
Disposable dual sensor array for simultaneous determination of chlorogenic acid and caffeine from coffee†
Ioana Vasilescua,
Sandra A. V. Eremia*a,
Ramona Penua,
Camelia Albua,
Antonio Radoi b,
Simona C. Litescua and
Gabriel-Lucian Radua
b,
Simona C. Litescua and
Gabriel-Lucian Radua
aCentre of Bioanalysis, National Institute of Research and Development for Biological Sciences, 296 Splaiul Independentei, 060031 Bucharest, Romania. E-mail: sandraeremia@gmail.com; Fax: +40 4021 2200 900; Tel: +40 4021 2200 900
bNational Institute for Research and Development in Microtechnology (IMT-Bucharest), 126 A Erou Iancu Nicolae, 077190 Bucharest, Romania
First published on 24th November 2014
Abstract
In this work a novel sensor array platform based on a dual carbon screen-printed electrode was developed for the simultaneous determination of chlorogenic acid and caffeine. One of the carbon working electrodes was modified with platinum nanoparticles, reduced graphene oxide and laccase (C-SPE/Pt-NPs/RGO/lacc-biosensor) for chlorogenic acid determination and the second carbon working electrodes was modified with reduced graphene oxide and Nafion (C-SPE/RGO/Nafion-sensor) for caffeine determination. Cyclic voltammetry was used to characterise and optimise the dual sensor array while chronoamperometry was used to investigate the bioelectrocatalytic response. The C-SPE/Pt-NPs/RGO/lacc for biosensing chlorogenic acid exhibited a sensitivity of 0.02 μA μM−1 and a detection limit of 2.67 μM whereas the C-SPE/RGO/Nafion used for sensing caffeine has showed a sensitivity of 1.38 μA μM−1 and a detection limit of 0.22 μM. The developed sensor array was used to determine these two major coffee bean compounds from real coffee samples. Due to its simplicity, feasibility and accessibility, the developed dual sensor array could represent the basis of a valuable analytical tool able to screen both chlorogenic acid and caffeine content from coffee samples offering important information about the phytochemical composition of the samples.
Introduction
Coffee is a highly popular drink that is rich in hydroxycinnamic acids such as caffeic, chlorogenic, coumaric, ferulic and sinapic acids, and other active compounds having significant antioxidant potential such as caffeine, nicotinic acid, trigonelline, cafestol and kaheol.1 In recent years people have become more aware about the importance of eating habits and try to adopt healthy diets. Coffee moderate consumption has positive benefits to human health due to the high content in antioxidant compounds. Antioxidants represent important compounds that should be present in everyone diet and therefore scientists have developed numerous methods for determining antioxidant capacity. The antioxidant properties of coffee were evaluated using several methods such as: 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) and 2,2-diphenyl-1-picrylhydrazyl (DPPH) assays,2–4 oxygen radical absorbance capacity (ORAC) assay,5 ferric-reducing antioxidant power (FRAP) assay6 and Folin–Ciocalteu test.3,4Coffee is considered to have the highest amount of phenolics, their content being influenced by the coffee beans origin. Among these polyphenols, chlorogenic acids are the most abundant, representing up to 12% of dry matter of coffee beans.1 Chlorogenic acid is a naturally occurring compound being an ester of caffeic acid and quinic acid, found in raw coffee beans and it is usually destroyed when coffee beans are roasted.7,8 Chlorogenic acids are usually characterised using high performance liquid chromatography (HPLC) or liquid chromatography-mass spectrometry (LC-MS).9,10 However there are also several electrochemical methods developed for the determination of chlorogenic acid. Yardim et al. have developed two electrochemical methods for the determination of chlorogenic acids using boron-doped diamond electrode.11,12 Santos et al. have also developed a molecularly imprinted electrochemical sensor for the selective detection of chlorogenic acid onto gold bare electrode surface and successfully applied for the determination of chlorogenic acid from coffee samples.13 Literature survey has revealed the construction of two biosensors for chlorogenic acid determination, the first one based on ionic liquid containing iridium nanoparticles and polyphenol oxidase obtained from the sugar apple tissue,14 while the second biosensor was being based on bean sprout homogenate immobilised into chitosan.15
Another compound present in coffee that has significant pharmacological effects is caffeine. Caffeine is a purine alkaloid known for its various health effects when consumed with moderation: it increases energy availability, it decreases fatigue, it boosts physical performance, it increases alertness and wakefulness, it increases the ability to focus, etc.16 However, when consumed in large doses it can cause many undesired effects such as cardiovascular diseases, depression, oversensitivity, anxiety, irritability and even addiction, etc.17 Various methods have been described for the detection of caffeine from coffee: ultraviolet spectrophotometry,18 high performance liquid chromatography with appropriate detection (e.g. diode array),19,20 gas chromatography21 and electrochemical methods.22–24
Therefore, considering the health effects and the unwanted side effects of caffeine and the beneficent antioxidant character of chlorogenic acid the simultaneous determination of caffeine and chlorogenic acid from coffee samples could be of great importance for food safety and food and beverages industry. A literature review has revealed the existence of several methods able to concomitantly determine chlorogenic acid and caffeine from coffee. De Maria et al. have developed a method for the simultaneous detection of chlorogenic acid and caffeine in green coffee using high performance gel filtration chromatography.25 One other method for the simultaneous detection of chlorogenic acid and caffeine was developed by Yardim et al. and it is based on adsorptive stripping voltammetry.12
In this context there is the demand for simple, low cost, low reagent consumption and rapid analytical methods or instruments to determine chlorogenic acids and caffeine from coffee samples. Therefore, in the present work we have developed a simple and cost effective dual electrochemical platform for the simultaneous detection of chlorogenic acid and caffeine from coffee samples.
When dealing with electrochemical detection the electrode surface has a key role in obtaining the advantages offered against classical methods. An important aspect concerning electrochemical sensors and enzyme-based biosensors is obtaining signal amplification and therefore higher sensitivities and lower detection limits could be achieved. The authors have already developed a laccase based biosensor having very low limits of detection and high sensitivity when using caffeic acid as laccase substrate, due to the use of platinum nanoparticles as support for enzyme immobilisation.26 Platinum nanoparticles when deposited onto carbon surfaces maximises the electrocatalytic surface area and therefore improve the electrocatalytic activity. When used to immobilise enzymes, platinum nanoparticles enhance the electron transfer between enzyme and electrode surface without the need of using a mediator.27 Graphene oxides and reduced graphene oxides are used frequently in combination with noble metal nanoparticles in order to form nanocomposites/derivatives with improved electrocatalytic properties ascribed to enhanced catalytic activity of noble metal nanoparticles.28 Reduced graphene oxide, an inexpensive material having good mechanical and thermal properties, represents a novel platform for enzyme immobilisation due to its ability to adsorb the enzyme by hydrophobic interactions.29
This paper shows the preparation of an electrochemical array consisting of a laccase based biosensor and a modified sensor for the simultaneous amperometric determination of chlorogenic acids and caffeine. The analytical tool was constructed on disposable dual screen-printed carbon electrodes by modifying one working electrode with platinum nanoparticles, reduced graphene oxides and laccase for chlorogenic acid determination while the other working electrode was modified with reduced graphene oxide and Nafion for caffeine detection. Once the dual sensor was optimised it was successfully applied for the determination of chlorogenic acids and caffeine from coffee real samples. To the best of our knowledge this is the first attempt to develop an amperometric dual sensor for the detection of active compounds present in coffee samples.
Experimental
Materials
Laccase from Trametes versicolor (EC 1.10.3.2, activity 0.5 U mg−1 according to the supplier) was provided by Sigma-Aldrich, Steinheim, Germany. The enzyme activity was checked spectrophotometrically using the unspecific substrate ABTS, also supplied by Sigma-Aldrich. Chlorogenic acid, caffeic acid, caffeine, (−)-catechin, formic acid, Nafion perfluorinated ion-exchange resin, 10 wt% dispersion in water, sodium acetate, ethylene glycol (EG), H2PtCl6, acetic acid and potassium chloride were supplied by Sigma-Aldrich, Steinheim, Germany. Potassium hexacyanoferrate(III) and ortho-phosphoric acid were obtained from Merck, Darmstadt, Germany while acetonitrile (ACN) was supplied by Biosolve, Netherlands, respectively. The electrolyte consisted of 0.1 M acetate buffer pH 5.00 in 0.1 M KCl. Graphite flakes (SP1) were from Bay Carbon, Inc., USA. All reagents were of analytical grade or chromatographic grade and all the aqueous solutions were prepared with Milli Q ultra-pure water.Coffee samples preparation
Seven samples of coffee were obtained from local markets; 5 g of each coffee sample was prepared in 50 mL hot water–ethanol (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 v/v) enabling the extraction of active principles for 30 minutes. Then the coffee samples were diluted 1:20 (v/v) (Carte Noir Café Long Délicat) and 1:40 (v/v) (Jacobs Espresso, Segafredo Intermezzo, Davidoff Espresso, Grandia Classic, Davidoff Rich Aroma) – in the case of chlorogenic acid electrochemical assay. When caffeine content was electrochemically tested, Carte Noir Café Long Délicat and Segafredo Intermezzo samples were dilutes 20 times and the remaining coffee samples were diluted 1:40 (v/v). Dilution was achieved in 0.1 M acetate buffer pH 5.00.
:30 v/v) enabling the extraction of active principles for 30 minutes. Then the coffee samples were diluted 1:20 (v/v) (Carte Noir Café Long Délicat) and 1:40 (v/v) (Jacobs Espresso, Segafredo Intermezzo, Davidoff Espresso, Grandia Classic, Davidoff Rich Aroma) – in the case of chlorogenic acid electrochemical assay. When caffeine content was electrochemically tested, Carte Noir Café Long Délicat and Segafredo Intermezzo samples were dilutes 20 times and the remaining coffee samples were diluted 1:40 (v/v). Dilution was achieved in 0.1 M acetate buffer pH 5.00.
Apparatus
Voltammetric and amperometric measurements were performed using a potentiostat Auto-Lab PGSTAT 302N (Utrecht, Netherlands) equipped with Nova (1.10) (Utrecht, Netherlands) software. The UV-Vis spectrophotometric measurements were carried out on a Thermo Evolution 260 Bio (Thermo Fischer Scientific). The chromatographic measurements were performed using a complete HPLC Shimadzu system (Kyoto, Japan) equipped with two pumps LC-20ADsp, an auto-sampler SIL-20AC, a column oven CTO-20AC, a degasser DGU-20A5, a photodiode array detector SPDM20A and LC solution software. All the measurements were carried out at room temperature.The micrographs were obtained using a Nova NanoSEM 630 (FEI Company, USA) Scanning Electron Microscope (SEM).
The electrochemical cell consisted of dual screen-printed carbon electrodes (DRP-C1110, DropSens). The dual sensor have two elliptical carbon working electrodes (6.3 mm2 each one), a carbon counter electrode and a Ag pseudo-reference electrode, all printed onto a ceramic support (3.4 cm × 1.0 cm).
Preparation of the sensor array
Platinum nanoparticles (Pt-NPs) were obtained by using a slightly modified protocol inspired from Bragaru et al.30 Briefly, 20 mL of ethylene glycol (EG) and 1 mL of H2PtCl6 solution 0.2 M prepared in distilled water were allowed to react at 60 °C, under agitation (600 rpm), during 30 minutes. After that the pH was adjusted to 11.00 using NaOH solution 1 M and the temperature was increased up to 130 °C and nitrogen was purged during 4 hours, the reaction being allowed to proceed under continuous stirring. After, the reaction mixture was cooled off, centrifuged at 6000 rpm, during 60 minutes, and the supernatant was collected and used further to modify the electrodes.Reduced graphene oxide (RGO) was obtained by chemically reducing graphene oxide obtained according to Hummers method31 with hydrazine according to Park et al. method.32 The procedure is detailed in Eremia et al.26
The dual screen-printed carbon electrodes were modified accordingly (Fig. 1):
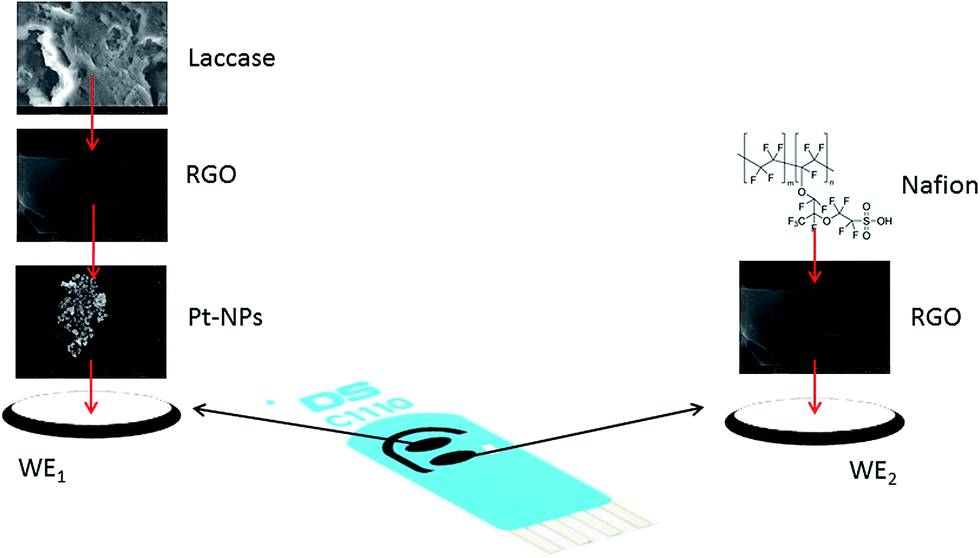 | ||
| Fig. 1 Schematic representation of the disposable dual sensor array. | ||
- the first working electrode (WE1) was modified by simply adding 3 μL Pt-NPs directly onto the surface of the carbon screen-printed electrode, then 3 μL of RGO (2 mg mL−1) were added over the Pt-NPs layer and finally 3 μL of laccase solution was adsorbed ensuring an enzymatic activity of approximately 250 mIU. The electrodes were dried at 20 °C between the deposition steps. This modified electrode will be named C-SPE/Pt-NPs/RGO/lacc electrode or laccase biosensor and it was optimised for caffeic acid determination in the previous mentioned work of Eremia et al.26
- the second working electrode (WE2) was modified by adding 3 μL RGO (2 mg mL−1) directly onto the surface of the carbon screen-printed electrode and then allowed to dry at 20 °C followed by the deposition of 3 μL Nafion 0.05 wt% obtained from the proper dilution of Nafion 10 wt% in Milli Q ultra-pure water.
The obtained sensor array was stored at 4 °C on silica gel layer in order to eliminate moistening.
Electrochemical methods
All the cyclic voltammetry and amperometric measurements were performed at room temperature (24 ± 0.5 °C), in drop mode. 0.1 M acetate buffer pH 5.00 in 0.1 M KCl was used as supporting electrolyte. Cyclic voltammograms were recorded in the range 0.6 and −0.4 V vs. Ag pseudo-reference electrode with a scan rate of 50 mV s−1 for K3[Fe(CN)6] and chlorogenic acid, while for caffeine the cyclic voltammograms were recorded between 1.0 and 1.6 V with a scan rate of 50 mV s−1.For the amperometric measurements the WE1 was polarised at −0.05 V vs. Ag pseudo-reference electrode for the determination of chlorogenic acid, while the WE2 was polarised at +1.3 V vs. Ag pseudo-reference electrode for caffeine determination. The same conditions were applied for real samples, too.
HPLC method
For the quantification of chlorogenic acid from coffee extracts, a HPLC-PDA method was used for measuring polyphenols published by Cristea et al.33 The mobile phase consisted of formic acid in water (pH 3.00) as solvent A and formic acid in ACN (pH 3.00) as solvent B. The polyphenolic compounds separation was performed using binary gradient elution: 0 min 5% solvent B; 0.01–20 min 5–30% solvent B; 20–25 min 30% solvent B; 25.01–28 min 30–5% solvent B and 28–30 min 5% solvent B. The flow rate was: 0–5 min 0.1 mL min−1; 5.01–15 min 0.2 mL min−1; 15.01–30 min 0.1 mL min−1. Compounds were separated on a Nucleosil 100-3.5 C18, KROMASIL, 100 × 2.1 mm column. The column was equilibrated for 1 hour before starting the injections. The analyses were performed at 20 °C (temperature column) for a period of 30 minutes and the injection volume was 20 μL. Then the column was washed over a period of 10 minutes with mobile phase using the flow rate 0.1 mL min−1 and 5% solvent B.Caffeine was quantified using a reversed-phase HPLC-PDA method34 using a mobile phase prepared from 0.1% ortho-phosphoric acid (v/v) in water as solvent A and ACN as solvent B and a Nucleosil 100-10 C18, KROMASIL, 250 × 4.6 mm column. The analyses were performed at 35 °C (temperature column) and the injection volume was 20 μL. For the separation an isocratic elution (solvent A:solvent B, 90:10) was used, with a flow rate of 1 mL min−1, during 30 minutes. The caffeine was detected and quantified at 272 nm wavelength.
All the coffee samples were filtrated before injection using PTFE Syringe Driven Filter Unit 0.2 μm (Macherey-Nagel) and analysed by direct injection in HPLC-PDA-MS system without any prior purification of the sample.
Results and discussion
The main breakthrough of this work was the development of the sensor for caffeine and the optimisation and application of the final dual amperometric sensor for coffee real samples. The laccase based biosensor was previously developed by our research group for caffeic acid determination26 and in this work it is optimised and used for chlorogenic acid determination, chlorogenic acids being a major active compound found in coffee.The electrochemical behaviour of caffeine was studied by cyclic voltammetry using different architectures of the WE2 accomplished by immobilising one of the following: (a) Pt nanoparticles, (b) RGO, (c) Nafion, (d) RGO adsorbed onto Pt-NPs and (e) RGO covered by Nafion. The choice of these modifiers will be motivated in the following section.
The influence of surface modifiers on sensor performance
In a first step Pt nanoparticles, RGO and Pt-NPs/RGO were used as modifiers. Cyclic voltammetry was used to assess the possible improvement of the surface of the carbon working electrode after modification. The redox electrochemical properties of the modified electrodes were tested using 5 mM [Fe(CN)6]3−/4− solution and it was observed that the obtained cyclic voltammograms had the same trend as for the previously developed laccase-based biosensor26 (Fig. 2). It is worth mentioning, that the peak potential separation decreased from ΔE = 170 mV (in the case of the unmodified screen-printed carbon electrode) to ΔE = 92 mV (for Pt-NPs/RGO modified electrodes) and to ΔE = 77 mV (for Pt-NPs modified electrodes).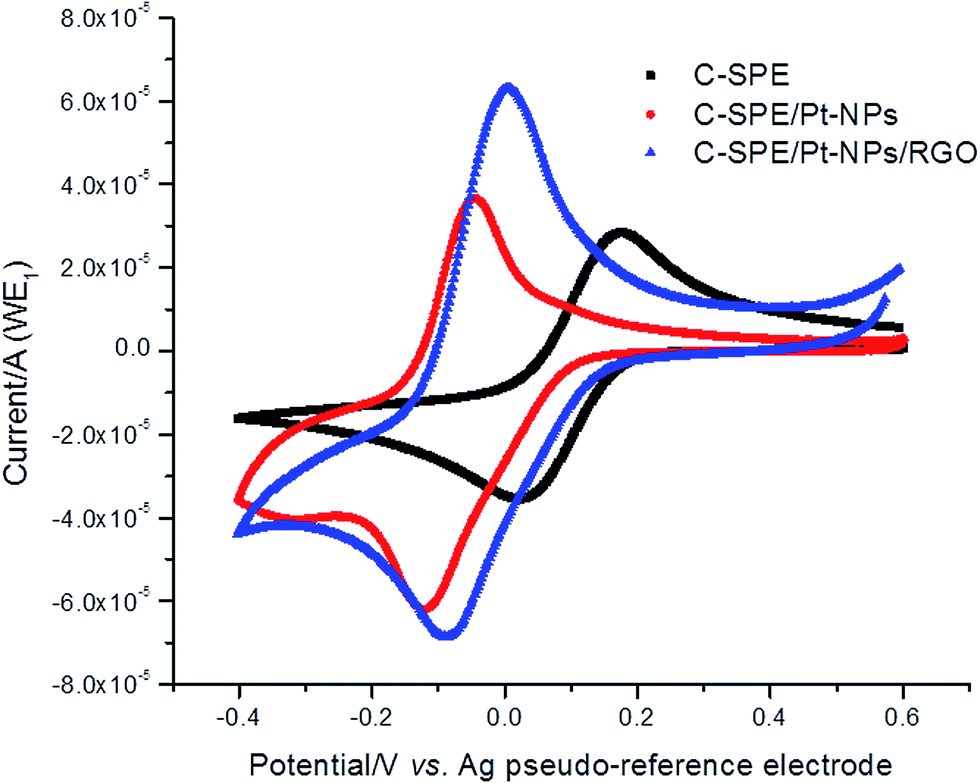 | ||
| Fig. 2 Cyclic voltammograms of 5 mM [Fe(CN)6]3−/4− in 0.1 M KCl on bare carbon screen-printed electrode (black), on carbon modified with Pt-NPs (red) and carbon modified with Pt-NPs and RGO (blue); scan rate of 50 mV s−1. | ||
However, when standards (chlorogenic acid and caffeine) were tested by cyclic voltammetry at the modified electrodes it was noticed that, in the case of caffeine, the modification of the electrode has not improved the transport between the analyte and the electrode surface and rather capacitive phenomena manifested through increased area under the very large oxidation peak, especially in the case of the carbon electrode modified Pt-NPs and RGO (see ESI S1†). In the case of the chlorogenic acid the cyclic voltammetry suggests that the surface modification has improved the analytical performances of the WE1, peak separation decreasing to ΔE = 50 mV (for C-SPE/Pt-NPs/RGO/lacc electrode), from an initial ΔE = 110 mV in the case of the unmodified electrode (see ESI S2†). As reported by Brownson et al.35 the peak potential separation (ΔE) depends on the amount of graphene deposited on the surface of the underlying electrode, reduced peak-to-peak separation marking improved electron kinetic ascribed to electrochemically active edge plane sites in “Zone II”. Therefore, the surface of the second working electrode was modified using only RGO, only Nafion 0.05 wt% or RGO and Nafion 0.05 wt%. Casting the electrode with Nafion was inspired by the work of Torres et al.36 and Zhao et al.37 Fig. 3 shows the cyclic voltammograms of 3 mM caffeine in the presence of different modifiers, used to improve the electron transfer rate between the analyte and the surface of WE2. It can be observed that when Nafion 0.05 wt% was adsorbed onto the electrode surface the signal for caffeine reached 41 μA, more than twice in intensity when compared to the signal obtained only on bare carbon screen-printed electrode (Ipa ∼ 19 μA), despite the findings of Zhao et al. that stated that there is no signal at the bare glassy carbon electrode, fact also disapproved by Torres et al. Furthermore, when RGO was deposited onto the surface of the WE2 a higher peak current (Ipa ∼ 32 μA) for caffeine was obtained also, with respect to the unmodified carbon screen-printed electrode. When Nafion 0.05 wt% was casted over the RGO modified carbon electrode, the oxidation current intensity reached 44 μA, thus a synergy between RGO and Nafion being revealed, meaning that the RGO improved the electron transfer of caffeine due to its increased electrical conductivity and the negatively charged Nafion has allowed the caffeine ions to preconcentrate at the electrode film surface.37 Nafion film also prevented the electrode surface fouling and therefore it represents an advantage when determining caffeine, a compound that easily adsorbs at the electrode surface. Peak potential was almost unchanged, ranging from +1.34 V in the case of C-SPE/RGO/Nafion electrode to +1.36 V for C-SPE/RGO electrode, for the unmodified carbon screen-printed electrode (C-SPE) and the Nafion 0.05 wt% modified screen-printed electrode (C-SPE/Nafion) the anodic peak potential being +1.35 V vs. Ag pseudo-reference electrode.
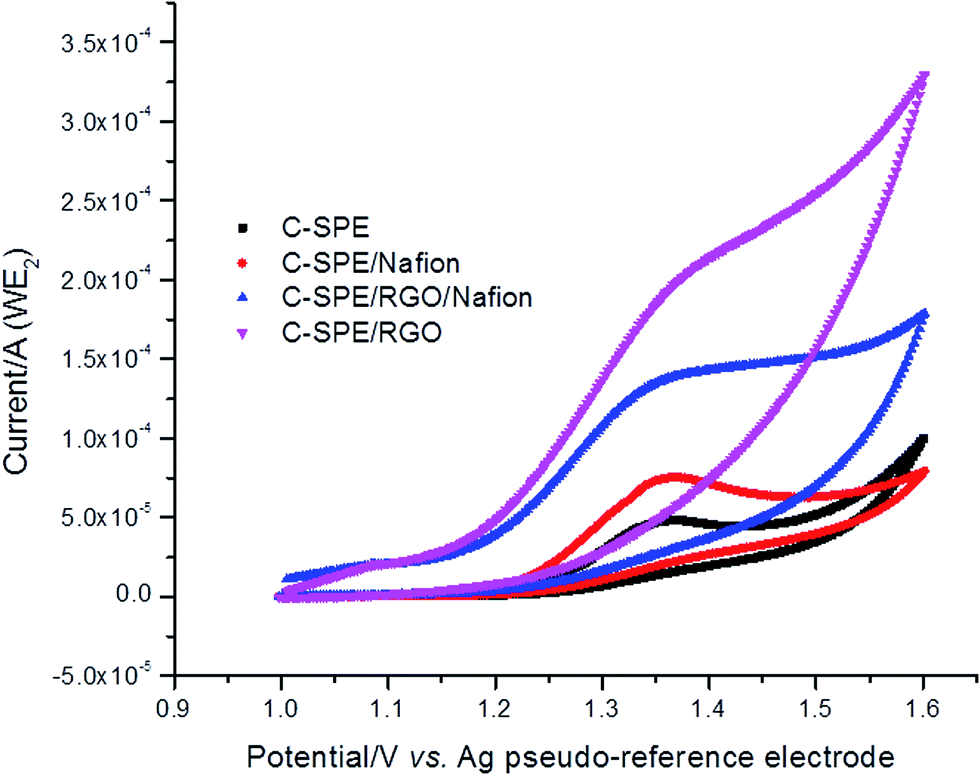 | ||
| Fig. 3 Cyclic voltammograms of 1 mM caffeine in 0.1 M acetate buffer pH 5.00 (with 0.1 M KCl) on bare carbon screen-printed electrode (black), on carbon modified with Nafion 0.05 wt% (red), on carbon modified with RGO (magenta) and on carbon modified with RGO and Nafion 0.05 wt% (blue); scan rate of 50 mV s−1. | ||
Caffeine electrochemistry was characterised by an oxidation peak at around +1.3 V vs. Ag pseudo-reference electrode and no reduction peak on the reverse scan indicating that the electrode process is irreversible.36
In conclusion, in order to obtain the optimum analytical signal the WE1 surface was modified using Pt-NPs, RGO and laccase (C-SPE/Pt-NPs/RGO/lacc), while WE2 surface was modified with RGO and Nafion 0.05 wt% (C-SPE/RGO/Nafion).
Analytical characteristics of the developed sensing array
Firstly the laccase based biosensor was optimised in terms of applied potential and buffer pH, the enzyme loading being kept at 250 mIU. The effect of applied potential on the biosensor response was studied for 10 μM chlorogenic acid in the potential window −200 mV up to +100 mV vs. Ag pseudo-reference electrode (Fig. 4a). As noticed from the figure, the highest sensitivity of the laccase based biosensor was obtained for an applied potential of −50 mV vs. Ag pseudo-reference electrode and therefore it was used for further experiments.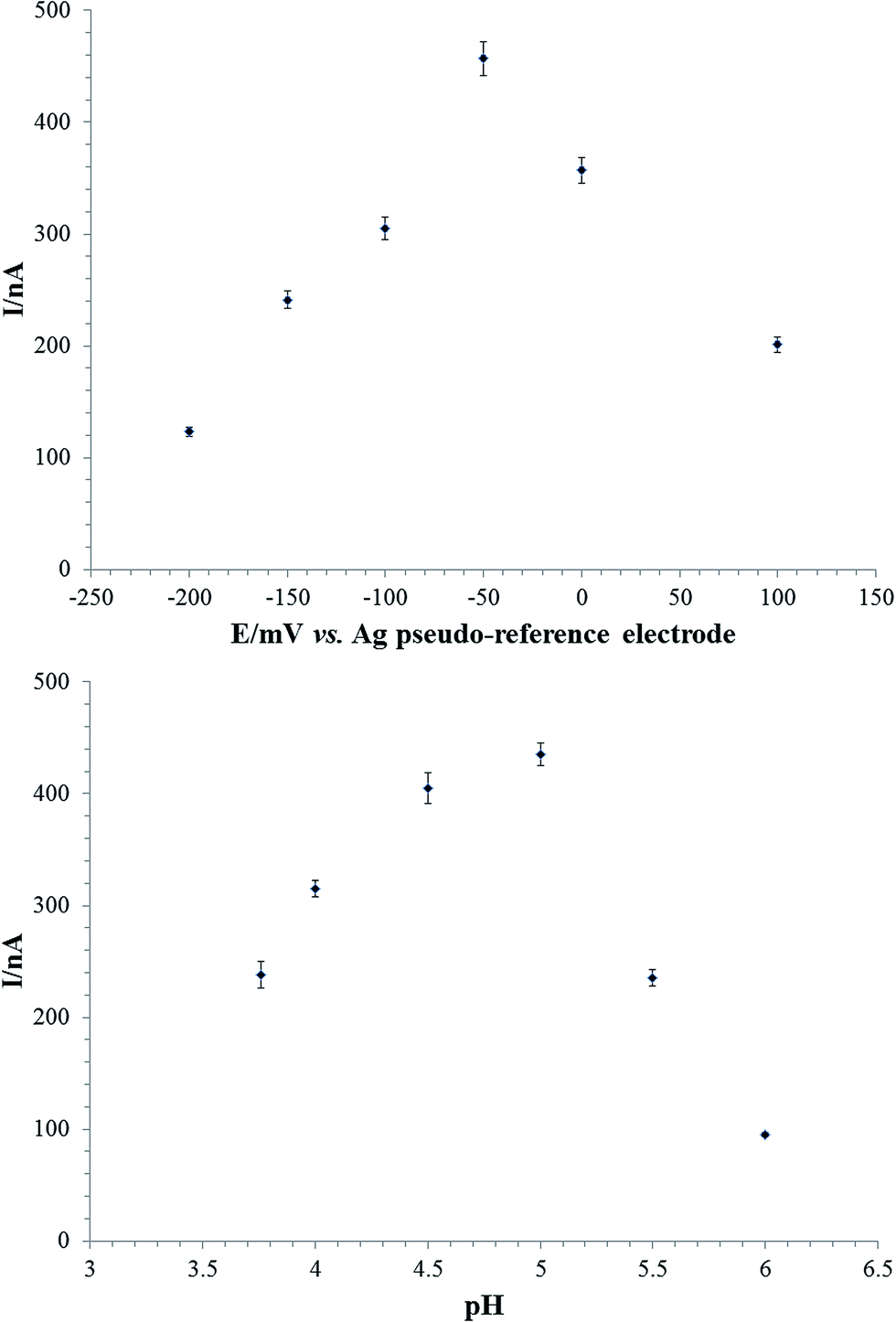 | ||
| Fig. 4 (a) The effect of the applied working potential on the amperometric response of 10 μM chlorogenic acid at C-SPE/Pt-NPs/RGO/lacc in 0.1 M acetate buffer pH 5.00 (with 0.1 M KCl); (b) the effect of pH on the amperometric response of 10 μM chlorogenic acid in 0.1 M acetate buffer pH 5.00 (with 0.1 M KCl), the applied potential being −0.05 V vs. Ag pseudo-reference electrode. | ||
The effect of buffer pH on the amperometric response of the laccase based biosensor for chlorogenic acid was tested in acetate buffer at pH values ranging from 3.76–5.50 (the acetate pH range is given by the acetate pKa that is 4.75) (Fig. 4b). The amperometric response was analysed for 10 μM chlorogenic acid at an applied potential of −50 mV vs. Ag pseudo-reference electrode. Fig. 4b shows a maximum current intensity at pH 5.00, like in our previous paper,26 and therefore this pH value will be further used in our experiments.
Secondly the caffeine sensor was optimised only with respect to applied potential as the working buffer pH was already settled for the laccase based biosensor. Moreover, considering the target of our work, the determination of caffeine and chlorogenic acid from different coffee samples a slightly acidic buffer is very close to the coffee real samples solutions. From Fig. 5 it can be noticed that the optimum working potential for the caffeine determination was +1.3 V vs. Ag pseudo-reference electrode.
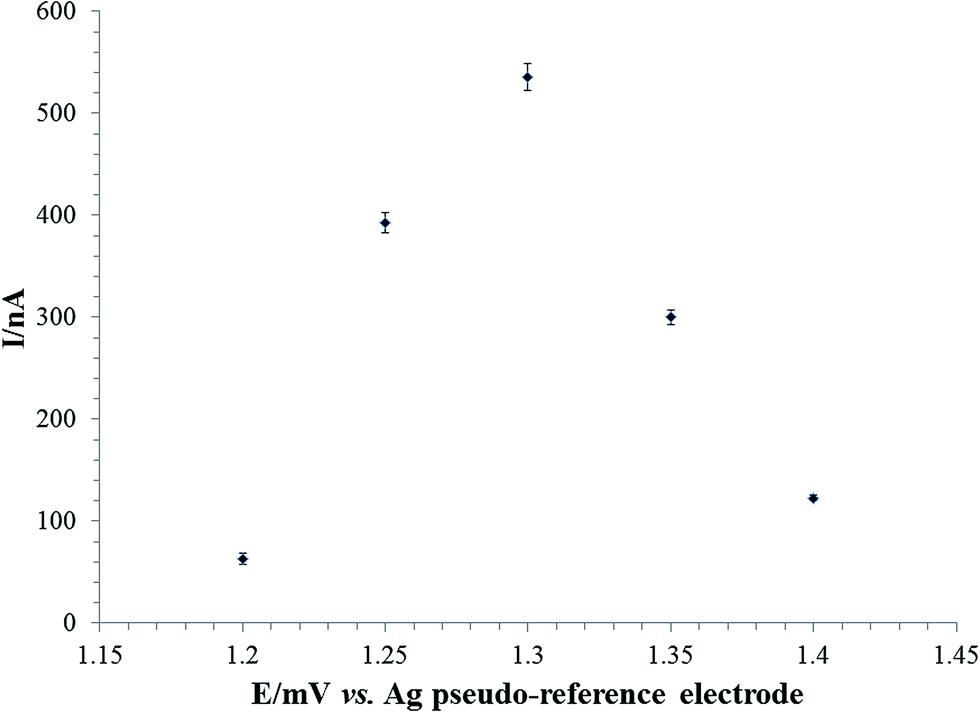 | ||
| Fig. 5 The effect of the applied working potential on the amperometric response of 1 μM caffeine at C-SPE/RGO/Nafion in 0.1 M acetate buffer pH 5.00 (with 0.1 M KCl). | ||
Cyclic voltammetry was used to rationalize on the nature of the electrode process, the effect of scan rate on the electrocatalysis of chlorogenic acid and caffeine being measured. Fig. S4a and b (see ESI S4†) depicts the relationships between the peak current intensity and the square root of the scan rate for 0.5 mM chlorogenic acid at the C-SPE/Pt-NPs/RGO/lacc and for 1 mM caffeine at the C-SPE/RGO/Nafion in 0.1 M acetate buffer pH 5.00 (with 0.1 M KCl). It can be concluded that Ip is directly proportional with v1/2 for both investigated compounds at the surface of the developed sensor array meaning that the caffeine oxidation and the chlorogenic acid reduction are diffusion controlled processes (with a slight adsorptive contribution).
The concomitant detection of chlorogenic acid and caffeine at the developed sensor surface was tested under the optimal working conditions and the obtained responses are depicted in S3 as well as the calibration plots for both tested compounds in Fig. 6a and b (see ESI S3†). The linear relationships between the concentrations of chlorogenic acid and caffeine, respectively, and current responses are given in Table 1 together with other significant analytical parameters. The linear response range for chlorogenic acid is 2.91 × 10−6 to 2.64 × 10−5 M with a regression equation of ip (μA) = 0.017–0.024C (μM); for caffeine it is 2.90 × 10−7 to 2.58 × 10−6 M with a regression equation of ip (μA) = −0.257 + 1.383C (μM). The limits of detection (LoD) were calculated as 3.3 × intercept standard error/sensitivity. The obtained limits of detection for both tested compounds are comparable to the ones found in literature, (Table 2). The dynamic linear range obtained by us for caffeine is narrower when compared to those reported in literature, this limiting factor being ascribed to the nature of the surface of the electrode used in amperometry for detecting caffeine. This compromise was also motivated by the development of a dual sensor array, able to perform chlorogenic acid and caffeine analysis in real coffee sample. However the linearity domain for caffeine was well suited when the sensor array was challenged against coffee samples.
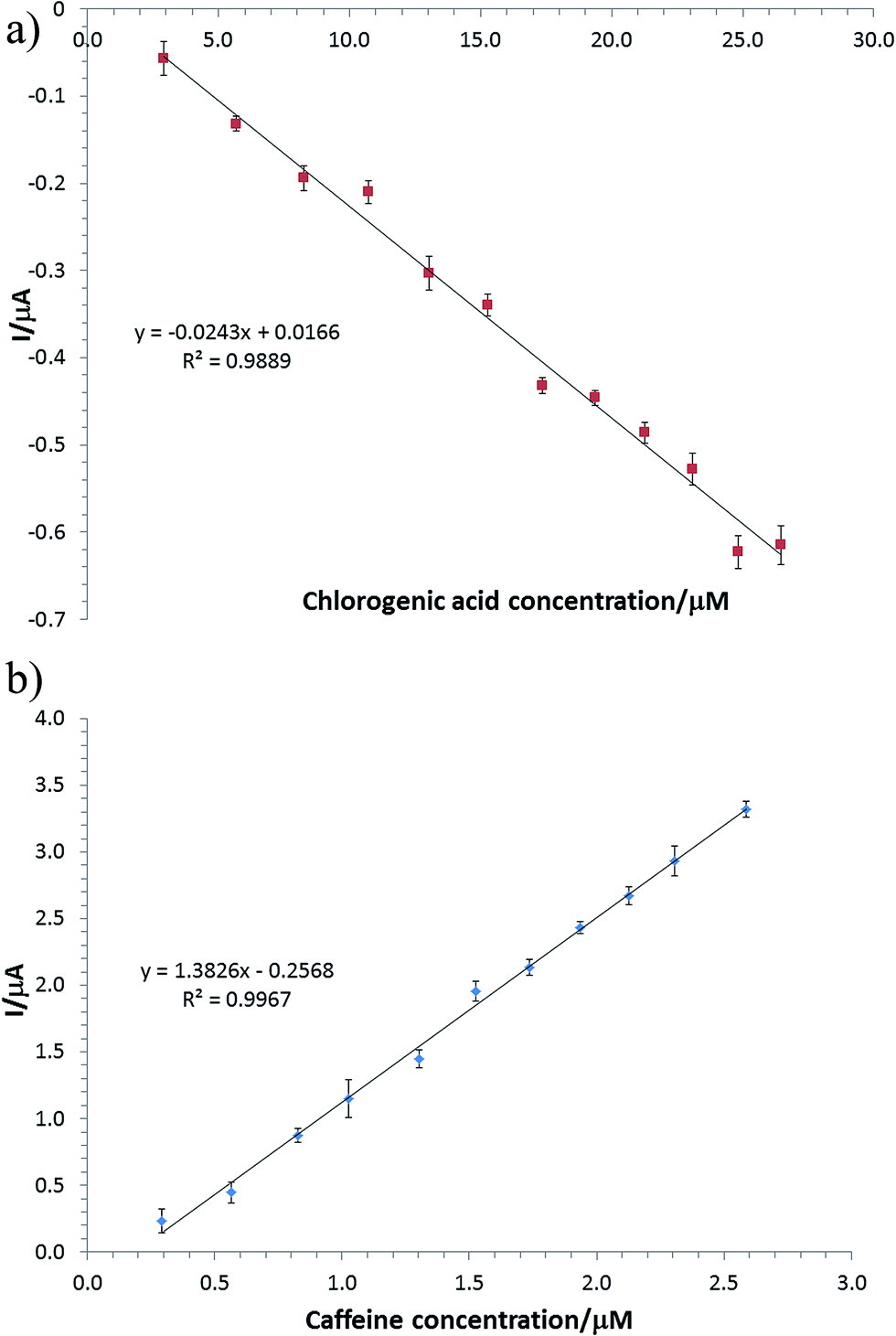 | ||
| Fig. 6 (a) Calibration curve for chlorogenic acid at the C-SPE/Pt-NPs/RGO/lacc, working potential: −0.05 V vs. Ag pseudo-reference electrode in 0.1 M acetate buffer pH 5.00 (with 0.1 M KCl); (b) calibration curve for caffeine at C-SPE/RGO/Nafion, working potential +1.3 V vs. Ag pseudo-reference electrode in 0.1 M acetate buffer pH 5.00 (with 0.1 M KCl). | ||
| Compound | Sensitivity (μA μM−1) | Linear range (μM) | LoD (μM) | R2 |
|---|---|---|---|---|
| Chlorogenic acid | 0.02 | 2.91–26.47 | 2.67 | 0.9889 |
| Caffeine | 1.38 | 0.29–2.58 | 0.22 | 0.9967 |
| Standard | Modified electrode | Technique | Eapp (V) | Linear working range (μM) | LoD (μM) | Ref. |
|---|---|---|---|---|---|---|
| Chlorogenic acid | Ir-BMI.PF6-PPOx | SWV | — | 3.48–49.50 | 0.91 | 38 |
| Tyrosinase alumina sol–gel/sonogel-carbon electrode | Amperometry | −0.30 vs. Ag/AgCl reference electrode | 5.00–30.00 | 0.61 | 39 | |
| Lacc–CS–MWCNT | Amperometry | −0.20 vs. Ag/AgCl reference electrode | 0.79–6.71 | 0.16 | 40 | |
| Au-SPE-lacc-Nafion | Amperometry | −0.20 vs. Ag pseudo-reference electrode | 3.00–15.00 | 2.80 | 41 | |
| C-SPE/Pt-NPs/RGO/lacc-array | Amperometry | −0.05 vs. Ag pseudo-reference electrode | 2.91–26.47 | 2.67 | This work | |
| Caffeine | ENGR–NCNTs/GCE | SWV | — | 0.06–50.00 | 0.02 | 17 |
| MIS/MWCNTs–VTMS/GCE | DPV | — | 0.75–40.00 | 0.22 | 42 | |
| PAHNSA/GCE | SWV | — | 0.06–40.00 | 0.13 | 23 | |
| GO-Nafion/GCE | DPV | — | 0.40–80.00 | 0.20 | 37 | |
| C-SPE/RGO/Nafion-array | Amperometry | +1.30 vs. Ag pseudo-reference electrode | 0.29–2.58 | 0.22 | This work |
The reproducibility of the developed sensor array is important and therefore assessed. Seven repetitive simultaneous measurements were carried out at the surface of the array under the optimal working conditions and the relative standard deviations were calculated 6.4% for chlorogenic acid and 8.7% for caffeine. The storage stability of the developed sensor array was studied by measuring the current response during storage in the optimised conditions. The sensor array maintained the initial response, in terms of current intensity, during the first two weeks, while after two months a response of about 80% from its initial value was obtained for the laccase-based electrode and approximately 90% for C-SPE/RGO/Nafion, respectively.
Analysis of real coffee samples
Chlorogenic acid and caffeine was determined from several coffee samples. The chlorogenic acid and caffeine amounts found in the real samples were calculated using the equations of the calibration curves for the two tested compounds. The results obtained with the sensor array were plotted against the results obtained by HPLC and fitted with a R2 = 0.9939 for chlorogenic acid and R2 = 0.9700 for caffeine, respectively.Table 3 reveals higher values for chlorogenic acid using the developed array than the values obtained by HPLC due to the fact that it is very well-known that the laccase based biosensors are not specific and they are also responding to other polyphenolic compounds that could be also present in the coffee samples.
| Sample | Chronoamperometry (μM) | HPLC (μM) | ||
|---|---|---|---|---|
| Chlorogenic acid | Caffeine | Chlorogenic acid | Caffeine | |
| Carte Noir Café Long Délicat | 92.67 | 9.75 | 123.20 | 7.05 |
| Jacobs Espresso | 256.46 | 13.89 | 236.45 | 10.99 |
| Segafredo Intermezzo | 247.90 | 10.59 | 176.50 | 10.95 |
| Davidoff Espresso | 169.22 | 26.74 | 143.28 | 26.69 |
| Grandia Classic | 297.28 | 16.32 | 246.28 | 15.55 |
| Davidoff Rich Aroma | 223.21 | 15.77 | 177.67 | 15.85 |
Conclusions
This work provides a dual sensor array for the simultaneous determination of chlorogenic acid and caffeine, the major constituents found in coffee samples. Chlorogenic acid was determined at the surface of a C-SPE/Pt-NPs/RGO/lacc at −0.05 V vs. Ag pseudo-reference electrode while caffeine was determined at the surface of a C-SPE/RGO/Nafion at +1.30 V vs. Ag pseudo-reference electrode. The developed sensor array could represent a simple alternative to the more elaborate spectrophotometric and chromatographic methods for the individual determination of chlorogenic acid and caffeine. The easy to use, feasibility and accessibility of the developed dual sensor array pave the way to the construction of an analytical tool able to screen both chlorogenic acid and caffeine content from coffee samples this being of great importance for food safety and food and beverages industry.Acknowledgements
This work was supported by two grants of the Romanian National Authority for Scientific Research, CNCS – UEFISCDI, Project no. PN-II-RU-TE-2011-3-0037 and Project no. PN-II-PT-PCCA-2011-3.1-1809.Notes and references
- D. Komes and A. Bušić, Processing and Impact on Antioxidants in Beverages, 2014, p. 25 Search PubMed.
- I. A. Ludwig, L. Sánchez, M. P. De Peña and C. Cid, Food Res. Int., 2014, 61, 67 CrossRef CAS PubMed.
- M. Pérez-Martínez, B. Caemmerer, M. P. De Peña, C. Cid and L. W. Kroh, J. Agric. Food Chem., 2010, 58, 2958 CrossRef PubMed.
- S. E. W. Opitz, S. Smrke, B. A. Goodman and C. Yeretzian, Processing and Impact on Antioxidants in Beverages, 2014, p. 253 Search PubMed.
- S. J. V. Vicente, Y. S. Queiroz, S. L. D. Gotlieb and E. A. F. S. Torres, Braz. Arch. Biol. Technol., 2014, 57, 110 CrossRef CAS PubMed.
- T. Niseteo, D. Komes, A. Belščak-Cvitanović, D. Horžić and M. Budeč, Food Chem., 2012, 134, 1870 CrossRef CAS PubMed.
- M. F. Matei, R. Jaiswal and N. Kuhnert, J. Agric. Food Chem., 2012, 60, 12105 CrossRef CAS PubMed.
- C. E. Mills, M. J. Oruna-Concha, D. S. Mottram, G. R. Gibson and J. P. E. Spencer, Food Chem., 2013, 141, 3335 CrossRef CAS PubMed.
- W. Mullen, B. Nemzer, B. Ou, A. Stalmach, J. Hunter, M. N. Clifford and E. Combet, J. Agric. Food Chem., 2011, 59, 3754 CrossRef CAS PubMed.
- A. N. Gloess, B. Schönbächler, B. Klopprogge, L. D'Ambrosio, K. Chatelain, A. Bongartz, A. Strittmatter, M. Rast and C. Yeretzian, Eur. Food Res. Technol., 2013, 236, 607 CrossRef CAS.
- Y. Yardim, J. Food Sci., 2012, 77, C408 CrossRef CAS PubMed.
- Y. Yardim, E. Keskin and Z. Şentürk, Talanta, 2013, 116, 1010 CrossRef CAS PubMed.
- W. D. J. R. Santos, M. Santhiago, I. V. P. Yoshida and L. T. Kubota, Anal. Chim. Acta, 2011, 695, 44 CrossRef CAS PubMed.
- S. C. Fernandes, S. K. Moccelini, C. W. Scheeren, P. Migowski, J. Dupont, M. Heller, G. A. Micke and I. C. Vieira, Talanta, 2009, 79, 222 CrossRef CAS PubMed.
- S. K. Moccelini, A. Spinelli and I. C. Vieira, Enzyme Microb. Technol., 2008, 43, 381 CrossRef CAS PubMed.
- M. J. Glade, Nutrition, 2010, 26, 932 CrossRef CAS PubMed.
- L. Jiang, Y. Ding, F. Jiang, L. Li and F. Mo, Anal. Chim. Acta, 2014, 833, 22 CrossRef CAS PubMed.
- O.-W. Lau, S.-F. Luk, O.-M. Cheng and T. P. Y. Chiu, Analyst, 1992, 117, 777 RSC.
- P. D. Tzanavaras and D. G. Themelis, Anal. Chim. Acta, 2007, 581, 89 CrossRef CAS PubMed.
- S. Casal, M. Beatriz Oliveira and M. A. Ferreira, Food Chem., 2000, 68, 481 CrossRef CAS.
- J. W. Dove, G. Buckton and C. Doherty, Int. J. Pharm., 1996, 138, 199 CrossRef CAS.
- J. Zen, Y.-S. Ting and Y. Shih, Analyst, 1998, 123, 1145 RSC.
- M. Amare and S. Admassie, Talanta, 2012, 93, 122 CrossRef CAS PubMed.
- W. Y. H. Khoo, M. Pumera and A. Bonanni, Anal. Chim. Acta, 2013, 804, 92 CrossRef CAS PubMed.
- C. A. B. De Maria, L. C. Trugo, R. F. A. Moreira and M. Petracco, Food Chem., 1995, 52, 447 CrossRef CAS.
- S. A. V. Eremia, I. Vasilescu, A. Radoi, S.-C. Litescu and G.-L. Radu, Talanta, 2013, 110, 164 CrossRef CAS PubMed.
- X. Guo, B. Liang, J. Jian, Y. Zhang and X. Ye, Microchim. Acta, 2014, 181, 519 CrossRef CAS.
- M.-J. Song, J.-H. Kim, S.-K. Lee, D.-S. Lim, S. W. Hwang and D. Whang, Electroanalysis, 2011, 23, 2408 CrossRef CAS.
- L. Baptista-Pires, B. Pérez-López, C. C. Mayorga-Martinez, E. Morales-Narváez, N. Domingo, M. J. Esplandiu, F. Alzina, C. M. Sotomayor-Torres and A. Merkoçi, Biosens. Bioelectron., 2014, 61, 655 CrossRef CAS PubMed.
- A. Bragaru, E. Vasile, C. Obreja, M. Kusko, M. Danila and A. Radoi, Mater. Chem. Phys., 2014, 146, 538 CrossRef CAS PubMed.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019 CrossRef CAS PubMed.
- V. Cristea, C. Deliu, B. Oltean, A. Brummer, C. Albu and G. L. Radu, Acta Horticulture, 2009, 843, 157 Search PubMed.
- V. Sharma, A. Gulati, S. D. Ravindranath and V. Kumar, J. Food Compos. Anal., 2005, 18, 583 CrossRef CAS PubMed.
- D. A. C. Brownson, L. J. Munro, D. K. Kampouris and C. E. Banks, RSC Adv., 2011, 1, 978 RSC.
- A. Carolina Torres, M. M. Barsan and C. M. A. Brett, Food Chem., 2014, 149, 215 CrossRef CAS PubMed.
- F. Zhao, F. Wang, W. Zhao, J. Zhou, Y. Liu, L. Zou and B. Ye, Microchim. Acta, 2011, 174, 383 CrossRef CAS.
- S. C. Fernandes, S. K. Moccelini, C. W. Scheeren, P. Migowski, J. Dupont, M. Heller, G. A. Micke and I. C. Vieira, Talanta, 2009, 79, 222 CrossRef CAS PubMed.
- H. Zejli, J. L. Hidalgo-Hidalgo de Cisneros, I. Naranjo-Rodriguez, B. Liu, K. R. Temsamani and J. L. Marty, Anal. Chim. Acta, 2008, 612, 198 CrossRef CAS PubMed.
- M. Diaconu, S. C. Litescu and G. L. Radu, Sens. Actuators, B, 2010, 145, 800 CrossRef CAS PubMed.
- S. C. Litescu, S. A. V. Eremia, A. Bertoli, L. Pistelli and G.-L. Radu, Anal. Lett., 2010, 43, 1089 CrossRef CAS.
- W. D. J. R. Santos, M. Santhiago, I. V. P. Yoshida and L. T. Kubota, Sens. Actuators, B, 2012, 166–167, 739 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra14464c |
| This journal is © The Royal Society of Chemistry 2015 |