
Metallohalide perovskite–polymer composite film for hybrid planar heterojunction solar cells†
Abstract
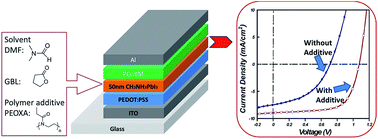
A polymer with tailored chemical functionality was introduced as a processing additive to control the film formation of the CH3NH3PbI3 perovskite structure, leading to enhanced photovoltaic performance in a PEDOT:PSS/perovskite/PCBM-based planar heterojunction solar cell under optimized conditions. By adjusting the polymer doping content and the processing solvent, the grain size, film coverage and the optical properties of the perovskite films can be effectively tuned. At optimized conditions, the planar heterojunction solar cell composed of a thin layer of perovskite–polymer film (∼50 nm) exhibits an average PCE of 6.16% with a Voc of 1.04 V, a Jsc of 8.85 mA cm−2 and a FF of 0.65, which are much higher than those of the control device with a pristine perovskite film. The higher performance was attributed to improved morphology and interfaces of the perovskite–polymer films, which reduced the undesired contact between PEDOT:PSS and PCBM and minimized the shunting paths in the device. In addition, since the fabrication process for the perovskite solar cells can be performed at low temperature, flexible cells built on plastic substrates can therefore be realized with a PCE of 4.35%.
 Please wait while we load your content...
Please wait while we load your content...

