
Nanoparticle mediated delivery of a GST inhibitor ethacrynic acid for sensitizing platinum based chemotherapy
Abstract
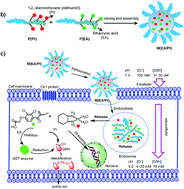
Drug resistance is a major obstacle for platinum based chemotherapy, and one of the most common reasons is GSH-mediated detoxification. To overcome this problem, we proposed ethacrynic acid (EA), a GST inhibitor, to reduce the conjugation of GSH with platinum agents so as to sensitize cancer cells to chemotherapy. We constructed biodegradable nanoparticles to co-deliver EA and DACHPt, a precursor of oxaliplatin. In vitro studies showed that the hybrid nanoparticles could release EA and DACHPt faster in intracellular-like environments than in blood circulation, enhancing the cellular Pt accumulation and Pt–DNA adduct formation. Most importantly, it showed a synergy effect between EA and DACHPt, resulting in an enhancement of up to 4.6 fold of the anticancer efficacy. We also verified this approach in vivo, taking into account both anticancer efficacy and systemic toxicity, and the nanoparticles were much better than the simple combination of free drugs. The results demonstrated that the nanoparticles we constructed could be a promising approach to overcome the drug resistant problem in cancer chemotherapy.
 Please wait while we load your content...
Please wait while we load your content...

