
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct C–H amination and C–H chloroamination of 7-deazapurines†
Nazarii Sabatab,
Martin Klečkaab,
Lenka Slavětínskáb,
Blanka Klepetářováb and
Michal Hocek*ab
aDepartment of Organic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 8, CZ-12843 Prague 2, Czech Republic
bInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Gilead & IOCB Research Center, Flemingovo nam. 2, CZ-16610 Prague 6, Czech Republic. E-mail: hocek@uochb.cas.cz
First published on 12th November 2014
Abstract
Protocols for selective Pd–Cu-catalyzed direct C–H amination or C–H chloroamination of 7-deazapurines with N-chloro-N-alkyl-arylsulfonamides have been developed leading either to 8-(arylsulfonyl)methylamino-7-deazapurines or to 7-chloro-8-(arylsulfonyl)methylamino-7-deazapurines. The scope and limitations of the methods, as well as synthesis of a small series of 6,8,9-tri- and 6,7,8,9-tetrasubstituted 7-deazapurines and deprotection of the sulfonamide are presented.
Introduction
Modified pyrrolo[2,3-d]pyrimidine (7-deazapurine)1 bases and nucleosides display a variety of biological effects. Substituted 7-deazapurine bases induct neurogenesis2 or exert antitumor3 or antiinflammatory4 activity, whereas 6- or 7-aryl-7-deazapurine nucleosides are potent cytostatics.5,6 Therefore, development of regioselective synthesis of 7-deazapurines bearing multiple substituents is a worthwhile goal. Some regioselective cross-couplings of di- or trihalogenated pyrrolo[2,3-d]pyrimidines were recently reported for the synthesis of di- or triaryl derivatives.7,8 We have developed regioselective C–H borylation9 and C–H sulfenylation10 leading to 8-B- or 8-S-substituted 7-deazapurines. Here we report on complementary C–H aminations.Metal-catalyzed direct C–H aminations are increasingly popular reactions for modifications of arenes and heterocycles.11–14 One of the most efficient reagents are N-chloro-sulfonamides under Pd/Cu catalysis.13 Inspired by related C–H aminations of indoles,13 we decided to study the C–H aminations of pyrrolo[2,3-d]pyrimidines (7-deazapurines). The only previously reported C–H amination of 7-deazapurine was performed through hypervalent iodanes and proceeded at position 7.14
Results and discussion
For our initial study, we selected easily accessible 6-phenyl-9-benzyl-7-deazapurine 1a.9 We started testing its reaction with N-chloro-N-methyl-tosylamide 2 under literature13 conditions in presence of Pd(OAc)2, Cu(acac)2, 2,2′-bipyridine (bpy), Na2CO3 in dioxane (Scheme 1 and Table 1). The reaction with 2 equiv. of 2 in presence of 2 equiv. of Na2CO3 gave the desired 8-tosylamino product 5a in 13% only (entry 1). The use of a larger excesses of the base (5–7 equiv.) and of reagent 2 (3 equiv.) led to only low increase of yields (18–29%). Only the use of large excess (5 equiv.) of 2 gave product 5a in acceptable preparative yields of 68%.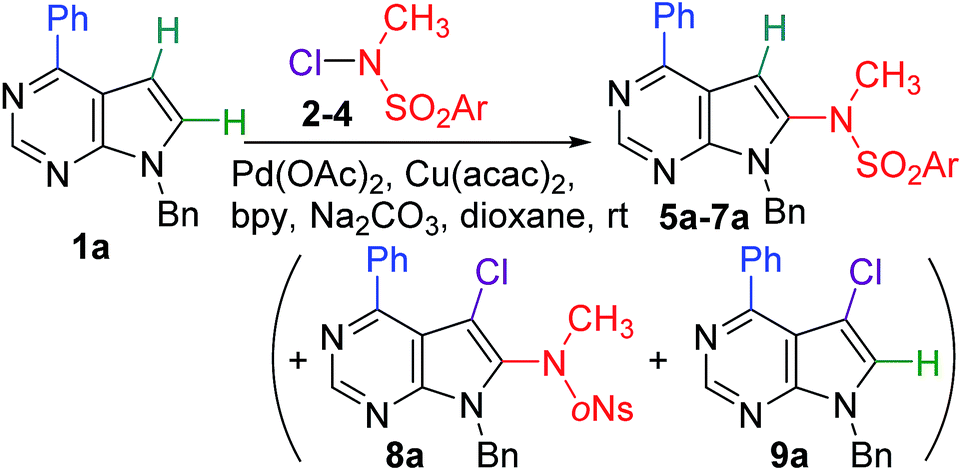 | ||
| Scheme 1 C–H aminations of 1a. | ||
| Entry | Ar | 2–4 (equiv.) | Na2CO3 (equiv.) | Product(s) (yield) |
|---|---|---|---|---|
| a Reagents and conditions: Pd(OAc)2 (5%), Cu(acac)2 (10%), bpy (10%), Na2CO3, 1,4-dioxane, Ar, rt, 24 h.b Reaction time 72 h. | ||||
| 1 | 4-MePh | 2 (2) | 2 | 5a (13%) |
| 2 | 4-MePh | 2 (2) | 5 | 5a (18%) |
| 3 | 4-MePh | 2 (3) | 5 | 5a (25%) |
| 4 | 4-MePh | 2 (3) | 7 | 5a (29%) |
| 5b | 4-MePh | 2 (5) | 7 | 5a (68%) |
| 6 | 4-NO2Ph | 3 (3) | 7 | 6a (47%) |
| 7 | 2-NO2Ph | 4 (5) | 5 | 7a (28%) + 8a (33%) + 9a (25%) |
| 8 | 2-NO2Ph | 4 (3) | 7 | 7a (60%) |
In order to have a choice of some more easily cleavable N-protecting groups,15 we also tested 4-nitrophenylsulfonyl(p-nosyl, pNs) and 2-nitrophenylsulfonyl (o-nosyl, oNs) chloroamides 3 and 4. The reaction of 1a with pNs reagent 3 (3 equiv.) gave the 8-p-nosylamino product 6a in acceptable 48% yield (entry 6). The reactions of 1a with oNs chloroamide 4 (2–3 equiv.) gave very low conversions (see Table S1 in ESI†), whereas the reaction with 5 equiv. of 4 gave a mixture of the desired product of 8-amination 7a (28%) accompanied by 7-chloro-8-amino- 8a and 7-chloro-7-deazapurine 9a as side-products. Apparently, the chloroamide 4 in larger excess can act as an electrophilic chlorination reagent which halogenates the deazapurine at position 7 (similarly as it was shown in indoles13). Therefore, we performed a detailed optimization of this reaction using different ratios of reagents, catalysts and additives and different conditions (see Table S1 in ESI†). The optimum protocol for aminations used 3 equiv. of 4 in presence of large excess of Na2CO3 (7 equiv.) to give the desired product 7a in 60% yield (Table 1, entry 8).
The detailed optimization also revealed some ratios of reagents and conditions under which the chloroamination proceeds. Also inspired by the related work on indoles,13 we employed CuCl as copper source, Ag2CO3 as base and LiCl as additive (Table S1 in ESI†) to find an optimum protocol leading exclusively to chloroamination,13,16 employing 4 (3 equiv.) in presence of Pd(OAc)2, CuCl (10 mol%), LiCl (2 equiv.) and Ag2CO3 (2 equiv.).
The next step was the study of the scope and limitation of the methods. A series of five 9-benzyl-7-deazapurine derivatives 1a–1e bearing phenyl, methoxy, methyl, chloro or amino group at position 6 was tested in the amination and chloroamination protocols (Scheme 2 and Table 2). The preparative aminations were performed with choroamide 4 (3 equiv.) in presence of Pd(OAc)2, Cu(acac)3, bpy and 7 equiv. of Na2CO3. The reactions of 6-phenyl, -methoxy and -methyl deazapurines proceeded smoothly to give desired 8-(o-nosyl)methylamino-7-deazapurines 7a–7c in acceptable yields of 41–62%. Conversely, analogous reaction of 6-chloro- and 6-amino-derivatives 1d,e led to very complex inseparable mixtures.
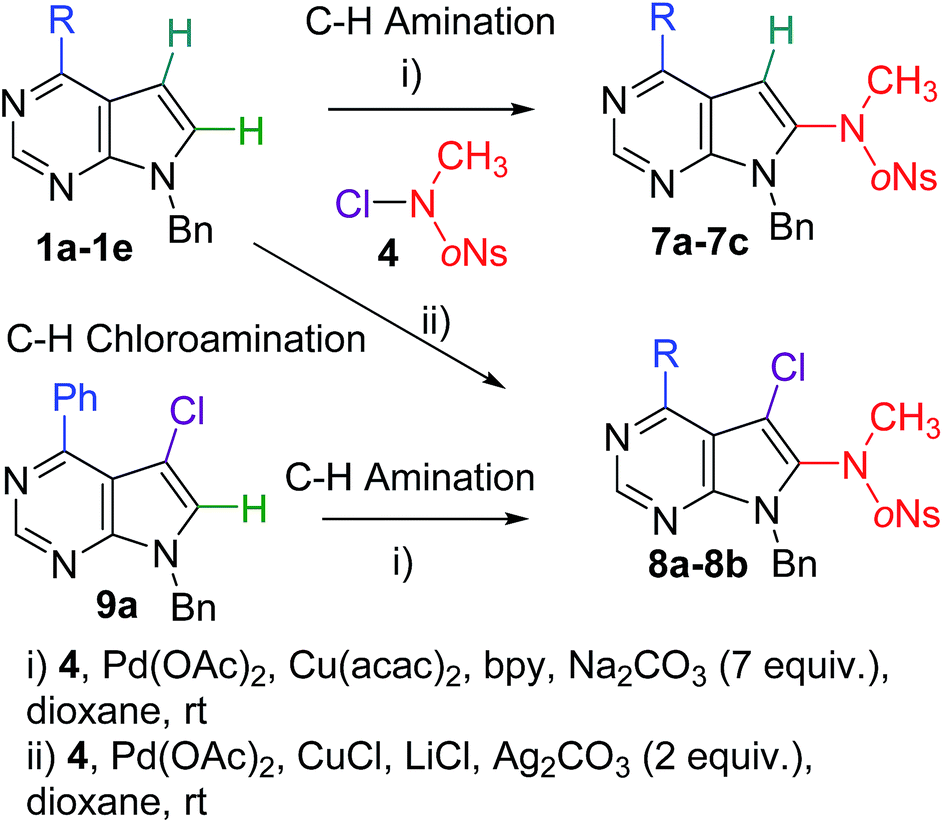 | ||
| Scheme 2 C–H aminations and chloroaminations of 7-deazapurines. | ||
| Entry | Starting compd | R | Product (yield) |
|---|---|---|---|
| 1 | 1a | Ph | 7a (62%) |
| 2 | 1b | OMe | 7b (60%) |
| 3 | 1c | Me | 7c (41%) |
| 4 | 1d | Cl | Complex mixture |
| 5 | 1e | NH2 | Complex mixture |
| 6 | 1a | Ph | 8a (51%) |
| 7 | 1b | OMe | 8b (42%) |
| 8 | 1c | Me | Low conversion, complex mixture |
| 9 | 1d | Cl | Complex mixture |
| 10 | 1e | NH2 | Complex mixture |
| 11 | 9a | Ph | 8a (41%) |
Then we tested the chloroamination protocol on the same series of deazapurines 1a–1e. The reactions with 4 (3 equiv.) were performed in presence of Pd(OAc)2, CuCl, LiCl and Ag2CO3. The reactions of 6-phenyl and 6-methoxy derivatives 1a,b proceeded well to get desired 7-chloro-8-(oNs)MeNH-7-deazapurines 8a,b in acceptable yields of 51 and 42%, whereas the reaction of 6-methyl derivative 1c gave low conversion to inseparable mixture containing products of chlorination and chloroamination. Similarly, the reactions of 6-chloro- and 6-aminodeazapurines 1d,e gave complex inseparable mixtures. Finally, 6-phenyl-7-chloro-7-deazapurine 9a was also converted to 7-chloro-8-aminated derivative 8a in 41% yield showing that the chlorine at position 7 is better tolerated (as it is less reactive toward nucleophiles) than the chlorine at position 6.
The last goal in this study was to test a deprotection of the sulfonamides and the stability of the corresponding 8-amino-7-deazapurines (2-aminoindoles are prone to tautomerization17 and oxidation18). Any attempt to cleave the Ts- or pNs-groups in compounds 5a or 6a according to literature15 either did not work or led to decomposition of the heterocycles. Therefore, major part of this study was performed with oNs-group which is more easily cleavable.15 The deprotection of compound 7a was successfully performed using thiophenol and cesium carbonate15d to afford 8-methylamino-7-deazapurine 10a in 75% yield (Scheme 3). We performed also one-pot C–H amination deprotection sequence to furnish the desired compound 10a directly in 35% for two steps. The 8-(methylamino)-7-deazapurine 10a was reasonably stable under neutral conditions but quickly decomposed when exposed to even traces of acid (e.g. in chlorinated solvents).
 | ||
| Scheme 3 Deprotection of 7a. | ||
All new compounds were fully characterized by NMR spectroscopy including assignment of all signals. In addition, to confirm the regioselectivity of the reactions, single-crystal X-ray diffraction was performed with compounds 5a, 7a, 7b and 8a. Fig. 1 shows the crystal structures of compounds 7a and 8a (for structures of 5a and 7b, see ESI†).
 | ||
| Fig. 1 An ORTEP view of compounds 7a (CCDC 1014820) and 8a (1014817) shown with 50% probability displacement ellipsoids. Structures 5a (1014819) and 7b (1014818) are shown in ESI.† | ||
Conclusions
In conclusion, we have developed selective protocols for palladium/copper-catalyzed direct C–H amination and C–H chloroamination of 7-deazapurines with N-chloro-N-alkyl-arylsulfoneamides. Reactions proceed under mild conditions regioselectively at position 8 of 7-deazapurines (in analogy to aminations at position 2 of indoles13) and are applicable to 6-aryl, -alkyl and -alkoxy 7-deazapurine derivatives. On the other hand, they are not compatible with 6-amino- and 6-chloro derivatives. Apart from the potential for the synthesis of series of 8-(arylsulfonyl)methylamino-7-deazapurines (and their 7-chloro-derivatives), when using oNs sulfonamides, the deprotection is possible to 8-methylamino-7-deazapurines. These protocols nicely complement the current toolbox of reactions for modifications of these privileged heterocycles and will be used for generation of libraries of compounds for biological activity screening.Experimental
General procedure for C–H amination of 7-deazapurines
7-Deazapurine 1a–1e (0.5 mmol), Pd(OAc)2 (0.025 mmol), Cu(acac)2 (0.05 mmol), bpy (0.05 mmol), Na2CO3 (3.5 mmol) and chlorosulfonamide (1–1.75 mmol) were placed in a vial which was purged with argon. Then 1,4-dioxane (2 mL) was added and the reaction mixture was then stirred for 24 h at rt, then quenched with H2O (2 mL), extracted with ethyl acetate (3 × 20 mL) and washed with brine (2 mL). The organic phases were combined and dried over sodium sulphate, filtered, and evaporated under vacuum. The crude product was purified by column chromatography on silica gel, eluting with hexanes/EtOAc (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:2) to afford the corresponding product.
:1 to 1:2) to afford the corresponding product.
N-(7-benzyl-4-phenyl-7H-pyrrolo[2,3-d]pyrimidin-6-yl)-N-methyl-2-nitrobenzenesulfonamide (7a)
1a (285 mg, 1 mmol) and N-chloro-N-methyl-2-nitrobenzenesulfonamide 4 (877 mg, 3.5 mmol) were used as starting compounds to give product 7a (310 mg, 62%) as colourless crystals after chromatography with hexanes/EtOAc 5:1 to 1:1 and crystallization from EtOAc/hexane. M.p. 102–103 °C. 1H NMR (500.0 MHz, CDCl3): 2.94 (s, 3H, CH3N); 5.67 (bs, 2H, CH2Ph); 6.45 (s, 1H, H-5); 7.22 (m, 2H, H-o-Bn); 7.27 (m, 1H, H-p-Bn); 7.30 (m, 2H, H-m-Bn); 7.50–7.53 (m, 3H, H-m,p-Ph); 7.61 (ddd, 1H, J5,6 = 8.1, J5,4 = 7.5, J5,3 = 1.3, H-5-C6H4NO2); 7.67 (ddd, 1H, J3,4 = 8.0, J3,5 = 1.3, J3,6 = 0.5, H-3-C6H4NO2); 7.76 (ddd, 1H, J6,5 = 8.1, J6,4 = 1.4, J6,3 = 0.5, H-6-C6H4NO2); 7.77 (ddd, 1H, J4,3 = 8.0, J4,5 = 7.5, J4,6 = 1.4, H-4-C6H4NO2); 7.97 (m, 2H, H-o-Ph); 9.10 (s, 1H, H-2). 13C NMR (125.7 MHz, CDCl3): 40.53 (CH3N); 45.29 (CH2Ph); 96.40 (CH-5); 113.91 (C-4a); 124.12 (CH-3-C6H4NO2); 127.65 (CH-o-Bn); 127.89 (CH-p-Bn); 128.85 (CH-m-Ph, CH-o-Bn); 128.94 (CH-m-Ph); 130.16 (C-1-C6H4NO2); 130.64 (CH-p-Ph); 131.26 (CH-5-C6H4NO2); 132.23 (CH-6-C6H4NO2); 134.62 (CH-4-C6H4NO2); 136.54 (C-i-Bn); 136.86 (C-i-Ph); 137.04 (C-6); 148.55 (C-2-C6H4NO2); 150.56 (C-7a); 152.40 (CH-2); 157.48 (C-4). IR(KBr): 2821, 1545, 1376, 1368, 1360, 1343, 1318, 1309, 1180, 1163, 924. HRMS (ESI) calculated for C26H22N5O4S: 500.1387; found 500.1387.
General procedure for C–H chloroamination of 7-deazapurines
7-Deazapurine 1a–1e (0.5 mmol), Pd(OAc)2 (0.0125 mmol), CuCl (0.05 mmol), LiCl (1.0 mmol), Ag2CO3 (1.0 mmol) and chlorosulfonamide (1.5–1.75 mmol) were placed in a vial which was purged with argon. Then 1,4-dioxane (2 mL) was added and the reaction mixture was stirred for 24 h at room temperature, then quenched with H2O (2 mL), extracted with ethyl acetate (3 × 20 mL) and washed with brine (2 mL). The organic phases were combined and dried over sodium sulphate, filtered, and evaporated under vacuum. The crude product was purified by column chromatography on silica gel, eluting with hexanes/EtOAc (5:1 to 1:2) to afford the corresponding product.
N-(7-benzyl-5-chloro-4-phenyl-7H-pyrrolo[2,3-d]pyrimidin-6-yl)-N-methyl-2-nitrobenzenesulfonamide (8a)
1a (285 mg, 1 mmol) and 4 (877 mg, 3.5 mmol) were used as starting compounds to give product 8a (273 mg, 51%) as white crystals after chromatography with hexanes/EtOAc 5:1 to 1:1 and crystallization from EtOAc/hexane. M.p. 215–216 °C. 1H NMR (500 MHz, CDCl3): 2.91 (s, 3H, CH3N); 5.42 (d, 1H, Jgem = 15.3 Hz, CH2a-Ph); 6.16 (d, 1H, Jgem = 15.3 Hz, CH2b-Ph); 7.26–7.31 (m, 3H, H-o,p-Bn); 7.33 (m, 2H, H-m-Bn); 7.42–7.50 (m, 3H, H-m,p-Ph); 7.58–7.63 (m, 2H, H-3,5-C6H4NO2); 7.71 (m, 2H, H-o-Ph); 7.73 (ddd, 1H, J4,3 = J4,5 = 7.7 Hz, J4,6 = 1.4 Hz, H-4-C6H4NO2); 7.84 (m, 1H, H-6-C6H4NO2); 9.09 (s, 1H, H-2). 13C NMR (125.7 MHz, CDCl3): 38.28 (CH3–N); 45.68 (CH2-Ph); 103.08 (C-5); 112.02 (C-4a); 124.00 (CH-3-C6H4NO2); 127.84 (CH-m-Ph); 128.09 (CH-p-Bn); 128.11 (CH-o-Bn); 128.95 (CH-m-Bn); 129.86 (CH-p-Ph); 130.20 (CH-o-Ph); 131.45 (CH-5-C6H4NO2); 131.62 (C-1-C6H4NO2); 131.65 (CH-6-C6H4NO2); 131.89 (C-6); 134.49 (CH-4-C6H4NO2); 136.48 (C-i-Bn); 136.62 (C-i-Ph); 148.56 (C-2-C6H4NO2); 148.78 (C-7a); 153.18 (CH-2); 160.20 (C-4). IR(KBr): 3050, 1583, 1545, 1374, 1345, 1165, 826, 558. HRMS (ESI) calculated for C26H21N5O4SCl: 534.0998; found: 534.0997.
Acknowledgements
This work was supported by the institutional support of the Charles University and Academy of Sciences of the Czech Republic (RVO: 61388963), by the Czech Science Foundation (P207/12/0205) and by Gilead Sciences, Inc.Notes and references
- Review: S. Tumkevicius and J. Dodonova, Chem. Heterocycl. Compd., 2012, 48, 258–279 CrossRef CAS.
- S. Ding, T. Y. H. Wu, A. Brinker, E. C. Peters, W. Hur, N. S. Gray and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7632–7637 CrossRef CAS PubMed.
- X. Y. Jiao, J. D. Kopecky, J. S. Liu, J. Q. Liu, J. C. Jaen, M. G. Cardozo, R. Sharma, N. Walker, H. Wesche, S. Li, E. Farrelly, S. H. Xiao, Z. Wang and F. Kayser, Bioorg. Med. Chem. Lett., 2012, 22, 6212–6217 CrossRef CAS PubMed.
- N. Chakka, H. Bregman, B. Du, H. N. Nguyen, J. L. Buchanan, E. Feric, J. Ligutti, D. Liu, J. S. McDermott, A. Zou, S. I. McDonough and E. F. Di Mauro, Bioorg. Med. Chem. Lett., 2012, 22, 2052–2062 CrossRef CAS PubMed.
- P. Nauš, R. Pohl, I. Votruba, P. Džubák, M. Hajdúch, R. Ameral, G. Birkuš, T. Wang, A. S. Ray, R. Mackman, T. Cihlar and M. Hocek, J. Med. Chem., 2010, 53, 460–470 CrossRef PubMed.
- (a) A. Bourderioux, P. Nauš, P. Perlíková, R. Pohl, I. Pichová, I. Votruba, P. Džubák, P. Konečný, M. Hajdúch, K. M. Stray, T. Wang, A. S. Ray, J. Y. Feng, G. Birkus, T. Cihlar and M. Hocek, J. Med. Chem., 2011, 54, 5498–5507 CrossRef CAS PubMed; (b) P. Nauš, O. Caletková, P. Konečný, P. Džubák, K. Bogdanová, M. Kolář, J. Vrbková, L. Slavětínská, E. Tloušťová, P. Perlíková, M. Hajdúch and M. Hocek, J. Med. Chem., 2014, 57, 1097–1110 CrossRef PubMed.
- (a) S. Tumkevicius, J. Dodonova, K. Kazlauskas, V. Masevisius, L. Skardziute and S. Jursenas, Tetrahedron Lett., 2010, 51, 3902–3906 CrossRef CAS PubMed; (b) R. V. Urbonas, V. Poskus, J. Bucevicius, J. Dodonova and S. Tumkevicius, Synlett, 2013, 24, 1383–1386 CrossRef CAS PubMed.
- V. Prieur, J. Rubio-Martinez, M. Font-Bardia, G. Guillaumet and M. D. Pujol, Eur. J. Org. Chem., 2014, 1514–1524 CrossRef CAS.
- M. Klečka, R. Pohl, B. Klepetářová and M. Hocek, Org. Biomol. Chem., 2009, 7, 866–868 Search PubMed.
- M. Klečka, R. Pohl, J. Čejka and M. Hocek, Org. Biomol. Chem., 2013, 11, 5189–5193 Search PubMed.
- Reviews: (a) M. Zhang, Synthesis, 2011, 21, 3408–3417 CrossRef PubMed; (b) M.-L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901–910 RSC.
- Examples of C–H Amination of azoles and indoles: (a) D. Monguchi, T. Fujiwara, H. Furukawa and A. Mori, Org. Lett., 2009, 11, 1607–1610 CrossRef CAS PubMed; (b) Q. Wang and S. L. Schreiber, Org. Lett., 2009, 11, 5178–5180 CrossRef CAS PubMed; (c) S. H. Cho, J. Y. Kim, S. Y. Lee and S. Chang, Angew. Chem., Int. Ed., 2009, 48, 9127–9130 CrossRef CAS PubMed; (d) T. Kawano, K. Hirano, T. Satoh and M. Miura, J. Am. Chem. Soc., 2010, 132, 6900–6901 CrossRef CAS PubMed; (e) M. Miyasaka, K. Hirano, T. Satoh, R. Kowalczyk, C. Bolm and M. Miura, Org. Lett., 2011, 13, 359–361 CrossRef CAS PubMed; (f) N. Matsuda, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2011, 13, 2860–2863 CrossRef CAS PubMed; (g) T. Froehr, C. P. Sindlinger, U. Kloeckner, P. Finkbeiner and B. J. Nachtscheim, Org. Lett., 2011, 13, 3754–3757 CrossRef CAS PubMed; (h) Y. Li, H. Wang, S. Ali, X. Xia and Y. Liang, Chem. Commun., 2012, 48, 2343–2345 RSC; (i) W.-B. Wu and J.-M. Huang, Org. Lett., 2012, 14, 5832–5835 CrossRef CAS PubMed; (j) J. Xu, J. Li, Z. Wei, Q. Zhang and D. Shi, RSC Adv., 2013, 3, 9622–9624 RSC; (k) K. Foo, E. Sella, I. Thome, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5279–5282 CrossRef CAS PubMed; (l) S. L. McDonald, C. E. Hendrick and Q. Wang, Angew. Chem., Int. Ed., 2014, 53, 4667–4670 CrossRef CAS PubMed.
- X.-Y. Liu, P. Gao, Y.-W. Shen and Y.-M. Liang, Org. Lett., 2011, 13, 4196–4199 CrossRef CAS PubMed.
- I. Sokolovs, D. Lubriks and E. Suna, J. Am. Chem. Soc., 2014, 136, 6920–6928 CrossRef CAS PubMed.
- (a) G. Sabitha, B. V. S. Reddy, S. Abraham and J. S. Yadav, Tetrahedron Lett., 1999, 40, 1569–1570 CrossRef CAS; (b) R. C. Roemmele and H. Rapoport, J. Org. Chem., 1988, 53, 2367–2371 CrossRef CAS; (c) D. A. Alonso and P. G. Andersson, J. Org. Chem., 1998, 63, 9455–9461 CrossRef CAS; (d) T. Fukuyama, C.-K. Jow and M. Cheung, Tetrahedron Lett., 1995, 36, 6373–6374 CrossRef CAS.
- Apart from ref. 13, the only example of C–H haloamination of indoles: (a) A. John and K. Nicolas, Organometallics, 2012, 31, 7914–7920 CrossRef CAS . Most haloaminations proceed as additions to multiple bonds, e.g.; (b) X.-Y. Liu, P. Gao, Y.-W. Shen and Y.-M. Liang, Adv. Synth. Catal., 2011, 17, 3157–3160 CrossRef.
- P. Diana, P. Barraja, A. Lauria, A. M. Almerico, G. Dattolo and G. Cirrincione, Tetrahedron, 2000, 56, 5177–5183 CrossRef CAS.
- T. Hino, M. Nakagawa, T. Hashizume, N. Yamaji and Y. Miwa, Tetrahedron Lett., 1970, 11, 2205–2208 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental part and characterization of all compounds, additional figures of X-ray structures, copies of NMR spectra. CCDC 1014817–1014820. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra13143f |
| This journal is © The Royal Society of Chemistry 2014 |