
Formation of dications bearing a S(OH)2+ group from long-lived 9,9-dimethyl-10 R-phenanthrenium cations in FSO3H–SbF5/SO2ClF/SO2: a mechanistic study†
George E. Salnikov,
Alexander M. Genaev*,
Vladimir A. Bushmelev,
Andrey A. Nefedov and
Vyacheslav G. Shubin
N. N. Vorozhtsov Novosibirsk Institute of Organic Chemistry, Academician Lavrent'ev Ave., 9, Novosibirsk 630090, Russian Federation. E-mail: genaev@nioch.nsc.ru; Fax: +7-383-330-97-52
First published on 14th October 2014
Abstract
1H and 13C NMR studies have shown that the long-lived 9,9-dimethyl-10-R-phenanthrenium cations (R = PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, Me, OH) generated in FSO3H–SbF5/SO2ClF/SO2/CD2Cl2 transform into long-lived 7-dihydroxysulfonio-9,9-dimethyl-10-R-phenanthrenium dications. The effect of the 10-R substituents on the reaction rate suggests that a key step in the reaction mechanism is addition of SO2 to protonated phenanthrenium cations.
C, Me, OH) generated in FSO3H–SbF5/SO2ClF/SO2/CD2Cl2 transform into long-lived 7-dihydroxysulfonio-9,9-dimethyl-10-R-phenanthrenium dications. The effect of the 10-R substituents on the reaction rate suggests that a key step in the reaction mechanism is addition of SO2 to protonated phenanthrenium cations.
Introduction
It is known that aromatic compounds in superacids, such as HF–SbF5 and FSO3H–SbF5, containing SO2, transform into protonated sulfinic acids.1–3 The mechanism of this reaction may consist of addition of SO2 to arenium cations formed under protonation of aromatics or electrophilic attack of the cation SO2H+ or complex of SO2 with SbF5 on the aromatics involved in equilibrium with arenium ions.1,4
These mechanisms suggest the opposite effect of the substituent R on the reaction rate. While studying the long-lived 9,9-dimethyl-10-R-phenanthrenium cations5,6 we have found that these cations are convenient model structures for study of carbocation reactions mechanisms. In the present work we have used these cations to determine the mechanism of sulfination reaction.
Results and discussion
We have found that the long-lived 9,9-dimethyl-10-phenylethynylphenanthrenium cation (1a)5 generated from 9-hydroxy-10,10-dimethyl-9-phenylethynyl-9,10-dihydrophenanthrene (2a) (Scheme 1) in FSO3H–SbF5/SO2ClF/SO2/CD2Cl2 at −95 °C, at −71 °C quickly (t1/2 = 3 min) and totally turns into another long-lived cationic particle. The structure of this particle, as unambiguously determined from 2D COSY, ROESY, HSQC and HMBC correlations (ESI, pp. S2–S6†), corresponds to the structure of the original cation (1a) in which the atom H7 is replaced with a substituent containing no carbon atoms. The 1H NMR spectrum of the new particle (Fig. 1) shows an additional singlet signal at δ 9.43 ppm of 2H intensity area, which correlates via long range spin–spin coupling with the 13C NMR signal of C7 in the 2D HMBC spectrum. The protons corresponding to this signal are involved in slow exchange with acid protons and the NOE cross-relaxation is observed between these protons and H6 and H8 signals. By analogy to the literary data1–3,7 according to which the protons of S(OH)2+ group in cations Ar–S(OH)2+ do not exhibit fast exchange with acidic media and show distinct signals with chemical shifts in the range δ 9.12–9.63 ppm we believe that the new signal in our case belongs to S(OH)2+ group and the new formed particle is 7-dihydroxysulfonio-9,9-dimethyl-10-phenylethynylphenanthrenium dication 3a (Scheme 1). Additional evidences for its dicationic nature are downfield shifts of all signals in the 1H NMR spectrum (Fig. 1), particularly of the signals of H6 and H8, relative to that of the original cation (1a), and instability of its solutions containing lock substance CD2Cl2 at the temperatures above −60 °C, cf. ref. 8. The remarkable features of the sulfination reaction are its high rate and practically full regioselectivity. The rate constant of dication 3a formation is 4 × 10−3 s−1 at −71 °C. | ||
| Scheme 1 | ||
 | ||
| Fig. 1 1H NMR spectra (aromatics area) of cation 1a in FSO3H–SbF5/SO2ClF/CD2Cl2 at −91 °C and dication 3a in FSO3H–SbF5/SO2ClF–SO2/CD2Cl2 at −81 °C. | ||
If the same superacid system does not contain SO2, cation 1a remains stable up to −40 °C, at higher temperatures giving cyclization product dication 4 (Scheme 2).5b
 | ||
| Scheme 2 | ||
An analogous sulfination reaction occurs with the long-lived 9,9,10-trimethylphenanthrenium cation 1b9 (Scheme 1). At −71 °C a mixture of this cation and 7-dihydroxysulfonio-9,9,10-trimethylphenanthrenium dication 3b is formed (ESI, pp. S10–S15†), the content of the latter being 34%, and maintaining practically not increased upon standing or by adding an additional amount of SO2. As in the case of 3a, all 1H NMR signals of 3b are downfielded relative to that of the precursor monocation 1b. The downfield displacement of NMR signals of the phenanthrene fragment of 3b is more pronounced than that of 3a (ESI, p. S23†), which is apparently due to lower opportunity for positive charge delocalization. Sulfination of 1b proceeds faster than that of 1a. The rate constant of dication 3b formation measured at −100 °C is 6 × 10−4 s−1 (0.2 s−1 for −71 °C as recalculated using the Eyring equation).
9,9-Dimethyl-10-hydroxyphenanthrenium cation 1c10 (Scheme 3) also undergoes sulfination reaction (ESI, pp. S16–S17†). This cation exists as a mixture of exchanging E- and Z-isomers (equilibrium constant Z/E is 0.3, exchange rate constant Z → E is 30 s−1 at −81 °C). ROESY spectrum shows cross peaks due to NOE cross relaxation of hydroxyl proton with protons of methyl groups for the E-isomer, and with the H1 proton for the Z-isomer. Hydroxyl proton of the Z-isomer is strongly downfielded relative to that of the E-isomer (δ 11.92 ppm vs. 11.31 ppm) due to deshielding anisotropy of the aromatic ring. Significant chemical shifts difference for H1 proton of E- and Z-isomers is observed (δ 8.73 ppm and 8.28 ppm, respectively). The probable reason of the downfield shift of H1 of the E-isomer is its spatial proximity to the lone pair of oxygen atom, cf. ref. 11.
 | ||
| Scheme 3 | ||
Sulfination of cation 1c results in dication 3c formation as the main product (ESI, pp. S18–S19†). Dication 3c also exists as an equilibrium mixture of slowly exchanging E- and Z-isomers (equilibrium constant Z/E is 0.4, exchange rate constant Z → E is 30 s−1 at −71 °C). All the 1H NMR signals of dications 3c are downfielded relative to the corresponding signals of cations 1c, the differences between 1H NMR spectra of E- and Z-isomers being analogous to those observed for E- and Z-isomers of cation 1c. In this case the sulfination reaction is slower than that of 1a and 1b. The rate constant of dication 3c formation is 7 × 10−5 s−1 at −71 °C.
Both cations 1d and 1e (Scheme 1) do not yield observable amounts of sulfination products. Cation 1d5a,6 at −71 °C transforms into a complex mixture of unidentifiable products (ESI, p. S21†). On the contrary, cation 1e5a,6 is quite stable up to −39 °C, content of the sulfination products being less than 2%. At higher temperatures this cation is decomposed (ESI, p. S22†).
Possible mechanisms of the sulfinated dications formation are presented in Schemes 4–6, cf. ref. 1,4. The mechanism, in which the protonated SO2 is an attacking electrophile (Scheme 4), seems to be unlikely, as this mechanism assumes an approach of two positively charged particles to the distance necessary for the formation of a chemical bond. In Scheme 5 complex SO2·SbF5 (ref. 12) serves as electrophile. Its presence in the reaction mixture has been established by 19F NMR spectroscopy (ESI, p. S8†), cf. ref. 13. Both Schemes 4 and 5 assume that cation 1 is a nucleophilic particle. The nucleophilicity of the aromatic fragment should be in line with the electron donating properties of 10-R substituent. Apparently the nucleophilicity should decrease in the order R = OH > CCPh > Me. This assumption is confirmed by chemical shifts of aromatic fragments of cations 1a,b,c in 1H and 13C NMR spectra (ESI, p. S23†). The rate of dications 3 formation according to Scheme 4 or 5, as determined by the nucleophilicity of cations 1, would decrease in the order 3c > 3a > 3b. However, the rate decreases just in the opposite order: 3b > 3a > 3c (the rate constants ratio is approx. 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.02 at −71 °C). Moreover, neither of Schemes 4 and 5 can explain the observed regioselectivity of the reaction, as according to these mechanisms an electrophile has to attack the C7 atom bearing a partial positive charge.
:1:0.02 at −71 °C). Moreover, neither of Schemes 4 and 5 can explain the observed regioselectivity of the reaction, as according to these mechanisms an electrophile has to attack the C7 atom bearing a partial positive charge.
 | ||
| Scheme 4 | ||
 | ||
| Scheme 5 | ||
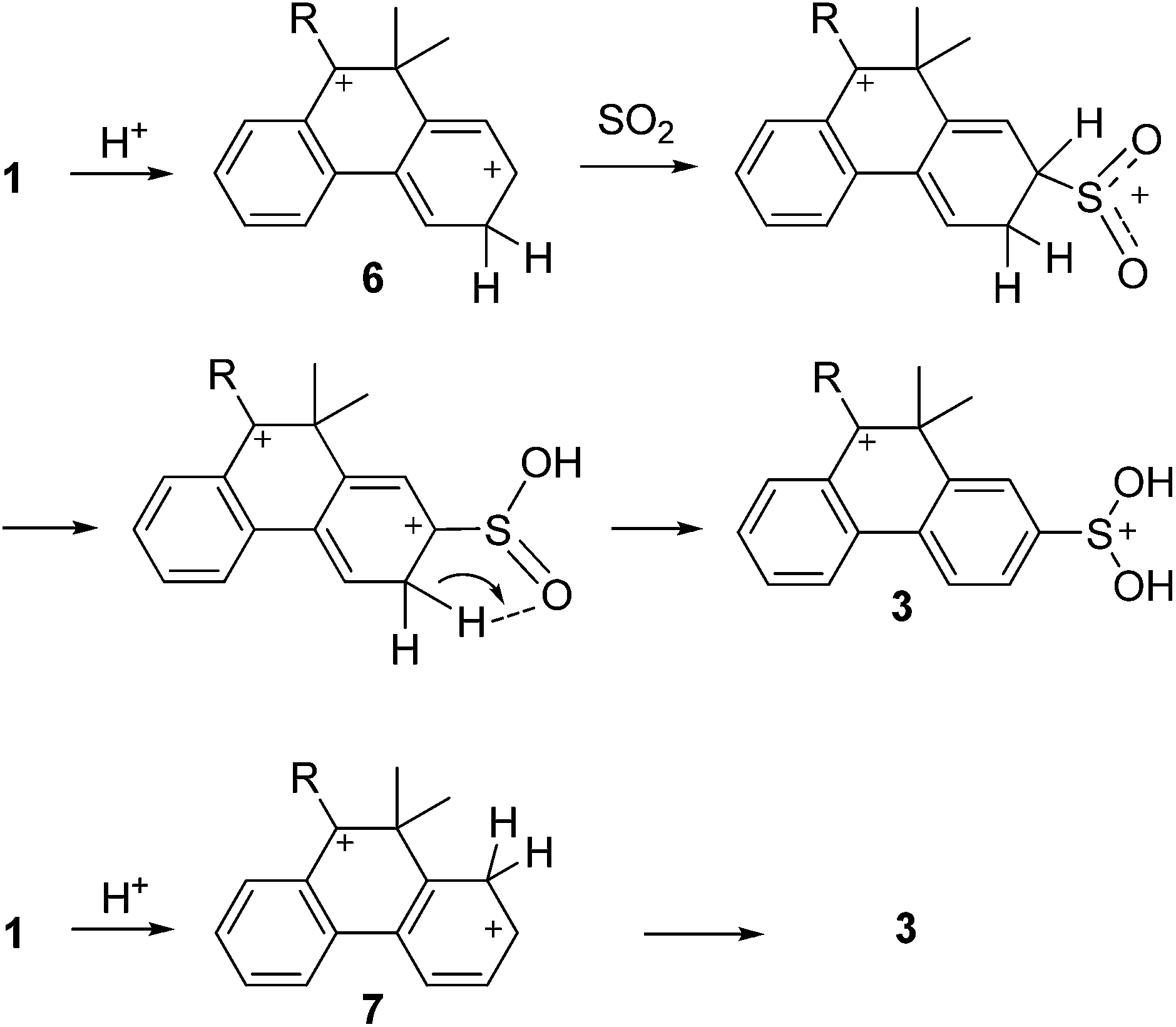 | ||
| Scheme 6 | ||
Scheme 6 assumes that the superelectrophilic intermediates – dications 6 and/or 7 play a key role in the reaction. The inactivity of cation 1e in the sulfination reaction may result from a low concentration of the respective dication intermediates owing to low basicity of C8 and C6 atoms of cation 1e.
In order to verify the possibility of intermediates 6 and 7 formation, we carried out the sulfination reaction of 1a in FSO3D–SbF5/SO2ClF/SO2 (ESI, p. S9†). The methyl groups of both the precursor 1a and the product 3a do not undergo deuteration, as unambiguously follows from the absence of the corresponding signals in the 2H NMR spectrum. The 2H NMR signals in the aromatics region are too broad and indistinguishable from each others because of severe overlaps. Integrals of 1H NMR signals of phenanthrene rings taken relative to the integrals of methyl group signals clearly indicate that protons belonging to the more positively charged ring (protons H1−4 of both 1a and 3a) do not take part in deuteration to any visible degree. On the contrary, integrals of proton signals of the other phenanthrene ring both in the precursor monocation 1a and in the sulfinated product 3a (protons H5−8 of 1a and protons H5,6,8 of 3a) are significantly reduced due to their partial deuteration. Deuteration of 1a into the positions H5−8 proceeds practically immediately even at −100 °C, i.e. much faster than sulfination. Therefore dications 6 and 7 in superacid FSO3H–SbF5 can be intermediates in the sulfination reaction. The regioselectivity of sulfination can be explained by the higher stability of the dications sulfinated at C7 in comparison with other sulfination products. In this case the second formal positive charge (at C7–S(OH)2+) is located most distantly along the delocalized π bond system from the first one (at C10).
It is important to note that keeping the monocation precursors in FSO3D in the absence of SbF5 even up to −15 °C did not lead to any deuteration, and the reaction with SO2 under these conditions also does not proceed. Therefore we believe that the role of SbF5 in the sulfination reaction consists solely in ensuring high acidity of the medium necessary for formation of the dication intermediates 6 and/or 7.
In 1H DNMR and ROESY spectra (ESI, pp. S14–S15†) of dication 3b chemical exchange between 9- and 10-methyl groups is observed (k = 6.4 s−1 at −61 °C, DNMR), the signals of aromatic ring protons being intact. Probable mechanisms of this exchange are shown in Schemes 7 and 8. As the exchange rate between protons of S(OH)2+ group of dication 3b and protons of superacid medium (k = 0.2 s−1 at −61 °C, ROESY, pseudomonomolecular approximation) is significantly less than the rate of methyl groups exchange, Scheme 8 cannot be the preferred one. The chemical exchange between 9- and 10-methyl groups of dication 3b is slower than that of monocation 1b (k = 12.3 s−1 at −61 °C, DNMR) probably due to lower stability of the dication intermediate 8 (Scheme 7) compared to dication 3b. In contrast to the latter monocation 1b undergoes degenerate rearrangement, so that there is no such a retarding effect.
 | ||
| Scheme 7 | ||
 | ||
| Scheme 8 | ||
Conclusions
Long-lived carbocations of the phenanthrene series are fruitful models for solving mechanistic problems of the organic reactions proceeding with formation of carbocation intermediates. The advantage of these models in study of the sufination reaction is the opportunity to explore kinetics and mechanism of the reaction, based on dependence of the rate on the substituents in aromatic ring.Experimental
General methods and materials
NMR spectra were obtained at the Chemical Service Centre of Siberian Branch of the Russian Academy of Sciences on Bruker AV-600, using the residual proton and carbon signals of deuterated methylene chloride as internal references (CHDCl2, δH 5.33 ppm; CD2Cl2, δC 53.6 ppm) and CFCl3 (δF 0 ppm) as external standard. For structure elucidation and NMR signal assignment 2D correlation spectra 1H–1H (COSY, ROESY) and 1H–13C (HSQC, HMBC) were used.Analysis of SO2ClF was performed on Hewlett-Packard G1800A device, equipped with gas chromatograph HP 5890 of II series having capillary column HP-5MS and mass selective detector HP 5971. The column temperature was 30 °C.
Doubly distilled FSO3H (b.p. 158–161 °C), freshly distilled SbF5, SO2 (from Na2S2O5 and H2SO4) drained by a transmission through conc. H2SO4, and CD2Cl2 drained by 4 Å molecular sieves were used. SO2ClF was prepared by the method of Woyski.14 According to GC-MS data it contained SO2 (8%). Pure SO2ClF was obtained by distillation from SbF5.15

7-Dihydroxysulfonio-9,9-dimethyl-10-phenylethynylphenanthrenium dication (3a)
Solution of carbinol 2a5a (20 mg, 0.062 mmol.) in 0.12 mL of CD2Cl2 was added dropwise to solution of FSO3H (130 mg, 1.3 mmol) and SbF5 (250 mg, 1.2 mmol) in 0.3 mL of SO2ClF–SO2 (8% SO2) placed into NMR tube under stirring at −95 °C. Formation of cation 1a5 was observed. The tube was kept for 10 min at −71 °C and then cooled down to −81 °C. 1H NMR spectrum (δ, ppm): 2.13 s (6H, 9-Me), 7.82 t (2H, Hm, J 7.6 Hz), 8.04 t (1H, Hp, J 7.5 Hz), 8.11 t (1H, H2, J 7.6 Hz), 8.21–8.26 m (3H, Ho, H6), 8.45 s (1H, H8), 8.61 t (1H, H3, J 7.6 Hz), 8.72 d (1H, H4, J 8.7 Hz), 8.88 d (1H, H5, J 9.2 Hz), 8.94 d (1H, H1, J 8.2 Hz), 9.43 s (2H, OH). 13C NMR spectrum: (δ, ppm): 31.7 q (9-Me), 52.2 s (C9), 105.5 s (Ca), 119.8 s (Ci), 126.2 d (C6), 126.3 d (C8), 126.9 d (C4), 128.7 d (C5), 130.7 d (Cm), 133.5 d (C2), 134.0 s (C4b), 134.7 s (C7), 135.5 s (C10a), 138.1 d (Co), 140.1 d (Cp), 140.1 d (C1), 141.4 s (C4a), 147.0 s (C8a), 150.5 d (C3), 161.2 s (Cb), 199.2 s (C10).Analogous NMR spectra were obtained for solution in SO2 at −61 °C (ESI, p. S7†). The signal of S(OH)2+ group is observed at δ 9.86 ppm. 19F NMR spectrum (δ, ppm): −101.8 d (4F, J 97 Hz), −134.9 quintet (1F, J 97 Hz) (SO2·SbF5). The 19F NMR assignments were made using COSY 19F–19F and the data of the paper.13
7-Dihydroxysulfonio-9,9,10-trimethylphenanthrenium dication (3b)
Solution of carbinol 2b9b (20 mg, 0.084 mmol.) in 0.12 mL of CD2Cl2 was added to solution of FSO3H (115 mg, 1.15 mmol) and SbF5 (235 mg, 1.22 mmol) in 0.25 mL of SO2ClF–SO2 (1:17 by volume) placed into NMR tube at −95 °C, the mixture was stirred and then warmed to −71 °C. Formation of the mixture of cation 1b9 and dication 3b was observed, the content of the latter being 34%. Addition of SO2 (0.1 mL) to the solution did not result in increase of the content. 1H NMR spectrum (δ, ppm): 2.01 s (6H, 9-Me), 3.65 s (3H, 10-Me), 8.16 t (1H, H2, J 7.7 Hz), 8.28 dd (1H, H6, J 8.8, 1.8 Hz), 8.58 d (1H, H8, J 1.8 Hz), 8.82 t (1H, H3, J 7.6 Hz), 8.89 d (1H, H4, J 8.1 Hz), 8.98 d (1H, H5, J 9.0 Hz), 9.03 d (1H, H1, J 8.3 Hz), 9.86 s (2H, OH). 13C NMR spectrum: (δ, ppm): 27.8 q (9-Me), 26.7 q (10-Me), 54.6 s (C9), 126.3 d (C6), 127.0 d (C8), 127.7 d (C4), 129.4 d (C5), 133.8 d (C2), 133.5 s (C4b), 135.7 s (C7), 133.8 s (C10a), 140.2 d (C1), 145.1 s (C4a), 145.2 s (C8a), 156.0 d (C3), 239.9 s (C10).
7-Dihydroxysulfonio-9,9-dimethyl-10-hydroxyphenanthrenium dication (3c)
Solution of ketone 516 (20 mg, 0.090 mmol.) in 0.12 mL of CD2Cl2 was added to solution of FSO3H (105 mg, 1.07 mmol) and SbF5 (230 mg, 1.06 mmol) in 0.25 mL of SO2ClF placed into NMR tube at −95 °C and the mixture was stirred. Formation of cation 1c10 (as an equilibrium mixture of E- and Z-isomers) was observed by NMR. Liquid SO2 (0.05 mL) was added into the NMR tube, the resulting solution was mixed, kept at −70 °C for 5 h and then at −61 °C for 2 h. The reaction results in formation of a multicomponent mixture. The content of (E)-3c is approx. 35% and that of (Z)-3c is approx. 15%.(E)-1c 1H NMR spectrum (δ, ppm, −91 °C): 1.92 s (6H, 9-Me), 7.72 t (1H, H6, J 7.3 Hz), 7.82 t (1H, H7, J 8 Hz), 7.85 d (1H, H8, J 8 Hz), 7.85 t (1H, H2, J 8 Hz), 8.44 t (1H, H3, J 7.7 Hz), 8.49 d (1H, H5, J 8.1 Hz), 8.61 d (1H, H4, J 8.5 Hz), 8.73 d (1H, H1, J 8.1 Hz), 11.31 s (OH). 13C NMR spectrum: (δ, ppm, −91 °C): 27.6 q (9-Me), 46.8 s (C9), 121.4 s (C10a), 125.5 d (C4), 125.9 d (C5), 126.0 s (C4b), 127.3 d (C8), 129.5 d (C6), 130.7 d (C2), 133.2 d (C1), 133.3 d (C7), 141.7 s (C8a), 148.2 s (C4a), 148.3 d (C3), 215.5 s (C10).
(Z)-1c 1H NMR spectrum (δ, ppm, −91 °C): 1.96 s (6H, 9-Me), 7.88 d (1H, H8, J 7.9 Hz), 7.92 t (1H, H2, J 7.5 Hz), 8.28 d (1H, H1, J 8.1 Hz), 8.67 d (1H, H4, J 8.3 Hz), 11.92 s (OH); signals of H3,5,6,7 are hidden by multiplets of the respective signals of E-isomer. 13C NMR spectrum: (δ, ppm, −91 °C): 28.9 q (9-Me), 47.8 s (C9), 120.2 s (C10a), 126.4 d (C4), 127.1 d (C8), 129.3 d (C1), 129.3 d (C6), 130.8 d (C2), 133.4 d (C7), 142.4 s (C8a), 147.6 d (C3), 147.9 s (C4a), 217.0 s (C10); signals of C4b,5 are hidden by the respective signals of E-isomer.
(E)-3c 1H NMR spectrum (δ, ppm, −91 °C): 2.00 s (6H, 9-Me), 8.04 t (1H, H2, J 8 Hz), 8.24 d (1H, H6, J 9 Hz), 8.34 s (H8), 8.57 t (1H, H3, J 8 Hz), 8.71 d (1H, H4, J 8 Hz), 8.83 d (1H, H5, J 9 Hz), 8.86 d (1H, H1, J 8 Hz), 9.72 s (2H, S(OH)2), 12.22 s (OH). 13C NMR spectrum: (δ, ppm, −91 °C): 27.1 q (9-Me), 47.4 s (C9), 122.4 s (C10a), 126.3 d (C8), 126.8 d (C4), 127.0 d (C6), 128.0 d (C5), 132.9 d (C2), 133.3 s (C4b), 134.3 d (C1), 134.9 s (C7), 142.8 s (C8a), 143.5 s (C4a), 148.6 d (C3), 216.1 s (C10).
(Z)-3c 1H NMR spectrum (δ, ppm, −91 °C): 2.00 (9-Me), 8.10 (H2), 8.24 (H6), 8.36 (H8), 8.57 (H3), 8.75 (H4), 8.80 (H5), 8.47 (H1), 9.71 (S(OH)2), 12.82 (OH). The 13C NMR assignments for (Z)-3c are unavailable because of its low concentration.
Data on generation and attempts to sulfinate cations 1d,e are given in ESI, p. S20.†
Acknowledgements
We thank the Russian Academy of Sciences (Project 5.1.4) for financial support.Notes and references
- G. A. Olah, R. H. Schlosberg, D. P. Kelly and G. D. Mateescu, J. Am. Chem. Soc., 1970, 92, 2546–2548 CrossRef CAS
.
- M. Brookhart, F. A. L. Anet and S. Winstein, J. Am. Chem. Soc., 1966, 88, 5657–5659 CrossRef CAS
- G. A. Olah and T. E. Kiovsky, J. Am. Chem. Soc., 1967, 89, 5692–5694 CrossRef CAS
- K. K. Laali and D. S. Nagvekar, J. Org. Chem., 1991, 56, 1867–1874 CrossRef CAS
-
(a) V. A. Bushmelev, A. M. Genaev, G. E. Sal'nikov and V. G. Shubin, Russ. J. Org. Chem., 2013, 49, 853–859 CrossRef CAS
- G. E. Salnikov, A. M. Genaev, V. A. Bushmelev and V. G. Shubin, Org. Biomol. Chem., 2013, 11, 1498–1501 CAS
- G. A. Olah, A. T. Ku and J. A. Olah, J. Org. Chem., 1970, 35, 3908–3912 CrossRef CAS
- C. Taeschler and T. S. Sorensen, Tetrahedron Lett., 2001, 42, 5339–5341 CrossRef CAS
-
(a) V. G. Shubin, D. V. Korchagina, A. I. Rezvukhin and V. A. Koptyug, Dokl. Akad. Nauk SSSR, 1968, 179, 119–122 CAS
- V. A. Bushmelev, A. M. Genaev, G. E. Sal'nikov and V. G. Shubin, Russ. J. Org. Chem., 2011, 47, 1057–1061 CrossRef CAS
- A. V. Afonin, I. A. Ushakov, S. V. Zinchenko, O. A. Tarasova and B. A. Trofimov, Magn. Reson. Chem., 2000, 38, 994–1000 CrossRef CAS
- E. E. Aynsley, R. D. Peacock and P. L. Robinson, Chem. Ind., 1951, 1117 CAS
- A. Commeyras and G. A. Olah, J. Am. Chem. Soc., 1969, 91, 2929–2942 CrossRef CAS
- M. M. Woyski, J. Am. Chem. Soc., 1950, 72, 919–921 CrossRef CAS
- V. P. Reddy, D. R. Bellew and G. K. S. Prakash, J. Fluorine Chem., 1992, 56, 195–197 CrossRef CAS
- T. Zincke and W. Tropp, Justus Liebigs Ann. Chem., 1908, 362, 242 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11673a |
| This journal is © The Royal Society of Chemistry 2014 |