
Effects of transition metal precursors (Co, Fe, Cu, Mn, or Ni) on pyrolyzed carbon supported metal-aminopyrine electrocatalysts for oxygen reduction reaction
Abstract
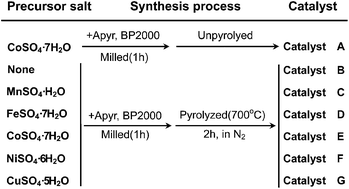
In the past four decades, non-precious metal catalysts (NPMCs) have been extensively studied as low-cost catalyst alternatives to Pt for the oxygen reduction reaction (ORR) in polymer electrolyte membrane (PEM) fuel cells. However, the role of transition metal playing in the catalysts' active sites is still a subject of controversy. In order to further clarify the nature of the active sites of NPMCs, in this work, using aminopyrine (Apyr) as the nitrogen precursor, Co-, Fe-, Cu-, Mn-, and Ni-incorporated nitrogen-containing electrocatalysts are synthesized for fuel cell ORR in alkaline media. The catalysts' ORR performance can be significantly improved by pyrolysis when the catalysts are incorporated by different transition metals. The observed catalytic activity order is: Co ≫ Fe ∼ Cu > Mn ≫ Ni. However, with respect to the electron transfer numbers (selectivity), the order is: Fe > Mn > Co ≫ Cu > Ni. XRD results reveal that Mn and Fe are more likely to be combined with S than Co, Ni and Cu. XPS analysis indicates that N concentration has a negative correlation with S concentration in the pyrolyzed catalysts, indicating a competitive mechanism between N and S on catalyst surfaces when metal sulfate is applied as the transition metal precursor. For ORR active site identification, the surface N species analysis reveals that catalyst containing more M–N group would give a higher catalytic ORR activity, while the metal incorporation is essential in the ORR active site structure, forming the M–Nx/C catalysts rather than just serving to catalyze the formation of N/C active sites.
 Please wait while we load your content...
Please wait while we load your content...

