
A two step protocol for the synthesis of highly substituted benzobicyclo[2.2.2]octadienone derivatives from 2-methoxyphenols†
Santhosh Kumar Chittimalla*,
Chennakesavulu Bandi and
Sireesha Putturu
Medicinal Chemistry Department, AMRI Singapore Research Centre, 61 Science Park Road, #05-01, The Galen, Science Park III, Singapore 117525, Singapore. E-mail: santhosh.chittimalla@amriglobal.com; chemcsk@gmail.com; Fax: +65-63985511
First published on 19th December 2014
Abstract
A rapid process for the synthesis of functionally rich benzobicyclo[2.2.2]octadienone derivatives has been realized through oxidative dimerization of commercially/readily available 2-methoxyphenols followed by an aromatization sequence.
Benzobicyclo[2.2.2]octadienyl derivatives are most often perceived as substrates for acid1a–f or base1g catalyzed or photochemical2 rearrangement reactions in the context of mechanistic studies. On the other hand, a variety of benzobicyclo[2.2.2]octene derivatives were synthesized and screened for their diverse biological activities. For example, benzobicyclo[2.2.2]octene derivatives were utilized as calcium channel blockers3 and cannabinoid receptor ligands4 (Fig. 1). 1-Piperidinobenzobicyclo[2.2.2]octene was synthesized as a rigid analogue of phencyclidine and was found to be selectively bound to σ receptor and not PCP receptor.5 2-Aminobenzobicyclo-[2.2.2]octene-2-carboxylic acids6a and 2-aminobenzobicyclo-[2.2.2]octenes6b were synthesized as conformationally restricted aminoacids and conformationally defined adrenergic agents respectively (Fig. 1).
 | ||
| Fig. 1 Few medicinally important benzobicyclo[2.2.2]octene derivatives. | ||
We envisaged that employing masked o-benzoquinones (MOBs, 2),11 in a tandem Michael addition and aromatization reaction sequence12 with oxygen or nitrogen nucleophiles as Michael donors should furnish corresponding O- or N-arylated products. During these investigations we noticed that imidazole did not react with in situ generated MOB 2a (from 2-methoxyphenol, 1a). In this reaction, the only product isolated was dimer 3a with no traces of desired N-arylimidazole I (Scheme 1A). Consequently, an alternative procedure to gain access to N-arylimidazole was envisioned. Thus, a solution of dimer 3a (1.0 equiv.) and imidazole (1.0 equiv.) in o-dichlorobenzene (o-DCB) was heated under microwave irradiation at 150 °C for 1 h (Scheme 1B). It was expected that Michael addition of imidazole to in situ generated MOB 2a (from retro-Diels–Alder reaction of dimer 3a)11d would produce Michael adduct which further aromatizes to yield the desired N-arylimidazole I. However, to our surprise this procedure delivered benzobicyclo[2.2.2]octadienone 4a in 78% isolated yield. We reasoned this observation to the aromatization of cyclohexenone unit in dimer 3a under the influence of imidazole. Interestingly, when dimer 3a was treated with HCl in methanol (or dioxane)12a,e or HClO4–SiO2 (ref. 12d) in 1,2-dichloroethane at room temperature, only starting material was recovered. On the other hand, the above reactions at elevated temperature (60–70 °C) generated complex reaction mixture with traces of benzobicyclo[2.2.2]octadienone 4a as indicated by 1H NMR of crude reaction mixture.13 Moreover, when a 1,4-dioxane solution of dimer 3a was refluxed in the presence of pTSA, 20% of product 4a was produced along with uncharacterized product mixture. As a result, the serendipitous imidazole mediated aromatization reaction to us was a valuable outcome and worth pursuing.
 | ||
| Scheme 1 Attempted synthesis of N-arylimidazole I. Unexpected formation of benzobicyclo[2.2.2]octadienone 4a. | ||
Literature survey indicates several innovative methodologies for the synthesis of benzobicyclo[2.2.2]octene systems. For instance, recently enantioselective synthesis of benzobicyclo[2.2.2]octenone derivatives was reported via cyclobutanone cleavage followed by intramolecular alkene insertion mediated by nickle7 or rhodium8 catalysis. Other protocols include interesting transformation of 2-cyanomethylbenzaldehyde9 and Diels–Alder reaction10 between cyclohexa-1,3-dienes and benzynes to give benzobicyclo[2.2.2]-octene derivatives. Believing aromatization of MOB dimers as a valuable alternative to the already existing literature protocols for the synthesis of benzobicyclo[2.2.2]octene derivatives, we wish to report initial results in this endeavour.
At the outset, a methanolic solution of 2-methoxyphenols 1a–k was separately treated with (diacetoxyiodo)benzene to furnish dimers 3a–k in high yields via Diels–Alder self-dimerization of in situ generated MOBs 2a–k (Scheme 2). Interestingly, aromatization reaction of MOB dimers with other nucleophilic organic bases such as DABCO14a and DBU14b,c were not effective in performing the above conversion. On the other hand, DMAP14d and N–Me imidazole14d,e could perform the above transformation to some extent (Scheme 1B) but were less effective than imidazole. Therefore, we reasoned the success with imidazole to its amphoteric nature (Fig. 2). We eventually identified that heating a o-DCB solution of 1.0 equiv. of dimer 3a, 2.5 equiv. of imidazole, under microwave irradiation at 150 °C for 1.5 h was optimal to affect complete conversion of dimer 3a providing benzobicyclo[2.2.2]octadienone 4a in 94% isolated yield (Scheme 1B). Under exactly same conditions, when benzimidazole was used in place of imidazole, benzobicyclo[2.2.2]octadienone 4a was produced in 40% yield indicating that imidazole is superior for this conversion (Scheme 1B). Though the reaction mechanism is not completely understood, initially it appeared to us that imidazole adds to the activated alkene followed by elimination of catalyst by abstraction of bridgehead hydrogen by another equivalent of imidazole to generate Int-I which subsequently looses methanol to provide the final aromatized product (Fig. 2, path A). To our surprise, under the optimized reaction condition dimer 3b furnished product 4b in 85% yield with no traces of product 5. All our attempts to capture the possible imidazole-MOB dimer adduct by lowering the temperature (160, 150 or 100 °C) failed and provided only recovery of starting material along with variable amounts of aromatized product. Consequently, an alternative reaction path B is proposed for the observed results, where imidazole first acts as base and then as a proton donor (Fig. 2, path B). To this end, the feasibility of base mediated aromatization was tested. Thus, when a THF solution of dimer 3a was treated with KOtBu at room temperature gave a complex reaction mixture from which 10% of product 4a could be isolated indicating reaction path B might be reasonable pathway for the imidazole mediated reaction.
 | ||
| Scheme 2 Synthesis of highly functionalized benzobicyclo[2.2.2]octadienones 4. | ||
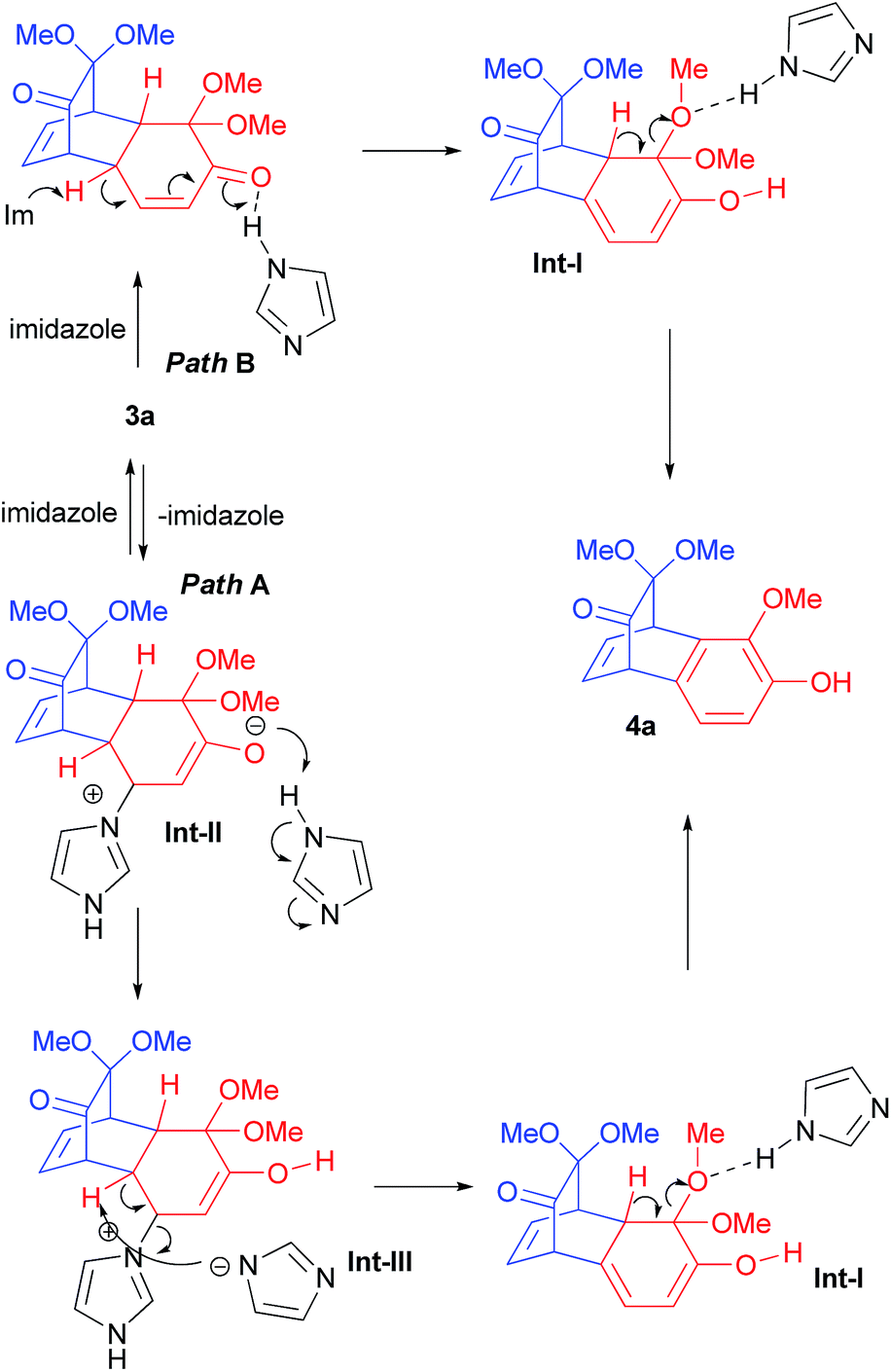 | ||
| Fig. 2 Possible reaction pathways for the formation of benzobicyclo[2.2.2]octadienone derivatives 4 from MOB dimers 3. | ||
Next, a o-DCB solution of dimer 3b–3d and 3g were separately subjected to microwave irradiation under different conditions (150 or 180 °C and 1.5 or 2.5 equiv. of imidazole, Scheme 2) and eventually found that 180 °C and 2.5 equiv. of imidazole is optimal and general condition to provide the desired benzobicyclo[2.2.2]octadienone 3b–3d and 3g in good yields. Consequently, the above condition was applied to remaining MOB dimers. Later, to extend the scope of the reaction methodology and to further understand if reaction follows path B, several other dimers, 3f, 3h, and 3j bearing substituents with varying size (at Michael addition prone site) were utilized.
Gratifyingly, all the dimers employed generated the desired products in high yields demonstrating that reaction path B might be operating. To our delight, under these conditions even sensitive functionalities like ketal- (3i, j), bromo (3b), and methoxy (3k) were stable and effectively furnished the corresponding highly functionalized benzobicyclo[2.2.2]octadie-none derivatives 4b, 4i–k in very good yields.
Subsequent objective was to study transformations of obtained benzobicyclo[2.2.2]octadienone derivatives. Our research group has been exploring the use of MOBs as substrates in other than Diels–Alder reaction, thereby greatly expanding their potential use in synthesis.12 As a result, we wished to convert the newly obtained bicyclo[2.2.2]octenone system fused 2-methoxyphenols 4 to novel MOBs. Accordingly, benzobicyclo[2.2.2]octadienone derivatives 4a, 4c, 4d and 4g were subjected to oxidation with (diacetoxyiodo)benzene in methanol at room temperature for 45 min. As expected novel MOBs 5a, 5c, 5d and 5g were furnished in near quantitative yield (Scheme 3).
 | ||
| Scheme 3 Synthesis of bicyclo[2.2.2]octenone fused MOBs 5. | ||
Benzoquinoxalinobarralenes are well-known substrates in the study of photochemical rearrangement reactions.15 Benzobicyclo[2.2.2]octadienones 4 can be viewed as protected form of benzobicyclo[2.2.2]octadiendiones which are precursors for the synthesis of benzoquinoxalinobarralenes. Consequently, we decided to prepare few benzoquinoxalinobarralenes from compound 4. At the outset, when products 4a and 4h were treated with 2 N aq. H2SO4 at 50 °C for 16 h, benzobicyclo[2.2.2]octadiendiones 6a and 6h were obtained in 82% and 92% yields, respectively (Scheme 4). Furthermore, when these dienediones 6a and 6h were treated with 1,2-diaminobenzene derivatives 7–9, highly functionalized benzoquinoxalinobarralenes 10–13 were obtained in up to 95% yields (Scheme 4).
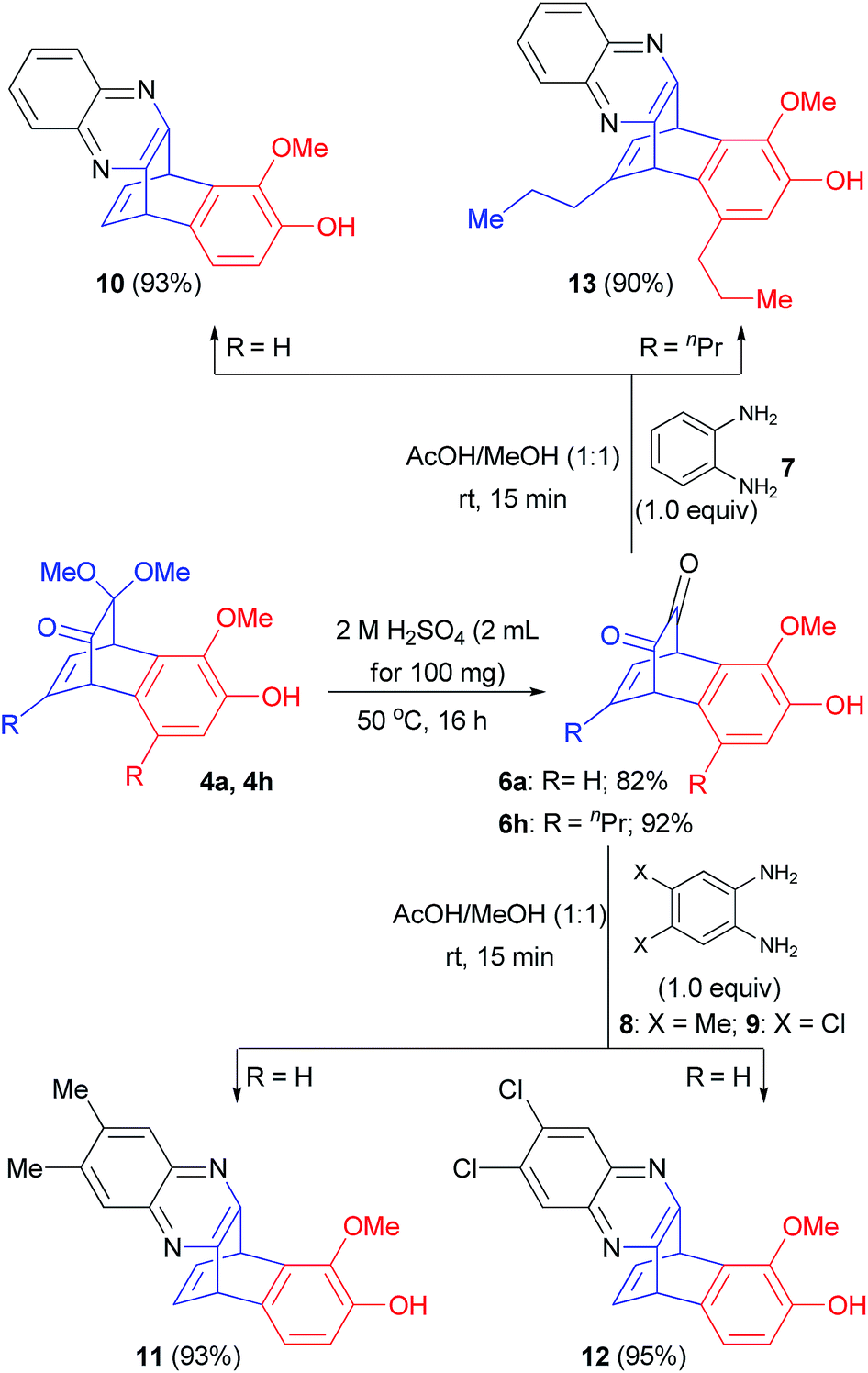 | ||
| Scheme 4 Preparation of benzoquinoxalinobarralenes 10–13. | ||
Next goal was to ensure if we could further functionalize these benzobicyclo[2.2.2]octadienone systems. For such a study, benzobicyclo[2.2.2]octadienone 4a was chosen (Scheme 5). At the outset, free hydroxyl group in benzobicyclo[2.2.2]octadienone 4a was easily modified by alkylating it with methyliodide, allylbromide and bromomethylacetate to give corresponding alkylated products 14, 15 and 16 in >90% isolated yield. Subsequently, O-arylation could be efficiently performed under Chan–Lam coupling16 conditions furnishing product 17 in 86% yield. Olefinic moiety could be effortlessly reduced under hydrogenation conditions providing benzobicyclo[2.2.2]octenone system 18 in 93% yield. Besides, when bromination reaction was performed on compound 4a, mono-brominated product 19 was obtained in 91% yield. Such halogenated products can be further utilized in cross-coupling reactions to introduce additional functionalities on these benzobicyclo[2.2.2]octadienone products. To this end, phenolic group in compound 19 was reacted with Michael acceptor ethylpropiolate to yield (E)-ethyl 3-phenoxyacrylate derivative 20 in 82% isolated yield. With a pre-installed bromo substitution in compound 20, intramolecular Heck type coupling17 was carried out to generate benzofuran fused bicyclo[2.2.2]octadienone derivative 21 in acceptable yield. Subsequently, to test the facial selectivity in nucleophilic addition on compound 4a, vinylmagnesium bromide was used as nucleophilic reagent. To a THF solution of compound 5 at −78 °C when vinylmagnesium bromide (4.0 equiv.) was added, products 22a and 22b were obtained as inseparable mixture of isomers. The ratio of products was determined from 1H NMR of crude reaction mixture. After the purification, this inseparable mixture of isomers up on 2D NOESY experiment indicated that the major isomer is product 22a. It is well-documented that the 1,2-addition of alkenyl anions to α,γ-unsaturated ketones is kinetically controlled.18a Consequently, the facial selectivity is most often governed by steric and electronic factors.18a Perhaps, the ketone face towards the aromatic ring is electronically rich and thus the vinyl nucleophile attacks from the opposite face providing the observed selectivity.18b
 | ||
| Scheme 5 Further functionalization of benzobicyclo[2.2.2]octadienone 4a. | ||
Conclusions
In conclusion, we have described a simple approach to highly functionalized benzobicyclo[2.2.2]octadienone derivatives from commercially/readily available 2-methoxyphenols in two steps. The obtained products were utilized in the synthesis of novel masked o-benzoquinones, benzoquinoxalinobarralenes and a benzobicyclo[2.2.2]octenone derivative to name a few. Moreover, the products obtained here contain handy substituents for further derivatization to elaborate the structural diversity and synthesize new molecular libraries. We believe that the methodology presented here is a valuable alternative to the already existing literature protocols for the synthesis of benzobicyclo[2.2.2]octadiene derivatives (Scheme 5).General procedure for the imidazole mediated aromatization reaction
A solution of dimer 3 (1.0 equiv.) and imidazole (2.5 equiv.) in o-dichlorobenzene (1 mL for 100 mg of dimer 3) was sealed and subjected to microwave irradiation [Biotage® microwave synthesizer] at appropriate temperature for an appropriate time to give corresponding benzobicyclo[2.2.2]octadienone products after purification by flash column chromatography using ethyl acetate–hexanes.4a
1H NMR (400 MHz, CDCl3) δ 6.90 (d, J = 8.0 Hz, 1H), 6.78 (d, J = 8.0 Hz, 1H), 6.67–6.58 (m, 2H), 4.64 (dd, J = 6.0, 2.4 Hz, 1H), 4.28 (dd, J = 5.6, 2.0 Hz, 1H), 3.90 (s, 3H), 3.41 (s, 3H), 3.26 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 195.8 (C), 148.3 (C), 143.5 (C), 133.3 (CH), 131.6 (CH), 131.1 (C), 128.8 (C), 121.1 (CH), 114.1 (CH), 91.8 (C), 62.4 (CH3), 56.8 (CH), 50.4 (CH3), 49.9 (CH3), 42.2 (CH). HRMS (ESI+): calcd for C15H16NaO5 [M + Na] 299.0890; found 299.0899.4h
1H NMR (400 MHz, CDCl3) δ 6.64 (s, 1H), 6.14 (dd, J = 6.4, 1.6 Hz, 1H), 4.49 (d, J = 6.4 Hz, 1H), 4.26 (d, J = 1.6 Hz, 1H), 3.87 (s, 3H), 3.40 (s, 3H), 3.24 (s, 3H), 2.55 (t, J = 7.6 Hz, 2H), 2.28–2.14 (m, 2H), 1.62–1.42 (m, 4H), 0.93 (t, J = 7.2 Hz, 3H), 0.85 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 196.8 (C), 147.5 (C), 145.7 (C), 141.3 (C), 134.7 (C), 131.9 (C), 126.8 (C), 125.4 (CH), 114.5 (CH), 92.3 (C), 62.4 (CH3), 57.8 (CH), 50.5 (CH3), 49.8 (CH3), 41.7 (CH), 35.9 (CH2), 34.2 (CH2), 24.6 (CH2), 20.2 (CH2), 13.8 (CH3), 13.4 (CH3). HRMS (ESI+): calcd for C21H28NaO5 [M + Na] 383.1829; found 383.1837.12
1H NMR (400 MHz, DMSO-d6) δ 9.44 (s, 1H), 8.17 (s, 1H), 8.13 (s, 1H), 7.25–7.15 (s, 2H), 7.08 (d, J = 8.0 Hz, 1H), 6.60 (d, J = 8.0 Hz, 1H), 5.52 (dd, J = 5.6, 1.6 Hz, 1H), 5.27 (dd, J = 6.0, 1.6 Hz, 1H), 3.79 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 159.4 (C), 158.9 (C), 148.6 (C), 143.8 (C), 138.9 (CH), 137.2 (CH), 137.1 (C), 134.4 (C), 133.0 (C), 131.5 (C), 131.4 (C), 129.0 (CH), 128.9 (CH), 120.3 (CH), 114.1 (CH), 61.0 (CH3), 50.1 (CH), 45.3 (CH). HRMS (ESI+): calcd for C19H13Cl2N2O2 [M + H] 371.0349; found 371.0359.17
1H NMR (300 MHz, CDCl3) δ 7.35–7.27 (m, 2H), 7.11–7.02 (m, 1H), 6.98–6.90 (m, 3H), 6.84 (d, J = 7.8 Hz, 1H), 6.69–6.59 (m, 2H), 4.82–4.74 (m, 1H), 4.36–4.29 (m, 1H), 3.91 (s, 3H), 3.42 (s, 3H), 3.29 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 195.9 (C), 157.7 (C), 147.9 (C), 147.8 (C), 134.2 (CH), 133.2 (C), 132.3 (C), 130.6 (CH), 129.6 (CH × 2), 122.7 (CH), 120.3 (CH), 120.1 (CH), 117.1 (CH × 2), 91.8 (C), 61.5 (CH3), 57.2 (CH), 50.4 (CH3), 50.1 (CH3), 41.9 (CH). HRMS (APCI+): calcd for C21H21O5 [M + H] 353.1384; found 353.1376.19
1H NMR (400 MHz, CDCl3) δ 7.27–7.28 (m, 1H), 7.16 (s, 1H), 6.63–6.58 (m, 2H), 5.80 (br s, 1H), 4.65 (dd, J = 4.4, 2.8 Hz, 1H), 4.24–4.21 (m, 1H), 3.93 (s, 3H), 3.40 (s, 3H), 3.25 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 195.3 (C), 145.5 (C), 144.0 (C), 133.7 (CH), 131.14 (C), 131.11 (CH), 129.6 (C), 123.6 (CH), 107.7 (C), 91.6 (C), 62.1 (CH3), 56.4 (CH), 50.4 (CH3), 50.1 (CH3), 42.0 (CH). HRMS (ESI+): calcd for C15H14BrO5 [M + H] 353.0030; found 353.0033.Notes and references
-
(a) K. Schötz, T. Clark and P. von Ragué Schleyer, J. Am. Chem. Soc., 1988, 110, 1394–1405 CrossRef
; (b) G. L. Grunewald, D. E. Walters and T. R. Kroboth, J. Org. Chem., 1978, 43, 3478–3481 CrossRef CAS
-
(a) C.-C. Liao and Y.-G. Ueng, Tetrahedron Lett., 1990, 31, 1563–1566 CrossRef CAS
- K. Hilpert, F. Hubler and D. Renneberg, PCT Int. Appl. WO 2010/046855A1, 2010
- M. Muthuppalantappan, K. Sukeerthi, G. Balasubramanian, S. Gullapalli, N. K. Joshi, S. Narayanan and P. V. Karnic, PCT Int. Appl. WO 2008/053341A2, 2008
- R. M. Moriarty, L. A. Enache, L. Zhao, R. Gilardi, M. V. Mattson and O. Prakash, J. Med. Chem., 1998, 41, 468–477 CrossRef CAS PubMed
-
(a) G. L. Grunewald, S. H. Kuttab, M. A. Pleiss and J. B. Mangold, J. Med. Chem., 1980, 23, 754–758 CrossRef CAS
- L. Liu, N. Ishida and M. Murakami, Angew. Chem., Int. Ed., 2012, 51, 2485–2488 CrossRef CAS PubMed
- L. Souillart and N. Cramer, Chem. Sci., 2014, 5, 837–840 RSC
- M. Panasiewicz, T. Zdrojewski, K. Chrulski, A. Wojtasiewicz and A. Jończyk, ARKIVOC, 2009, VII, 98–110 CrossRef
-
(a) T. Kaicharla, S. S. Bhojgude and A. T. Biju, Org. Lett., 2012, 14, 6238–6241 CrossRef CAS PubMed
-
(a) L. Pouysegu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235–2261 CrossRef CAS PubMed
-
(a) S. K. Chittimalla, R. Kuppusamy and C. Bandi, Synlett, 2014, 1991–1996 CrossRef CAS PubMed
-
(a) A. A. Patchett and B. Witkop, J. Org. Chem., 1957, 1477–1484 CrossRef CAS
-
(a) S. C. Stolze and M. Kaiser, Molecules, 2013, 18, 1337–1367 CrossRef CAS PubMed
-
(a) J. H. Boyer and K. G. Srinivasan, J. Chem. Soc., Perkin Trans. 1, 1976, 1583–1589 RSC
-
(a) D. M. T. Chan, K. L. Monaco, R.-P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933–2936 CrossRef CAS
-
(a) J. T. Link, Org. React., 2002, 60, 157–534 CAS
-
(a) W.-J. Juo, T.-H. Lee, W.-C. Liu, S. Ko, S. K. Chittimalla, C. P. Rao and C.-C. Liao, J. Org. Chem., 2007, 72, 7992–8004 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for all new compounds. See DOI: 10.1039/c4ra14219e |
| This journal is © The Royal Society of Chemistry 2015 |