
Hollow and mesoporous ZnTe microspheres: synthesis and visible-light photocatalytic reduction of carbon dioxide into methane†
Muhammad Fahad Ehsanab,
Muhammad Naeem Ashiqa and
Tao He*a
aLaboratory of Nanosystem and Hierarchical Fabrication, National Center for Nanoscience and Technology, Beijing 100190, China. E-mail: het@nanoctr.cn; Fax: +86-10-62656765; Tel: +86-10-82545655
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 15th December 2014
Abstract
Hollow and mesoporous microstructures have been found to be an attractive class of materials due to their superior physical properties and potential applications. In the present work, a hydrothermal method has been used to synthesize hollow and mesoporous ZnTe hierarchical microspheres. The as-synthesized microspheres are characterized by a variety of techniques, including X-ray diffraction, scanning electron microscopy, energy dispersive X-ray spectroscopy, transmission electron microscopy, high resolution transmission electron microscopy, X-ray photoelectron spectroscopy and ultraviolet-visible spectroscopy. Nitrogen adsorption–desorption measurements and the curve of relative pore size distribution via Brunauer–Emmett–Teller analysis confirm the existence of mesopores in the obtained nanomaterials. Different reaction parameters such as NaOH concentration, reaction temperature and time, and concentration of the tellurium precursor have been studied and the detailed growth mechanism has been proposed. These hollow and mesoporous microspheres are also used for the photoreduction of carbon dioxide into methane under visible-light illumination (λ ≥ 420 nm) with a solar energy conversion efficiency of 0.072%.
Introduction
Recent research in the field of materials chemistry has publicized that the physical and chemical properties of inorganic materials, in general, are strongly dependent on the morphology, particle size and structure.1–6 Therefore, the systematic control over these factors to efficiently tune their properties in a desirable way is a challenging task in the field of modern synthetic chemistry.7,8 In the past decades, several fruitful efforts have been made to investigate some complex superstructures, especially, three-dimensional (3D) hierarchical superstructures assembled by nanostructured building blocks such as nanorods, nanoparticles, nanoplates, and nanoribbons owing to their unique structures and properties.9–11 The synergistic effects of these nanometer-sized primary building blocks on the synthesis of micro-sized uniform hierarchical superstructures are desirable due to their unique properties. These 3D hierarchical architectures have grabbed considerable attention because of their widespread applications in the fields of efficient catalysis, energy storage, drug delivery, nanoreactors, sensors, and the like.12–17 Among these hierarchical architectures, hollow and mesoporous microstructures have received special attention because of their novel characteristics due to higher surface-to-volume ratio and special shape.18–23 Different methodologies involving the soft templates (e.g., macromolecules, microemulsions, micelles, oil droplets and gas bubbles)24–28 and hard templates (e.g., silica spheres, carbon, monodispersed polymer latex and reducing metal nanoparticles)29–32 have already been developed for the fabrication of hollow structured inorganic materials. However, the use of these templates carries several disadvantages related to their high cost and complicated synthetic procedures. Moreover, the removal of these templates is also very hard and often affects the integrity and quality of the final product, which limits its practical applications.33 Therefore, it is still a challenging task for the material scientists to develop a facile, cheap and clean synthetic procedure for the fabrication of hierarchical inorganic materials with definite shape and size.3D hierarchical architectures of many II–VI, III–VI and IV–VI semiconductors including ZnS,34 CdS,35 CdSe,36 PbTe37 and In2S3/In2O3 (ref. 38) have been extensively studied in the past decades. However, only a few reports are on ZnTe semiconductor. ZnTe is a p-type II–VI semiconductor with small direct band gap energy of ∼2.26 eV at 300 K and Bohr exciton radius of ∼6.2 nm. It is an attractive material because of its applications in optoelectronic devices, such as green light-emitting diodes and solar cells.39,40 Different structures of ZnTe including microspheres, nanoflowers, nanodots, nanorods, quasispheres and tetrahedron nanocrystals have been synthesized using different synthetic techniques.41–44 It is noted that all these techniques involve some toxic/poisonous organic chemicals that are environmentally hazardous. In addition, so far there are no reports on the preparation or growth mechanism of hollow and mesoporous ZnTe microstructures, neither its application in photoreduction of carbon dioxide (CO2), while the conversion of CO2 into value-added chemicals is a hot topic.
Here, we reported template-free hydrothermal synthesis of uniform hollow and mesoporous ZnTe microspheres, rather than the solid ZnTe microsphere as reported previously,41 The as-synthesized microspheres are formed in the presence of NaOH at a relatively low temperature of 160 °C. The growth mechanism of these microspheres has been investigated via changing different reaction parameters, such as concentration of NaOH and reaction precursors, reaction time and temperature. It is found that NaOH concentration plays a critical role in the formation of these microspheres and only irregular shaped ZnTe nanocrystals can be obtained in the absence of NaOH. In addition, these as-synthesized microspheres of ZnTe have been used for the visible-light induced (λ ≥ 420 nm) photoreduction of CO2, which results in the formation of methane (CH4) as the major product.
Experimental
Chemicals
All the chemicals used for the synthesis of ZnTe microspheres are of analytical grade and were used without further purification. Zinc nitrate (Zn(NO3)2·6H2O) (≥99%) and NaOH (≥96%) were purchased from Sinopharm Chemicals, absolute ethanol (≥99.7%) was purchased from Beijing Chemical Works, and sodium tellurite (Na2TeO3) (≥99.5%) and sodium borohydride (NaBH4) (≥96%) were bought from Alpha Aesar. Milli-Q water was used during all the synthesis.Synthesis of hollow and mesoporous ZnTe microspheres
In a typical procedure, 1.6 g of NaOH (0.33 M) was dissolved in 120 mL of Milli-Q water. Then 0.1485 g of Zn(NO3)2·6H2O (4 mM) and 0.1064 g of Na2TeO3 (1 mM) were added to this clear solution under constant stirring. Finally, 0.7566 g of NaBH4 (0.17 M) was added to the above mixture and was kept for stirring until a clear transparent solution was formed, followed by being transferred into a 150 mL Teflon-lined stainless steel autoclave. The autoclave was held at 160 °C for 24 h and then cooled in air at room temperature. The dark reddish brown precipitates of ZnTe were collected via centrifugation, and were washed several times with Milli-Q water and finally with absolute ethanol. The obtained ZnTe precipitates were then dried in an oven at 60 °C for 8 h under vacuum.Characterization
The crystal structure of as-synthesized products has been studied by X-ray diffraction (XRD) using Bruker D8 focus diffractometer with Ni-filtered Cu-Kα radiation. The diffractograms were recorded in the 2θ range of 20 to 80° at a scan rate of 0.1° min−1. Lambda 750 UV-visible spectrophotometer was used to investigate the optical properties using wavelength in the range of 250–800 nm at room temperature with BaSO4 as a reference. The hollow structure, morphology and composition of the synthesized ZnTe have been analyzed by using Hitachi S4800 field-emission scanning electron microscopy (FESEM) and Tecnai G2 F20 U-TWIN transmission electron microscopy (TEM), and both of them were equipped with energy dispersive X-ray (EDX) spectrometer. The pore size and mesoporous texture of the microspheres were determined by Brunauer–Emmett–Teller (BET) analysis (Micromeritics, tristar II 3020). X-ray Photoelectron Spectroscopy (XPS) has been used to determine the valence band position by using ESCALAB 250Xi X-ray photoelectron spectrometer.Photocatalytic reduction of CO2
The photoreduction system has been described previously.45 Highly purified CO2 (≥99.999%) gas was bubbled through the photocatalyst dispersed in Milli-Q water (10 mg/80 mL) in the quartz reactor attached with the photoreaction system to produce a mixture of CO2 and water. The positive pressure of CO2 inside the photoreaction system was maintained at 25 kPa and was assured before the irradiation that there was no leakage in the system. Before the visible-light irradiation (λ ≥ 420 nm), wet CO2 was allowed to flow through the photoreaction system loaded with the ZnTe microsphere for around 40 minutes to ensure the complete removal of air from the system, followed by the maximum adsorption of the gas molecules on the active sites of the photocatalyst. After CO2-flow was stopped, the 300 W xenon lamp with a 420 nm cut-off filter was turned on and the product yield as a function of irradiation time was determined for a total time period of 8 h. An approximate distance of 14.5 cm was maintained between the light source and the solution surface in the photoreactor. The system was constantly recycled with tap water (15 °C) through a water recycling machine during the entire course of the photoreaction so as to maintain the temperature.The CH4 produced as a result of photoreduction of CO2 via photocatalysts was identified by an Agilent 7890A gas chromatograph (GC) via an automated gas valve using helium (He) as the carrier gas. The GC was equipped with two (front and back) flame ionization detectors (FID) and one thermal conductivity detector (TCD). Four different control experiments were carried out to confirm that the CH4 being produced in the present work is from the photocatalysts under visible light and not from any impurities, i.e., blank reactor with and without irradiation, dark experiment with the photocatalyst, and with the same experimental conditions but using N2 instead of CO2. No CH4 was produced during these controlled reactions, indicating that the CH4 was indeed from the photoconversion of CO2 using the photocatalysts under visible-light irradiation and not from any impurities. The solar energy conversion efficiency can be determined by using the converted amount of energy contained in CH4 relative to the irradiated solar power (λ ≥ 420 nm). The latter was measured to be 1.3 W with ILT 950 spectrodiometer (International Light Technologies), and the former was calculated by using the amount of CH4 produced upon photoreduction of CO2 and the energy density of CH4 (55.6 MJ kg−1). To minimize the influence from the side reactions during the photoreduction (such as photocorrosion of the catalyst), the reaction time of half an hour was used for the calculation. The detailed calculation was presented in the ESI.†
Results and discussion
Structural analysis
The ZnTe is formed via a series of chemical reactions. Zinc hydroxide (Zn(OH)2) is produced once Zn(NO3)2 is added to an aqueous solution of NaOH, which is dissolved by the excess of NaOH in the reaction mixture, and is dissociated into respective zinc and hydroxyl ions. When Na2TeO3 is added into the above alkaline solution, it is dissociated into tellurite ions, which can be reduced to Te and Te2− by a strong reducing agent like NaBH4. The ZnTe is obtained at an optimum pH and reaction temperature via the reactions between the ions of zinc(II) and tellurium(II).The structural phase and purity of the as-synthesized products were studied by the XRD pattern. A typical XRD pattern of the as-synthesized ZnTe at 160 °C for 24 h is shown in Fig. 1a. All the diffraction peaks can be assigned to the cubic ZnTe with a lattice constant of 6.1273 Å, which is in good agreement with the available standard pattern (powder diffraction file (PDF) no. 15-0746). No impurity peaks (i.e., any metallic tellurium or zinc) were observed, which also confirms the purity of the product. The narrow and sharp peaks from the XRD patterns indicate the high crystalline nature of the obtained ZnTe. The purity and composition of the products are also confirmed by the EDX analysis. The EDX spectrum of single ZnTe is shown in Fig. 1b. Almost all the peaks in the spectrum correspond to tellurium and zinc with an atomic ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2, supporting the impurity free synthesis of the product. Two small peaks of carbon and copper appear in the EDX spectrum and are ascribed to come from the instrument and the sample preparing procedure, i.e., copper grid used for the sample preparation.
:1.2, supporting the impurity free synthesis of the product. Two small peaks of carbon and copper appear in the EDX spectrum and are ascribed to come from the instrument and the sample preparing procedure, i.e., copper grid used for the sample preparation.
 | ||
| Fig. 1 (a) XRD pattern and (b) EDX spectrum of the as-synthesized ZnTe microspheres at 160 °C for 24 h. | ||
The texture of the as-synthesized ZnTe has been investigated by the nitrogen adsorption–desorption analysis. Fig. 2 shows the nitrogen sorption isotherms and the corresponding pore size distribution curve of the as-prepared product. The BET sorption isotherms are found to be of reverse “S” shape, which displays type IV isotherm with H-3 hysteresis loop (at 0.2 < P/P0 < 1) in accordance with the IUPAC classification.46 This indicates the characteristics of the mesoporous materials with pore size ranging from 2 to 50 nm.47,48 The corresponding pore size distribution can also be confirmed by adsorption and desorption analysis using Barrett–Joyner–Halenda (BJH) method, as shown in the inset of Fig. 2. There are four characteristic peaks that indicate the existence of mesopores in the hollow microspheres. The BJH curve also shows the presence of some macropores of diameter <∼80 nm, while with very low density. The respective average pore diameter of BJH adsorption and desorption is around 16.3 and 20.9 nm, which are in good agreement with the SEM results discussed later. The BET surface area of the obtained ZnTe microspheres calculated from nitrogen adsorption is ∼5 m2 g−1 and the pore volume is 0.019 cm3 g−1. The relatively low surface area is due to the very highly crystalline nature and larger size of the microspheres. So the as-prepared ZnTe microspheres are mesoporous in texture.
 | ||
| Fig. 2 Typical N2 gas adsorption–desorption isotherm of ZnTe microspheres. Inset is the corresponding pore size distribution curve. | ||
Morphological analysis
Fig. 3 represents the morphological images of hydrothermally synthesized mesoporous ZnTe microspheres at 160 °C for 24 h in terms of SEM. From the low magnification image, it is evident that the ZnTe is uniformly composed of the microspheres with an average diameter of ∼1.6 μm with the absence of any other significant morphology (Fig. 3a). It can clearly be seen from the zoom-in image that the microspheres are hollow in structure, as shown by the arrow heads for some broken spheres with rough surface (Fig. 3b). It can be confirmed from the high magnification images that these microspheres are mesoporous in texture, specifically as indicated in Fig. 3d. The detailed mechanism for the formation of these 3D structures is discussed later. | ||
| Fig. 3 (a) Low-magnification, (b) zoom-in, (c) high-magnification SEM images of ZnTe hollow microspheres, and (d) that of a single microsphere. | ||
To further investigate the surface and structure of the products, the as-synthesized hollow and mesoporous ZnTe microspheres have been characterized with TEM images, as shown in Fig. 4. These results are found to be in good agreement with the above discussed SEM data. It can be clearly seen from Fig. 4a that for a single ZnTe microsphere, it is hollow in structure with a thick shell and rough surface, as already illustrated by the SEM images. Fig. 4b represents the selected area electron diffraction (SAED) pattern for the microspheres with diffractional rings, indicating that these hollow microspheres are polycrystalline in nature.
 | ||
| Fig. 4 (a) TEM micrograph for a single ZnTe microsphere, (b) SAED pattern, and (c) HRTEM image of the microsphere. | ||
Fig. 4c is the high resolution TEM (HRTEM) image taken from the rectangular fringe of a single microsphere in Fig. 4a. These data confirm the polycrystalline nature of these hollow microspheres too, representing the lattice interplanar spacing from different crystal planes of cubic ZnTe, as indicated in Fig. 4c. It also provides additional evidence for the ultra high crystalline nature of these 3D hierarchical hollow and mesoporous microspheres.
Growth mechanism
The detailed growth mechanism of these hollow and mesoporous microspheres has been investigated. It is suggested that NaOH plays a critical role in the formation of the hierarchical structures synthesized at 160 °C for 24 h. The effect of the concentration of NaOH has been shown in Fig. 5. No microspheres, i.e., only the aggregates of ZnTe nanocrystals with irregular morphology were formed in the absence of NaOH (Fig. 5a). The morphology of ZnTe hardly changes with increase in NaOH to 0.1 M (Fig. 5b), while some microspherical structures started appearing as the NaOH concentration increased to 0.2 M (Fig. 5c). When the NaOH concentration further increased to 0.33 M, the uniformly distributed hollow and mesoporous ZnTe microspheres were formed, as shown in Fig. 5d. The effect of NaOH might be attributed to the fact that it plays a crucial role during the nucleation process of these microstructures. | ||
| Fig. 5 FESEM micrographs of as-synthesized ZnTe with different concentrations of NaOH: (a) 0, (b) 0.1, (c) 0.2 and (d) 0.33 M. Inset is TEM image of the aggregates of ZnTe crystals. | ||
Another important factor that plays a significant role in the formation of these hierarchical microspheres is the reaction time. To figure out this formation mechanism, time-dependent reactions were carried out and their corresponding SEM and EDX results have been shown in Fig. 6 and 7, respectively. In the first half an hour of the reaction, only the tellurium nuclei in the form of rods were observed (Fig. 6a), for which the composition was confirmed by the EDX spectrum (Fig. 7a). The peak of Si in the EDX spectra (Fig. 7) is attributed to the Si substrate used for the sample preparation. The nucleation of this tellurium continues with increase in the reaction time to 1 h, while some of them change into spherical tellurium oxide (Fig. 6b). The elemental composition of one of the spheres was confirmed by the EDX analysis as shown in Fig. 7b, which supports the previous claim. It can be seen from Fig. 6c that the spheres of tellurium oxide became larger and more uniform with an increase in the reaction time to 2 h, whereas less rod-like tellurium based structures were observed. Again, the elemental composition of the product was confirmed by the EDX data (Fig. 7c).
 | ||
| Fig. 6 FESEM images of the obtained product prepared with different reaction time: (a) 0.5, (b) 1, (c) 2, (d) 4, (e) 6, (f) 12 and (g) 24 h. | ||
 | ||
| Fig. 7 EDX spectra of as-synthesized ZnTe microspheres with different reaction time. | ||
This nucleation process is followed by Ostwald ripening, where the zinc from Zn(OH)2 first dissolved in the excess of aqueous NaOH solution, re-depositing on to the large spherical and rod-like crystals of tellurium oxides, as indicated by the black outline of rods and spheres in the schematic illustration in Fig. 8. Accordingly, the bi-structural ZnTe was formed, i.e., rod-like structures and microspheres. This mechanism can be endorsed on the basis of the EDX spectrum of the sample shown in Fig. 7c, in which a small peak of zinc appeared for the first time after two-hour reaction and then increased with further increase in the reaction time to four hours, as shown in the Fig. 7d.
 | ||
| Fig. 8 Schematic illustration of the formation mechanism of hollow and mesoporous ZnTe hierarchical microspheres. | ||
Once ZnTe was formed, the peak corresponding to zinc became stable as shown in the Fig. 7(e and f). After 4 hours, the large rods were broken into small ones, followed by their self-assembly/dispersion on the spheres (Fig. 6d). These small rods then penetrated into the spheres as a result of the inward diffusion process as shown in Fig. 6e and 8, resulting in the release of oxygen gas and, thus, eventually making the spheres hollow and mesoporous in texture (Fig. 6f). The quality and uniformity of these hollow and mesoporous microspheres were improved with further increase in the reaction time and finally after 24 hours uniform hollow and mesoporous ZnTe microspheres are formed (Fig. 6g). The schematic illustration for the formation of these hollow and mesoporous microspheres are shown in Fig. 8.
The concentration of tellurium precursor (i.e., Na2TeO3) also plays an important role in controlling the size of the microspheres. The microspheres with different concentration of tellurium precursor, with respect to zinc, have been synthesized (Fig. 9). It is found that when tellurium is in equimolar ratio with zinc (i.e., 1:1), the size of these microspheres is around 3.5 to 4 μm, as shown in Fig. 9a. While the size of the microspheres decreased almost by 50% (Fig. 9b) when the tellurium concentration decreased to 25%. This may be explained by the already proposed mechanism. In case of high concentration of tellurium, more ZnTe rods could undergo inward diffusion process, leading to a larger size and vice versa.
 | ||
| Fig. 9 FESEM images of ZnTe microspheres with different concentration of Na2TeO3: (a) 4 and (b) 1 mM. | ||
The reaction temperature is another essential parameter that plays an important role in the formation of the microspheres. For the detailed investigation, these microspheres were synthesized at different reaction temperatures and the results are shown in Fig. 10. At a low temperature of 120 °C, the temperature was not high enough for complete reduction and the inward diffusion of tellurium based rod-like structures. Thus, both the structures of spheres and rods were visible at such a low temperature (Fig. 10a). As the temperature increased to 140 °C, the yield of the microspheres became relatively large, while the quality of the product in terms of uniformity was still not good (Fig. 10b). Homogeneous and uniform hollow and mesoporous microspheres were formed at a reaction temperature of 160 °C (Fig. 10c), which was found to be the optimum temperature in this study. As the temperature increased to 180 °C, it resulted in the nucleation of relatively large ZnTe crystals along with the microspheres (Fig. 10d). At a further high temperature of 200 °C, only large ZnTe crystals could be observed (Fig. 10e), which might be due to agglomeration at such an elevated temperature.
 | ||
| Fig. 10 FESEM images of ZnTe microspheres synthesized at different reaction temperature: (a) 120, (b) 140, (c) 160, (d) 180 and (e) 200 °C. | ||
Alignment of band energy levels
The photocatalytic properties of a catalyst are strongly dependent on the alignment of its energy levels, which can be determined in terms of valence band position and band gap energy. The XPS spectroscopy has been employed to calculate the valence band position of ZnTe. The work function of the XPS instrument in the present case was 4.62 eV and the result is shown in Fig. 11a. The inset of Fig. 11a represents zoom-in plot of the measured valence band position, which is 0.49 eV (0.51 V against standard hydrogen electrode (SHE)). | ||
| Fig. 11 (a) Valence band edge from the XPS analysis with zoom-in plot as the inset, (b) UV-visible spectrum with Tauc plot of the as-synthesized ZnTe microspheres as the inset. | ||
UV-visible diffuse reflectance spectroscopy was used to study the optical properties and the band gap of these microspheres. The Kubelka–Munk function was employed to convert the diffuse reflectance spectrum into the equivalent absorption coefficient.49 Fig. 11b is the UV-visible spectrum of the ZnTe microspheres, which clearly indicates that the ZnTe microspheres exhibit a strong absorption in the green region of the visible light at ∼559 nm. The optical band gap for the hollow and mesoporous ZnTe microspheres was determined to be ∼2.22 eV via the Tauc plot, as shown in the inset of Fig. 11b.
The alignment of energy levels for the microspheres can thus be drawn (Fig. 12). Clearly, the conduction band minimum (−1.71 V vs. SHE) is significantly negative than the redox potential of CO2/CH4 (−0.24 V vs. SHE). So the obtained microspheres are considered suitable photocatalyst for the photocatalytic reduction of CO2 into CH4.
 | ||
| Fig. 12 Alignment of energy levels of synthesized hollow and mesoporous ZnTe microspheres. | ||
Photoreduction of CO2
The as-synthesized hollow and mesoporous ZnTe microspheres (Fig. 3) and ZnTe nanocrystals (Fig. 5a) have been used to photoreduce CO2 into CH4 under visible-light irradiation (λ ≥ 420 nm). No CH4 was observed in all the four controlled experiments (Fig. 13), suggesting that the CH4 produced in this work is the direct result of photoconversion of CO2 over the photocatalysts under visible-light irradiation (λ ≥ 420 nm). As shown in Fig. 12, the electrons can be excited from the valence band of ZnTe into conduction band upon illumination of visible light, leaving holes behind in the valence band. Due to the suitable potential of the conduction band, the photogenerated electrons can reduce CO2 into superoxide radicals (˙CO2−) and reduce H+ to ˙H radicals, which can then react with each other and eventually lead to the formation of CH4.50–52 In addition, the peak intensity increases with time in terms of the GC plots for the reduction of CO2 over hollow and mesoporous microspheres after different time intervals (Fig. S1 in ESI†), indicating the continuous production of CH4. Since the potential of the holes left in the valence band may not be positive enough to oxidize the H2O, they may react with other reducible photoreduction product (such as CH3OH and HCHO) in the solution, as no O2 and such products are detected in this work. So the activity and, specifically, the selectivity could be improved greatly once upon the introduction of hole scavenger into the system. The oxidation of these reducible products may result in the generation of H+, which can participate in the photocatalytic conversion of CO2 into CH4. The H+ can also come from the acidic aqueous solution due to the dissolution of CO2 in water.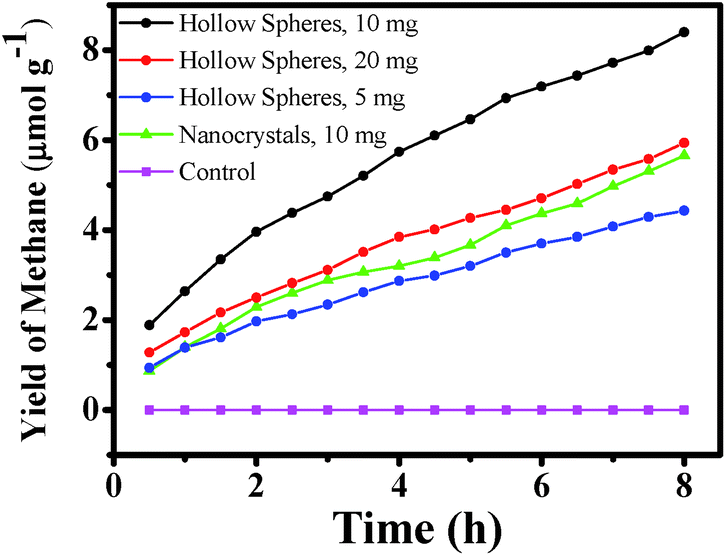 | ||
| Fig. 13 Production of CH4 as a function of time over ZnTe nanocrystals and different amount of microspheres. | ||
The maximum CH4 yield was ∼8.40 μmol g−1 after 8 h for the hollow and mesoporous microspheres, and was ∼5.60 μmol g−1 for the nanocrystals (Fig. 13). By using the converted amount of fuel energy (CH4) in the first half an hour relative to the irradiated solar power (λ ≥ 420 nm), the solar energy conversion efficiency was determined to be ∼0.072% and ∼0.034% for the microspheres and nanocrystals, respectively. The value for the microspheres is found to be similar to or higher than some of the already reported ones in the literature.51–58 The relatively high efficiency for the microspheres may be ascribed to the superior nature of hollow and mesoporous microspheres, such as the low density, large fraction of voids and the capability to store large amount of CO2 compared to the aggregated nanocrystals. Furthermore, the as-synthesized hollow microspheres may have some surface defects and step edges due to their surface roughness caused by the formation of mesopores, which are believed to be the active sites for photoreaction.59 This may also explain their relatively high photocatalytic activity. As for the nanocrystals, the agglomeration could seriously reduce the available catalytic active sites, leading to low yield of CH4 production. In addition, it is noted that the rate of CH4 production is relatively high in the initial stage, while decreases slightly thereafter. It may be due to the destruction of the spherical structures after photoreduction because of the photocorrosion of ZnTe upon the oxidization by photogenerated holes, as manifested in the SEM image of the spheres after photoreduction (Fig. 14). This also results in decreased photocatalytic activity, where a decrease of ∼15% in the CH4 production yield is observed after each run (Fig. S2 in ESI†).
 | ||
| Fig. 14 SEM micrographs of ZnTe microspheres (a) before and (b) after photoreduction. | ||
In addition, the influence of different amount of mesoporous photocatalysts on the production of CH4 has also been investigated (Fig. 13). The maximum yield of CH4 produced by using 5, 10 and 20 mg of the photocatalysts was ∼4.43, 8.40 and 5.94 μmol g−1, respectively. The system with 10 mg of the photocatalyst exhibited the highest yield, followed by the one with 20 mg of the photocatalyst. The system with 5 mg of the photocatalyst was the lowest. All these are closely related to the insufficiency and excess of the catalysts. When the amount of photocatalyst is insufficient, the yield would become lower; while it becomes lower too in the case of excess photocatalyst due to the possible agglomeration/poor dispersion of the large amount of photocatalysts. The difference in the maximum amount of CH4 produced with different amount of photocatalyst also confirms that the CH4 produced in the present case is the direct result of CO2 photoreduction over as-synthesized ZnTe photocatalysts, and not from any organic contaminations.
Conclusions
The hollow and mesoporous ZnTe microspheres have been successfully synthesized via the hydrothermal approach using NaOH as the structure directing agent. Some reaction parameters like the reaction temperature and time are found to play an important role in the formation of such microstructures. The as-synthesized microspheres exhibit relatively high photocatalytic activity for the photoconversion of CO2 into CH4 under visible-light irradiation (λ ≥ 420 nm) mainly due to the characteristic nature of the hollow and mesoporous nanomaterials.Acknowledgements
This work was supported by the Hundred-Talent Program of the Chinese Academy of Sciences.Notes and references
- Y. C. Zhu, T. Mei, Y. Wang and Y. T. Qian, J. Mater. Chem., 2011, 21, 11457–11463 RSC.
- W. Xiao, D. L. Wang and X. W. Lou, J. Phys. Chem. C, 2010, 114, 1694–1700 CAS.
- J. Zhang, L. D. Sun, J. L. Yin, H. L. Su, C. S. Liao and C. H. Yan, Chem. Mater., 2002, 14, 4172–4177 CrossRef CAS.
- X. Y. Li, Z. J. Si, Y. Q. Lei, X. N. Li, J. K. Tang, S. Y. Song and H. J. Zhang, CrystEngComm, 2011, 13, 642–648 RSC.
- L. Xu, C. L. Lu, Z. H. Zhang, X. Y. Yang and W. H. Hou, Nanoscale, 2010, 2, 995–1005 RSC.
- W. J. Zhou, X. Y. Liu, J. J. Cui, D. Liu, J. Li, H. D. Jiang, J. Y. Wang and H. Liu, CrystEngComm, 2011, 13, 4557–4563 RSC.
- H. Dong, X. J. Li, Q. Peng, X. Wang, J. P. Chen and Y. D. Li, Angew. Chem., Int. Ed., 2005, 44, 2782–2785 CrossRef PubMed.
- Y. Li, M. H. Cao and L. J. Feng, Langmuir, 2009, 25, 1705–1712 CrossRef CAS PubMed.
- S. Zhang, C. Wu, Z. Wu, K. Yu, J. Wei and Y. Xie, Cryst. Growth Des., 2008, 8, 2933–2937 CAS.
- L. Zhen, C. Y. Xu, L. Yang and W. Z. Shao, Cryst. Growth Des., 2008, 8, 1734–1740 Search PubMed.
- J. Yang, C. X. Li, X. M. Zhang, Z. W. Quan, C. M. Zhang, H. Y. Li and J. Lin, Chem.–Eur. J., 2008, 14, 4336–4345 CrossRef CAS PubMed.
- J. Li and H. C. Zeng, Angew. Chem., Int. Ed., 2005, 44, 4342–4345 CrossRef CAS PubMed.
- X. W. Lou, Y. Wang, C. L. Yuan, J. Y. Lee and L. A. Archer, Adv. Mater., 2006, 18, 2325–2329 CrossRef CAS.
- Y. F. Zhu, J. L. Shi, W. H. Shen, X. P. Dong, J. W. Feng, M. L. Ruan and Y. S. Li, Angew. Chem., Int. Ed., 2005, 44, 5083–5087 CrossRef CAS PubMed.
- X. M. Sun and Y. D. Li, Angew. Chem., Int. Ed., 2004, 43, 3827–3831 CrossRef CAS PubMed.
- X. L. Xu and S. A. Asher, J. Am. Chem. Soc., 2004, 126, 7940–7945 CrossRef CAS PubMed.
- X. L. Li, T. J. Lou, X. M. Sun and Y. D. Li, Inorg. Chem., 2004, 43, 5442–5449 CrossRef CAS PubMed.
- S. Ghosh and M. K. Naskar, RSC Adv., 2013, 3, 13728–13733 RSC.
- S. Ding, F. Huang, X. Mou, J. Wu and X. Lu, J. Mater. Chem., 2011, 21, 4888–4892 RSC.
- Z. Jin, F. Wang, F. Wang, J. Wang, J. C. Yu and J. Wang, Adv. Funct. Mater., 2013, 23, 2137–2144 CrossRef CAS.
- J. Zhou, Z. Hua, W. Wu, Z. Liu, Y. Zhu, Y. Chen and J. Shi, Dalton Trans., 2011, 40, 12667–12669 RSC.
- S. Rengaraj, S. Venkataraj, C.-W. Tai, Y. Kim, E. Repo and M. Sillanpaa, Langmuir, 2011, 27, 5534–5541 CrossRef CAS PubMed.
- X. Chen, K. Kierzek, Z. Jiang, H. Chen, T. Tang, M. Wojtoniszak, R. J. Kalenczuk, P. K. Chu and E. B. Palen, J. Phys. Chem. C, 2011, 115, 17717–17724 CAS.
- A. D. Dinsmore, M. F. Hsu, M. G. Nikolaides, M. Marquez, A. R. Bausch and D. A. Weitz, Science, 2002, 298, 1006–1009 CrossRef CAS PubMed.
- Y. S. Li, J. L. Shi, Z. L. Hua, H. R. Chen, M. L. Ruan and D. S. Yan, Nano Lett., 2003, 3, 609–612 CrossRef CAS.
- H. G. Yang and H. C. Zeng, Angew. Chem., 2004, 116, 5318–5321 (Angew. Chem., Int. Ed., 2004, 43, 5206–5209) CrossRef.
- D. H. M. Buchold and C. Feldmann, Nano Lett., 2007, 7, 3489–3492 CrossRef CAS PubMed.
- J. Zhou, W. Wu, D. Caruntu, M. H. Yu, A. Martin, J. F. Chen, C. J. O. Connor and W. L. Zhou, J. Phys. Chem. C, 2007, 111, 17473–17477 CAS.
- F. Caruso, R. A. Caruso and H. Mohwald, Science, 1998, 282, 1111–1114 CrossRef CAS.
- J. G. Yu and X. X. Yu, Environ. Sci. Technol., 2008, 42, 4902–4907 CrossRef CAS.
- S. W. Kim, M. Kim, W. Y. Lee and T. Hyeon, J. Am. Chem. Soc., 2002, 124, 7642–7643 CrossRef CAS PubMed.
- J. H. Gao, B. Zhang, X. X. Zhang and B. Xu, Angew. Chem., 2006, 118, 1242–1245 (Angew. Chem., Int. Ed., 2006, 45, 1220–1223) CrossRef.
- H. G. Yu, J. G. Yu, S. W. Liu and S. Mann, Chem. Mater., 2007, 19, 4327–4334 CrossRef CAS.
- F. Piret, C. Bouvy and B.-L. Su, J. Mater. Chem., 2009, 19, 5964–5969 RSC.
- C. Wei, W. Zang, J. Yin, Q. Lu, Q. Chen, R. Liu and F. Gao, ChemPhysChem, 2013, 14, 591–596 CrossRef CAS PubMed.
- S. Gullapalli, J. M. Grider, H. G. Bagaria, K.-S. Lee, M. Cho, V. L. Colvin, G. E. Jabbour and M. S. Wong, Nanotechnology, 2012, 23, 495605 CrossRef PubMed , 10pp.
- G. Zou, Z. Liu, D. Wang, C. Jiang and Y. Qian, Eur. J. Inorg. Chem., 2004, 43, 4521–4524 CrossRef.
- P. Zhao, T. Huang and K. Huang, J. Phys. Chem. C, 2007, 111, 12890–12897 CAS.
- Q. Wu, M. Litz and X. C. Zhang, Appl. Phys. Lett., 1996, 68, 2924–2926 CrossRef CAS PubMed.
- D. N. Bose and S. Bhunia, J. Cryst. Growth, 1998, 186, 535–542 CrossRef.
- X. Wu, S. Zhou, S. Lou, Y. Wang, X. Shi, T. Gao and Y. Liu, Mater. Lett., 2012, 83, 4–7 CrossRef CAS PubMed.
- Y. Li, Y. Ding and Z. Wang, Adv. Mater., 1999, 11, 847–850 CrossRef CAS.
- S. H. Lee, Y. J. Kim and J. Park, Chem. Mater., 2007, 19, 4670–4675 CrossRef CAS.
- J. Zhang, K. Sun, A. Kumbhar and J. Fang, J. Phys. Chem. C, 2008, 112, 5454–5458 CAS.
- M. F. Ehsan, M. N. Ashiq, F. Bi, Y. Q. Bi, S. Palanisamy and T. He, RSC Adv., 2014, 4, 48411–48418 RSC.
- M. Thommes, Chem. Ing. Tech., 2010, 82, 1059–1073 CrossRef CAS.
- Y. Y. Li, J. P. Liu, X. T. Huang and G. Y. Li, Cryst. Growth Des., 2007, 7, 1350–1355 CAS.
- F. Lu, W. P. Cai and Y. G. Zhang, Adv. Funct. Mater., 2008, 18, 1047–1056 CrossRef CAS.
- W. E. Vargas, J. Opt. A: Pure Appl. Opt., 2002, 4, 452–456 CrossRef.
- M. Setvin, X. F. Hao, B. J. Daniel, J. Pavelec, Z. Novotny, G. S. Parkinson, M. Schmid, G. Kresse, C. Franchini and U. Diebold, Angew. Chem., Int. Ed., 2014, 53, 4714–4716 CrossRef CAS PubMed.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- M. F. Ehsan, M. N. Ashiq, F. Bi, Y. Bi, S. Palanisamy and T. He, RSC Adv., 2014, 4, 48411–48418 RSC.
- W. J. Ong, M. M. Gui, S. P. Chai and A. R. Mohamed, RSC Adv., 2013, 3, 4505–4509 RSC.
- J. G. Yu, J. X. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS PubMed.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed.
- Q. Y. Li, L. L. Zong, C. Li and J. J. Yang, Appl. Surf. Sci., 2014, 314, 458–463 CrossRef CAS PubMed.
- S. Rani, N. Z. Bao and S. C. Roy, Appl. Surf. Sci., 2014, 289, 203–208 CrossRef CAS PubMed.
- T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6, 1274–1282 CAS.
- H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Gas chromatographic analysis and SEM analysis after photoreduction. See DOI: 10.1039/c4ra13593h |
| This journal is © The Royal Society of Chemistry 2015 |