativistic stabilization, and errors associated with the functional. This was achieved by setting the energy of transitions simulated for the antibonding 2e2u (5f–δ) orbitals to be equal to those in the experimental spectra, which resulted in energy shifts of +10.450 and +10.026 for (C8H8)2Th and (C8H8)2U, respectively.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, U.S. Department of Energy at LBNL (contract DE-AC02-05CH11231) and under the Heavy Element Chemistry Program at LANL (operated by Los Alamos National Security, LLC, for the National Nuclear Security Administration; contract DE-AC52-06NA25396). Beamline 11.0.2 at the Advanced Light Source was supported by the Director, Office of Science, Office of Basic Energy Sciences Division of Chemical Sciences, Geosciences, and Biosciences; and the Condensed Phase and Interfacial Molecular Sciences Program of the aforementioned Division of the U.S. Department of Energy at LBNL under Contract No. DE-AC02-05CH11231. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract no. DE-AC02-05CH11231. Parts of this work were supported at LBNL by the Berkeley Actinide Postdoctoral Fellowship (Minasian), and at LANL by a Glenn T. Seaborg Institute Postdoctoral Fellowship (Minasian) and a Director's Postdoctoral Fellowship (Keith).References
- A. Streitwieser Jr and U. Mueller-Westerhoff, J. Am. Chem. Soc., 1968, 90, 7364 CrossRef.
- A. Streitwieser Jr and N. Yoshida, J. Am. Chem. Soc., 1969, 91, 7528 CrossRef.
- D. G. Karraker, J. A. Stone, E. R. Jones and N. Edelstein, J. Am. Chem. Soc., 1970, 92, 4841–4845 CrossRef CAS.
- D. F. Starks, T. C. Parsons, A. Streitwieser Jr and N. Edelstein, Inorg. Chem., 1974, 13, 1307–1308 CrossRef CAS.
- K. D. Warren, Struct. Bonding, 1977, 33, 97–138 CrossRef CAS.
- N. Rösch and A. Streitwieser Jr, J. Organomet. Chem., 1978, 145, 195–200 CrossRef.
- P. Pyykkö and L. L. Lohr, Inorg. Chem., 1981, 20, 1950–1959 CrossRef.
- C. Levanda and A. Streitwieser Jr, Inorg. Chem., 1981, 20, 656–659 CrossRef CAS.
- J. C. Green, Struct. Bonding, 1981, 43, 37–112 CrossRef CAS.
- N. Rösch and A. Streitwieser Jr, J. Am. Chem. Soc., 1983, 105, 7237–7240 CrossRef.
- N. Rösch, Inorg. Chim. Acta, 1984, 94, 297–299 CrossRef.
- A. H. H. Chang and R. M. Pitzer, J. Am. Chem. Soc., 1989, 111, 2500–2507 CrossRef CAS.
- M. Pepper and B. E. Bursten, Chem. Rev., 1991, 91, 719–741 CrossRef CAS.
- M. Dolg, P. Fulde, H. Stoll, H. Preuss, A. Chang and R. M. Pitzer, Chem. Phys., 1995, 195, 71–82 CrossRef CAS.
- D. Seyferth, Organometallics, 2004, 23, 3562–3583 CrossRef CAS.
- C. J. Burns and M. S. Eisen, in The Chemistry of the Actinide and Transactinide Elements, ed. L. Morss, N. M. Edelstein and J. Fuger, Springer, Berlin, 3rd edn, 2006, vol. 5, pp. 2799–2910 Search PubMed.
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2012, 257, 394–406 CrossRef.
- A. Kerridge, Dalton Trans., 2013 10.1039/c3dt52279b.
- J. P. Clark and J. C. Green, J. Organomet. Chem., 1976, 112, C14–C16 CrossRef CAS.
- J. P. Clark and J. C. Green, J. Chem. Soc., Dalton Trans., 1977, 505–508 RSC.
- J. G. Brennan, J. C. Green and C. M. Redfern, J. Am. Chem. Soc., 1989, 111, 2373–2377 CrossRef CAS.
- E. I. Solomon, B. Hedman, K. O. Hodgson, A. Dey and R. K. Szilagyi, Coord. Chem. Rev., 2005, 249, 97–129 CrossRef CAS.
- S. A. Kozimor, P. Yang, E. R. Batista, K. S. Boland, C. J. Burns, D. L. Clark, S. D. Conradson, R. L. Martin, M. P. Wilkerson and L. E. Wolfsberg, J. Am. Chem. Soc., 2009, 131, 12125–12136 CrossRef CAS PubMed.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. D. Conradson, S. A. Kozimor, R. L. Martin, D. E. Schwarz, D. K. Shuh, G. L. Wagner, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2012, 134, 5586–5597 CrossRef CAS PubMed.
- R. G. Denning, J. C. Green, T. E. Hutchings, C. Dallera, A. Tagliaferri, K. Giarda, N. B. Brookes and L. Braicovich, J. Chem. Phys., 2002, 117, 8008–8020 CrossRef CAS.
- C. Fillaux, D. Guillaumont, J.-C. Berthet, R. Copping, D. K. Shuh, T. Tyliszczak and C. Den Auwer, Phys. Chem. Chem. Phys., 2010, 12, 14253–14262 RSC.
- J. A. Bradley, P. Yang, E. R. Batista, K. S. Boland, C. J. Burns, D. L. Clark, S. D. Conradson, S. A. Kozimor, R. L. Martin, G. T. Seidler, B. L. Scott, D. K. Shuh, T. Tyliszczak, M. P. Wilkerson and L. E. Wolfsberg, J. Am. Chem. Soc., 2010, 132, 13914–13921 CrossRef CAS PubMed.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, J. A. Bradley, S. R. Daly, S. A. Kozimor, W. W. Lukens, R. L. Martin, D. Nordlund, G. T. Seidler, D. K. Shuh, D. Sokaras, T. Tyliszczak, G. L. Wagner, T.-C. Weng and P. Yang, J. Am. Chem. Soc., 2013, 135, 1864–1871 CrossRef CAS PubMed.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, S. A. Kozimor, R. L. Martin, D. K. Shuh, T. Tyliszczak and L. J. Vernon, J. Am. Chem. Soc., 2013, 135, 14731–14740 CrossRef CAS PubMed.
- J.-C. Berthet, P. Thuéry, N. Garin, J.-P. Dognon, T. Cantat and M. Ephritikhine, J. Am. Chem. Soc., 2013, 135, 10003–10006 CrossRef CAS PubMed.
- A. Hervé, N. Garin, P. Thuéry, M. Ephritikhine and J.-C. Berthet, Chem. Commun., 2013, 49, 6304–6306 RSC.
- S. G. Minasian, J. L. Krinsky, J. D. Rinehart, R. Copping, T. Tyliszczak, M. Janousch, D. K. Shuh and J. Arnold, J. Am. Chem. Soc., 2009, 131, 13767–13783 CrossRef CAS PubMed.
- P. M. Boerrigter, E. J. Baerends and J. G. Snijders, Chem. Phys., 1988, 122, 357–374 CrossRef CAS.
- J. Li and B. E. Bursten, J. Am. Chem. Soc., 1998, 120, 11456–11466 CrossRef CAS.
- A. Kerridge, R. Coates and N. Kaltsoyannis, J. Phys. Chem. A, 2009, 113, 2896–2905 CrossRef CAS PubMed.
- H. Bluhm, K. Andersson, T. Araki, K. Benzerara, G. E. Brown, J. J. Dynes, S. Ghosal, M. K. Gilles, H. C. Hansen, J. C. Hemminger, A. P. Hitchcock, G. Ketteler, A. L. D. Kilcoyne, E. Kneedler, J. R. Lawrence, G. G. Leppard, J. Majzlan, B. S. Mun, S. C. B. Myneni, A. Nilsson, H. Ogasawara, D. F. Ogletree, K. Pecher, M. Salmeron, D. K. Shuh, B. Tonner, T. Tyliszczak, T. Warwick and T. H. Yoon, J. Electron Spectrosc. Relat. Phenom., 2006, 150, 86–104 CrossRef CAS.
- J. A. Bradley, K. T. Moore, G. van der Laan, J. P. Bradley and R. A. Gordon, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 205105 CrossRef.
- D. E. Bugaris, E. S. Choi, R. Copping, P.-A. Glans, S. G. Minasian, T. Tyliszczak, S. A. Kozimor, D. K. Shuh and J. A. Ibers, Inorg. Chem., 2011, 50, 6656–6666 CrossRef CAS PubMed.
- H. J. Nilsson, T. Tyliszczak, R. E. Wilson, L. Werme and D. K. Shuh, Anal. Bioanal. Chem., 2005, 383, 41–47 CrossRef CAS PubMed.
- G. van der Laan, K. T. Moore, J. G. Tobin, B. W. Chung, M. A. Wall and A. J. Schwartz, Phys. Rev. Lett., 2004, 93, 097401 CrossRef CAS PubMed.
- J. G. Tobin, K. T. Moore, B. W. Chung, M. A. Wall, A. J. Schwartz, G. van der Laan and A. L. Kutepov, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085109 CrossRef.
- K. T. Moore, G. van der Laan, R. G. Haire, M. A. Wall and A. J. Schwartz, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 033109 CrossRef.
- K. T. Moore and G. van der Laan, Rev. Mod. Phys., 2009, 81, 235–298 CrossRef CAS.
- K. T. Moore, G. van der Laan, M. A. Wall, A. J. Schwartz and R. G. Haire, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 073105 CrossRef.
- The final state is comprised of both an unpaired electron and a core hole, which can couple to form both triplet and singlet final states. Ignoring spin–orbit coupling, only transitions to the singlet final state are allowed.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, C. N. Christensen, D. L. Clark, S. D. Conradson, S. A. Kozimor, R. L. Martin, D. E. Schwarz, D. K. Shuh, G. L. Wagner, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2012, 134, 5586–5597 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- G. Cavigliasso and D. P. Chong, J. Chem. Phys., 1999, 111, 9485–9492 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 234109 CrossRef PubMed.
- Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed.
- A. Kerridge and N. Kaltsoyannis, J. Phys. Chem. A, 2009, 113, 8737–8745 CrossRef CAS PubMed.
- D. B. Beach, K. D. Bomben, N. M. Edelstein, D. C. Eisenberg, W. L. Jolly, R. Shinomoto and A. Streitwieser Jr, Inorg. Chem., 1986, 25, 1735–1737 CrossRef CAS.
- F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple Bonds Between Metal Atoms, 3rd edn, Springer Science, New York, NY, 2005 Search PubMed.
- T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long and P. P. Power, Science, 2005, 310, 844–847 CrossRef CAS PubMed.
- L. Gagliardi and B. O. Roos, Nature, 2005, 433, 848–851 CrossRef CAS PubMed.
- G. Menconi and N. Kaltsoyannis, Organometallics, 2005, 24, 1189–1197 CrossRef CAS.
- G. Cavigliasso and N. Kaltsoyannis, Dalton Trans., 2006, 5476–5483 RSC.
- G. Cavigliasso and N. Kaltsoyannis, Inorg. Chem., 2007, 46, 3557–3565 CrossRef CAS PubMed.
- F. A. Cotton, P. A. Kibala and W. A. Wojtczak, J. Am. Chem. Soc., 1991, 113, 1462–1463 CrossRef CAS.
- P. L. Diaconescu, P. L. Arnold, T. A. Baker, D. J. Mindiola and C. C. Cummins, J. Am. Chem. Soc., 2000, 122, 6108–6109 CrossRef CAS.
- P. L. Diaconescu and C. C. Cummins, J. Am. Chem. Soc., 2002, 124, 7660–7661 CrossRef CAS PubMed.
- W. J. Evans, S. A. Kozimor, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2004, 126, 14533–14547 CrossRef CAS PubMed.
- W. J. Evans, S. A. Kozimor and J. W. Ziller, Chem. Commun., 2005, 4681–4683 RSC.
- G. B. Nikiforov, P. Crewdson, S. Gambarotta, I. Korobkov and P. H. M. Budzelaar, Organometallics, 2007, 26, 48–55 CrossRef CAS.
- W. J. Evans, C. A. Traina and J. W. Ziller, J. Am. Chem. Soc., 2009, 131, 17473–17481 CrossRef CAS PubMed.
- D. P. Mills, F. Moro, J. McMaster, J. van Slageren, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2011, 3, 454–460 CAS.
- M. J. Monreal, S. I. Khan, J. L. Kiplinger and P. L. Diaconescu, Chem. Commun., 2011, 47, 9119–9121 RSC.
- D. Patel, F. Moro, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 50, 10388–10392 CrossRef CAS PubMed.
- P. L. Arnold, S. M. Mansell, L. Maron and D. McKay, Nat. Chem., 2012, 4, 668–674 CrossRef CAS PubMed.
- P. L. Diaconescu and C. C. Cummins, Inorg. Chem., 2012, 51, 2902–2916 CrossRef CAS PubMed.
- V. Mougel, C. Camp, J. Pecaut, C. Coperet, L. Maron, C. E. Kefalidis and M. Mazzanti, Angew. Chem., Int. Ed., 2012, 51, 12280–12284 CrossRef CAS PubMed.
- B. Vlaisayljevich, P. L. Diaconescu, W. L. Lukens Jr, L. Gagliardi and C. C. Cummins, Organometallics, 2013, 32, 1341–1352 CrossRef.
- T. L. Gianetti, G. Nocton, S. G. Minasian, N. C. Tomson, A. L. D. Kilcoyne, S. A. Kozimor, D. K. Shuh, T. Tyliszczak, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2013, 135, 3224–3236 CrossRef CAS PubMed.
- B. E. Bursten and G. A. Ozin, Inorg. Chem., 1984, 23, 2910–2911 CrossRef CAS.
- R. G. Hayes and N. Edelstein, J. Am. Chem. Soc., 1972, 94, 8688–8691 CrossRef CAS.
- I. D. Prodan, G. E. Scuseria and R. L. Martin, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 033101 CrossRef.
- I. Kirker and N. Kaltsoyannis, Dalton Trans., 2011, 40, 124–131 RSC.
- Chemical reduction of (C8H8)2Th generates trivalent [(C8H8)2Th]1− with an (a1g)1 electronic configuration: J. S. Parry, F. G. N. Cloke, S. J. Goles and M. B. Hursthouse, J. Am. Chem. Soc., 1999, 121, 6867–6871 CrossRef CAS.
- A. Streitwieser Jr, U. Mueller-Westerhoff, F. Mares, C. B. Grant, D. G. Morrell, T. J. Marks and S. S. Miller, Inorg. Synth., 1979, 19, 149–154 CrossRef.
- T.-C. Weng, G. S. Waldo and J. E. Penner-Hahn, J. Synchrotron Radiat., 2005, 12, 506–510 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- P. Fuentealba, H. Preuss, H. Stoll and L. Vonszentpaly, Chem. Phys. Lett., 1982, 89, 418–422 CrossRef CAS.
- W. Kuchle, M. Dolg, H. Stoll and H. Preuss, Mol. Phys., 1991, 74, 1245–1263 CrossRef.
- W. Kuchle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS.
- S. R. Daly, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. A. Kozimor and R. L. Martin, J. Am. Chem. Soc., 2012, 134, 14408–14422 CrossRef CAS PubMed.
- C. J. Cramer, Essentials of Computational Chemistry, 2nd edn, Wiley, Chichester, UK, 2004 Search PubMed.
- M. Segala and D. P. Chong, J. Electron Spectrosc. Relat. Phenom., 2010, 182, 141–144 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Background subtracted and normalized C K-edge XAS data for (C8H8)2An (An = Th, U), as well as Kohn–Sham orbital pictures and tables of spectral and computational data. See DOI: 10.1039/c3sc52030g |
| ‡ Current address: Department of Chemistry, Colgate University, 13 Oak Dr., Hamilton, NY 13346, USA. |
| This journal is © The Royal Society of Chemistry 2014 |