
Effect of two facile synthetic strategies with alterable polymerization sequence on the performance of N-vinyl carbazole-based conjugated porous materials†
Wei Huangab,
Chuantao Guab,
Ting Wanga,
Chunyang Guab,
Shanlin Qiao*a and
Renqiang Yang*a
aCAS Key Laboratory of Bio-based Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China. E-mail: yangrq@qibebt.ac.cn; Fax: +86 532 80662778; Tel: +86 532 80662700
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 5th November 2014
Abstract
Four (N-vinyl carbazole)-based polymers were prepared through two facile synthetic strategies including both free radical polymerization and oxidative polymerization in different sequences. N-vinyl carbazole containing an ethylenic bond and a carbazole group is a superior candidate to study the effects of polymerization methods and sequences on the performance of conjugated microporous materials (CMPs). P2 was obtained with a BET of 878.46 m2 g−1, 12.8 times that of P1 (68.65 m2 g−1) in path 1, whereas in path 2, P3 (621.18 m2 g−1) and P4 (660.62 m2 g−1) were obtained without obvious difference in surface areas and pore structure. The dominant pore of linear polymer P1 centered at 3.94 nm, which is consistent with the polymers of intrinsic microporous (PIMs) analogues, and the counterparts P2 (ultramicropores, 0.54 nm), P3 (1.13 nm), and P4 (1.17 nm) are typical CMPs. P2 shows the best gas uptake abilities and absorbancies for organic solvents among the polymers. The results demonstrate that polymerization methods and sequences can have a great influence on the performance of materials, and only by careful choice of polymerization method and fine adjustment of the polymerization sequence can one obtain conjugated porous materials with optimum performance. Path 1, which is cost effective, high yielding and pollution free, should be regarded as the optimum selection process to prepare the N-vinyl carbazole-based porous materials. The reusability of P2 and P4 shows their impressive stability after 5 cycles of use or acid/base treatment. It is worth noting that P2 and P4 with a high porosity and pore volume are promising materials in carbon dioxide uptake, and in methane and hydrogen storage.
Introduction
In recent years, microporous organic polymers (MOPs) have emerged as an important branch of advanced porous materials, showing great potential in energy-related areas including gas storage and separation and in heterogeneous catalysis.1–6 Compared to inorganic or inorganic–organic hybrid microporous materials, MOPs have intrinsic advantages, such as high stability, synthetic diversity and ultrahigh surface areas. MOPs can be separated into two branches, crystalline and amorphous polymers, by the crystal phase. Covalent organic frameworks (COFs)7,8 are the crystalline polymers in MOP materials, while the amorphous polymers include polymers of intrinsic microporous (PIMs),9,10 hyper-crosslinked polymers (HCPs)11 and conjugated microporous polymers (CMPs).12–15 CMPs combining π-conjugation with permanent microporosity have demonstrated a striking potential in wide applications such as gas uptake, selective adsorption, heterogeneous and other photoelectric related fields. Typically, the CMPs also have advantages such as low mass density, high chemical stability and numerous selectable monomers and synthetic methods due to their organic character. It has been reported that the porous properties of CMPs depend mainly on the length of the rigid connecting strut, topological structure design and synthetic technique. Up to now, most researches have focused on the design of new building blocks and topological investigations. Few studies have explored in detail the effect of synthesis methods and sequences on the CMPs. Typically, there are substantially four methods available to synthesize CMPs, such as the synthesis of CMP-1 to CMP-4 through Sonogashira–Hagihara cross reactions,13 P1 and P2 through Suzuki coupling reactions,16 TPE-CMP through Yamamoto coupling reactions,17 and CPOP-1, P-1 and P-2 by FeCl3 oxidative coupling polymerization.18,19 In the first three methods, heavy metals such as palladium or nickel, which are expensive and contaminative, are used to trigger the reaction, so taking this into consideration, the relatively non-polluting and cheap FeCl3 is adopted as the main catalyst in our work.The unique structure of the N-vinyl carbazole-containing ethylenic double bond and carbazole group is a superior candidate with which to study the synthesis methods and sequences effects on CMPs as there are two potential active sites. It is well-known that the N-substituted carbazole is a highly electro-active group with low oxidative potential which enables the effective coupling reaction to occur between its oxidative species (radical carbazyl cation) and produces only dimers.20 As the 3- and 6-positions of carbazole are oxidation sites, N-vinyl carbazole is a potential building block to construct conjugated microporous materials through FeCl3 oxidative coupling.18 As to the ethylenic double bond, free radical polymerization is employed to prepare the conventional poly(N-vinyl carbazole) (PVK),21 and its synthesis technology has reached the level of industrial production. It is worth pointing out that the source material N-vinyl carbazole is abundant and can be synthesized easily; meanwhile, the synthesis strategies can be carried out environmentally friendly without a toxic metal catalyst, and have high yield. These advantages make it possible to produce CMPs on a large scale and place practical application on the agenda. In addition, the low framework density, large pore volume and nitrogen-containing nature of the polymers make them candidates as adsorbents for CO2 capture and H2/CH4 storage.
Here, we designed two facile synthetic paths to prepare poly(N-vinyl carbazole) which can fully utilize the two important active sites of the structure with different sequences. In the first step of path 1 and path 2, free radical polymerization and oxidative polymerization are applied to give P1 and P3, where the ethylenic double bond and 3, 6-position in the carbazole group are connected to form backbones, respectively. And through the second step in path 1 and path 2, we aim to polymerize P1 and P3 further by utilizing their important remaining active sites (the 3, 6-position of carbazole in P1 and the ethylenic bond in P3). Thus, P2 and P4 were obtained from the same two polymerization methods but in different sequences. The detailed schematic is shown in Scheme 1. Finally, the effects of synthetic methods and sequences on the performance of porous materials were investigated.
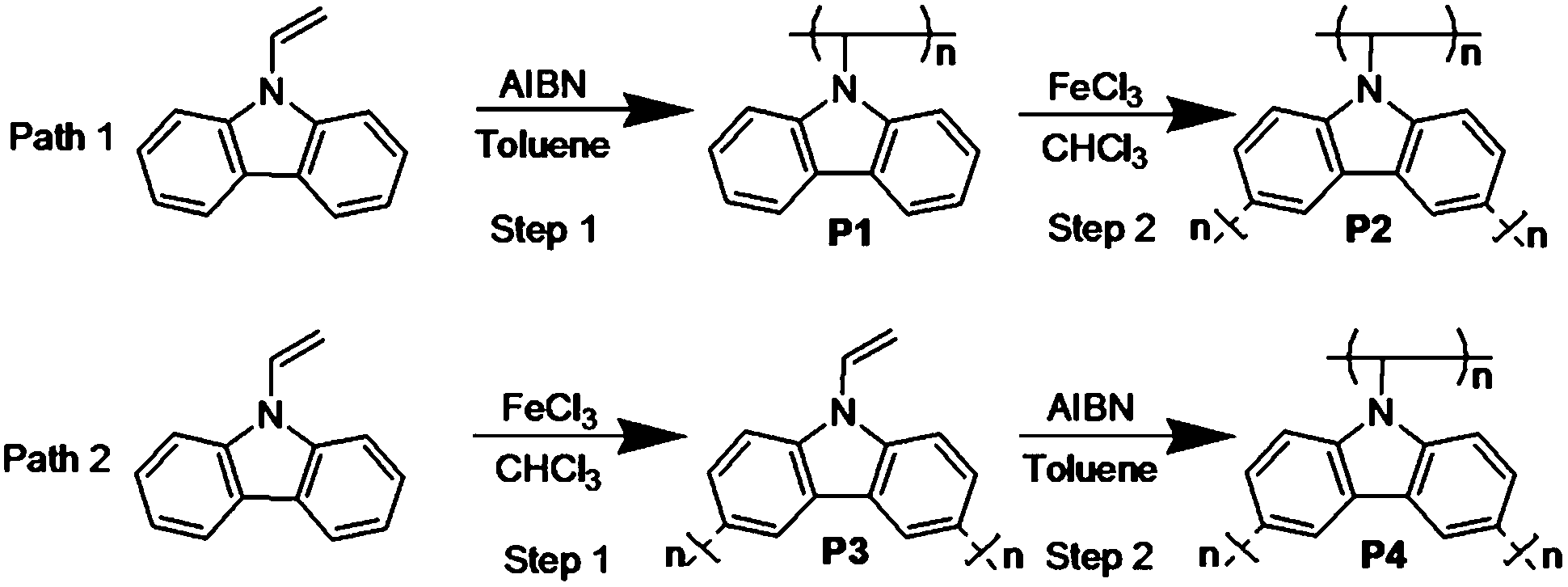 | ||
| Scheme 1 Synthetic routes of path 1 and path 2 towards the polymers P1 to P4. | ||
Experimental section
Materials
Toluene, tetrahydrofuran (THF) and chloroform were purified by distillation prior to use. 2,2′-Azobis-isobutyronitrile (AIBN) and N-vinyl carbazole (VCz) were purchased from J&K. Ferric chloride was obtained from Aldrich. All the reagents were used as received.Synthesis
P1: A typical procedure22 to prepare poly(N-vinyl carbazole) (PVK) was as follows: VCz (600 mg, 3.1 mmol) and AIBN (25 mg, 0.155 mmol) were dissolved in dry toluene. The mixture was heated to 70 °C and stirred for 6 h under a nitrogen atmosphere. Then 100 mL methanol was added to precipitate the PVK. The resulting precipitate was collected by filtration, washed with methanol three times and dried under vacuum at room temperature for 48 h to get a white solid (510 mg, 85% in yield). Calcd: C, 87.01; H, 5.74; N, 7.25. Found: C: 86.20; H, 5.32; N: 7.28.P2: P2 was straightforwardly synthesized by the FeCl3 oxidative coupling polymerization of P1 at room temperature.15 The solution of P1 (200 mg) dissolved in 30 mL anhydrous chloroform was dropwise transferred to a suspension of ferric chloride (1.38 g, 8.49 mmol) in 20 mL of anhydrous chloroform. The solution mixture was stirred for 24 h at room temperature under nitrogen protection, and then 100 mL of methanol was added to the mixture. The resulting precipitate was collected by filtration and washed with methanol and concentrated hydrochloric acid solution. After extraction in a Soxhlet extractor with methanol for 24 h, and then with THF for another 24 h, the desired polymer was collected as a brown solid and dried in a vacuum oven at 80 °C overnight (194 mg, 97% in yield). Calcd: C, 87.01; H, 5.74; N, 7.25. Found: C: 86.37; H: 5.38; N: 6.98.
P3: The solution of N-vinyl carbazole (600 mg, 3.10 mmol) dissolved in 30 mL of anhydrous chloroform was dropwise transferred to a suspension of ferric chloride (4.03 g, 24.84 mmol) in 20 mL of anhydrous chloroform. The solution mixture was stirred for 24 h at room temperature under nitrogen protection, and then 100 mL of methanol was added to the mixture. The resulting precipitate was collected by filtration and washed with methanol and concentrated hydrochloric acid solution. After extraction in a Soxhlet extractor with methanol for 24 h, and then with THF for another 24 h, the desired polymer was collected as a dark brown solid and dried in a vacuum oven at 80 °C overnight (190 mg, 95% in yield). Calcd: C, 87.01; H, 5.74; N, 7.25. Found: C: 86.93; H: 5.43; N: 6.87.
P4: P3 (200 mg) and AIBN (11 mg, 0.067 mmol) were dispersed in dry toluene. The solution was heated at 70 °C under magnetic stirring for 6 h under a nitrogen atmosphere. The resulting precipitate was collected by filtration and washed with methanol and concentrated hydrochloric acid solution. After extraction in a Soxhlet extractor with methanol for 24 h, and then with THF for another 24 h, the desired polymer was collected as a light brown solid and dried in a vacuum oven at 80 °C overnight (186 mg, 93% in yield). Calcd: C, 87.01; H, 5.74; N, 7.25. Found: C: 86.48; H: 5.01; N: 6.79.
Characterization
The structures of P1 to P4 were confirmed by 13C NMR spectroscopy and Fourier-transform IR (FTIR). P1 was measured by the Bruker Avance III model 600 MHz using CDCl3 as the solvent. 13C CP/MAS solid-state NMR measurements were carried out on a Bruker Avance III model 400 MHz NMR spectrometer at a MAS rate of 5 kHz to confirm the structure of P2, P3 and P4. FT-IR spectra were collected in the attenuated total reflection (ATR) mode on a Thermo Nicolet 6700 FT-IR Spectrometer. Thermogravimetric analysis (TGA) was carried out using a SDT Q600 V20.9 Build with a temperature ramp of 10 °C min−1 from 20 °C to 800 °C under N2 atmosphere. Scanning electron microscopy (SEM) was recorded using a Hitachi S-4800 with acceleration voltage 5.0 kV and working distance 8.9 mm (P1 and P2) and 8.8 mm (P3 and P4). Samples were coated with a thin layer of Au before investigation. Powder X-ray diffractometer (P-XRD) patterns were obtained using a Bruker X-ray diffractometer. Data were collected for 2θ from 5.00° to 60.00° (θ is the angle of diffraction). The polymers were degassed at 120 °C for 10 h under vacuum before analysis to remove residual moisture and other trapped gases. And all the samples were tested on an Autosorb-iQ-MP-VP volumetric adsorption analyzer with the same degassing procedure. The BET surface areas and pore size distributions of P1 to P4 were measured by nitrogen adsorption–desorption at 77 K in a liquid nitrogen bath. CO2 and CH4 uptake capacities were measured at 273 K and 298 K up to 1.1 bar. The reusability was tested by 5 cycles of adsorption and desorption measurements with the same degassing and test procedure above.Results and discussion
Four N-vinyl carbazole-based polymers were synthesized by different polymerization methods and sequences. Their synthesis procedures are shown in Scheme 1. The free radical polymerization was used in the polymerization of the ethylenic double-bond containing materials. And the FeCl3 oxidative polymerization of carbazole-based building block has been reported to give many kinds of CMPs with impressive performances. The two polymerization methods are both effective ways to synthesize N-vinyl carbazole-based polymers.FTIR and NMR spectra
The structures of the polymers were characterized by FT-IR and NMR. FTIR spectra (Fig. 1a), in which the strong absorption bands at 721 cm−1 and 744 cm−1 are assigned to the ring deformation of the substituted aromatic structure and CH2 deformation of the vinylidene group, respectively. The bands at 3050 cm−1 and 2930 cm−1 correspond to the stretching vibration of the C–H bonds in the aromatic ring and vinylidene group. The obvious absorption bands at 1450 cm−1 and 1330 cm−1 are attributed to the stretching vibration of C–H in the aromatic ring and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in the vinylidene group. The characteristic peak at 1219 cm−1 is the ring vibration of the N-vinyl carbazole moiety. It is obvious that the peaks in P2 are broader than those in P1, P3 and P4, which demonstrates the higher degree of polymerization in P1. The peak positions of the obtained polymers are consistent with the infrared PVK spectra reported previously.23
C in the vinylidene group. The characteristic peak at 1219 cm−1 is the ring vibration of the N-vinyl carbazole moiety. It is obvious that the peaks in P2 are broader than those in P1, P3 and P4, which demonstrates the higher degree of polymerization in P1. The peak positions of the obtained polymers are consistent with the infrared PVK spectra reported previously.23
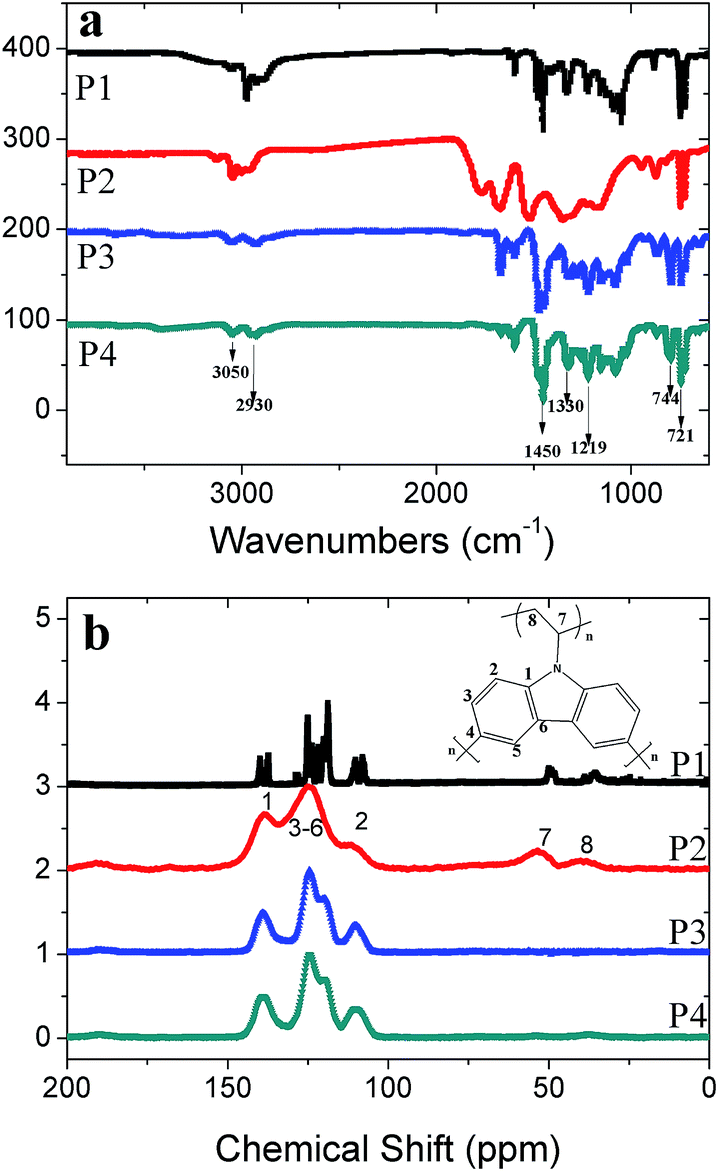 | ||
| Fig. 1 (a) FTIR spectra for P1 to P4 were collected in ATR mode (the transmittance of P1, P2 and P3 shifted by 100, 200 and 300); (b) solid state magic angle spinning 13C CP/MAS NMR spectra (the curves were shifted by 1, 2 and 3 units after normalization). | ||
It can be seen that in the NMR spectra (Fig. 1b) the four polymers have similar signal positions in the low-field region for the aromatic area. The signal at 139 ppm corresponds to the aryl-carbon binding with the carbazole nitrogen atom, while the broad peaks around 110 ppm, 120 ppm and 124 ppm are also typical characteristic peaks of N-vinyl carbazole. In the NMR spectra of P1 and P2, the peaks at 102 ppm and 129 ppm, which are assigned to the double bond of the vinylidene group (Fig. S2†), disappear or weaken in the polymers after the free radical polymerization in path 1. At the same time, the appearance of peaks at 35 ppm and 50 ppm is another proof that part of the vinylidene double bond has transformed into an alkyl group with a single bond. It is obvious that P2 is quite different from P1 as it has a higher degree of polymerization.24 P3 and P4, which involve path 2, have similar curve shapes and peak signal positions, although the peaks of P3 are a little sharper than those of P4. In contrast to path 1, there are no remarkable peaks at 35 ppm and 50 ppm in the spectra of P3 and P4. This may be explained in this way: after the first step of polymerization, the materials have obtained the highest phase separation and in the next step just a small amount of the crosslinking reaction takes place on the surface of the interparticles. It is notable that the peaks of P2 are broader than those of its analogues, which could be ascribed to the highest degree of polymerization amongst the four polymers.24 The results of NMR may show that path 1 is better than path 2, as the high crosslinking degree of monomers is essential to construct porous materials.
TGA, SEM and XRD analysis
The polymers obtained through path 1 and path 2 were also investigated by thermogravimetric analysis (TGA). The results are shown in Fig. 2a. A small weight loss observed in the initial stages was due to the residual solvents and the adsorbed moisture inside the polymers.25 As the temperature increased to 300 °C, P1 started to decompose or evaporate and the sample was totally gone when the temperature rose to 500 °C. The curve of P2 is quite different from P1 curve in path 1. As to path 2, it is obvious that the curves of P3 and P4 are quite similar in shape and turning point. The highest mass loss of P2 below 410 °C may be ascribed to the greatest BET surface and largest pore volume available to trap adsorbate. In all cases, P2, P3 and P4 are stable up to 400 °C. Scanning electron microscopy images (Fig. 2b) demonstrate that P1 to P4 are all aggregation morphologies showing solid sub-micron spheres. In path 1, P1 and P2 are composed of particles with different sizes, whereas, in the case of path 2, one can clearly see that the morphology of P3 is different from that of P4. P-XRD (Fig. S3†) shows that P1 has two diffraction peaks at around 7.5° and 23.5°, both characteristic peaks of PVK, in agreement with previous reports.26 The broad peak from 15° to 25° in P2 to P4 indicates that most of the N-vinyl carbazole was polymerized to form amorphous random materials. | ||
| Fig. 2 (a) Thermogravimetric analysis of P1 to P4; (b) field-emission scanning electron microscopy images of P1 to P4 (scale bar: 1 μm). | ||
Pore properties analysis
Nitrogen adsorption–desorption isotherms were measured to analyze the porous properties of microporous polymers.19,27,28 The surface areas of P1 to P4 calculated by the Brunauer–Emmett–Teller (BET) method in the relative pressure (P/P0) range from 0.05 to 0.20 are 68.65 m2 g−1, 878.46 m2 g−1, 621.18 m2 g−1 and 660.62 m2 g−1, respectively (Fig. 3b). The pore size distributions were calculated from the adsorption branches of the isotherms using the quenched solid density functional theory (QSDFT). As can be clearly seen, the pore width of the four polymers is 3.94 nm, 0.54 nm, 1.13 nm and 1.17 nm, respectively. Detailed information on the porosity structural parameters of P1 to P4 is shown in Table 1. | ||
| Fig. 3 a) Nitrogen adsorption–desorption isotherms of polymers measured at 77 K; (b) pore size distributions calculated using quenched solid density functional theory; the inset is the mesopore size distribution ranging from 2 to 35 nm. | ||
| Polymer | SBET [m2 g−1]a | SLang [m2 g−1] | V0.1 [cm3 g−1]b | Vtotal [cm3 g−1]c | Width [nm]d |
|---|---|---|---|---|---|
| a Surface area calculated from the N2 isotherm.b The micropore volume at P/P0 = 0.10.c Total pore volume at P/P0 = 0.99.d Dominant pore size calculated by quenched solid density functional theory (QSDFT). | |||||
| P1 | 69 | 137 | 0.10 | 0.60 | 3.94 |
| P2 | 878 | 1105 | 0.25 | 1.00 | 0.54 |
| P3 | 621 | 846 | 0.16 | 0.55 | 1.13 |
| P4 | 661 | 928 | 0.24 | 0.66 | 1.17 |
P1: The adsorption–desorption isotherm of P1 demonstrates that it is a mesoporous material, as there is little increase at a relatively low pressure and a steep gas uptake at a relatively high pressure (P/P0 = 0.9–1.0). The pore size distribution of P1 is another proof of its mesoporous nature, as its pore width is mainly 3.94 nm. P1 is a linear polymer which can be classified as polymer of intrinsic microporous (PIMs) analogues because the N-vinyl carbazole is not rigid enough to construct a microporous architecture by connecting only the ethylenic double bonds together.
As an ultramicropore conjugated polymer, among the four polymers P2 has the largest BET surface areas (878.46 m2 g−1) and narrowest pore size distribution centered at 0.54 nm. The isotherm of P2 shows a steep increase at a relatively low pressure, a slow increase at a relatively intermediate pressure, and a sharp gas uptake at a relatively high pressure. The high adsorption at low pressure can be interpreted as surface adsorption and micropore filling. And the gradual increase at relatively high pressure may be due to the meso/macrostructures and interparticular void in the polymer. The post-polymerization of P1 in the second step of path 1 prompts the 3, 6-position in the carbazole group to connect successfully, and the obtained P2 shows a narrower pore distribution with a great improvement in the BET surface areas and pore volume, which are beneficial for gas storage and separation. It can thus be seen that P2 as a post-polymer of P1 is far better than its initial parent polymer in all aspects.
It was found that P3 and P4 are both typical CMPs, possessing similar nitrogen adsorption and desorption isotherms. P3 and P4 share a nearly equivalent BET surface areas of 621.18 m2 g−1 and 660.62 m2 g−1, respectively. Furthermore, the two polymers exhibit the same dominant pore width centered at 1.13 nm and 1.17 nm. P4 shows a small increase of surface area and pore volume when compared with P3, which can be attributed to a little crosslinking reaction taking place on the surface of interparticles and the swelling effect in the second step of path 2. The accordant pore-size distributions of P3 and P4 demonstrate that the second step of path 2 does not change the pore width, which is determined by the stiff backbone. It also illustrates that the reaction happened on the surface of the interparticles only, but not internally in the P3.
Taking path 1 and path 2 in comparison, one can easily conclude that the former is superior to the latter, as P2 is much better than P4 in its porous properties. Path 1, which is cost effective, high yielding and pollution-free, should be regarded as the optimum choice to prepare the N-vinyl carbazole-based porous materials.
Gas uptake
 | ||
| Fig. 4 (a) CO2 adsorption–desorption properties of the polymers at 273 K; (b) CO2 isosteric enthalpies with the amount adsorbed (calculated by CO2 adsorption isotherms of the polymers at 273 K and 298 K); (c) reusability of P2 and P4 in five cycles and stability of P2 and P4 after treatment with NaOH (6 M) and HCl (6 M) for 1d. | ||
To further investigate the reusability and stability of polymers, P2 and P4 were chosen to go through five cycles and acid/base treatment because of their large BET surface areas and pore volume. As shown in Fig. 4c, the CO2 uptake of P2 and P4 does not decline after 5 cycles. In addition, the polymers are stable, even when treated with 6 M NaOH and 6 M HCl solutions. These results demonstrate that the pore structures of P2 and P4 are robust and that their performance will not weaken, even under rigorous conditions.
 | ||
| Fig. 5 (a) H2 adsorption at 77 K; (b) CH4 adsorption at 273 K; (c) reusability in uptake of H2 by P2 and P4 in five cycles and stability of P2 and P4 after treatment with NaOH (6 M) and HCl (6 M) for 1 day. (d) reusability in uptake of CH4 by P2 and P4 in five cycles and stability of P2 and P4 after treatment with NaOH (6 M) and HCl (6 M) for 1 day. | ||
Methane uptake
Although the combustion of methane produces CO2, it still has environmental advantages as a transitional fuel source with respect to other fossil fuels, due to its high ratio of hydrogen to carbon. Furthermore, it is available in large quantities, like combustible ice. Storing methane via adsorption on porous materials is the most promising near-term route because it allows operation at reasonable pressures (1–300 bar) and temperatures (7–298 K) and does not require extra energy input for conversion into higher hydrocarbons or methanol. Adsorption in nanoporous materials such as MOPs is very efficient and promising, because it can obtain methane densities comparable to those of the compressed phase at a much lower pressure and ambient temperature in lightweight carriers.40 The four polymers with a low framework density and large pore volume are alternatives in methane storage. The uptake capacities for methane of the prepared polymers were measured at 273 K from zero to 1.0 bar. As shown in Fig. 5b, methane uptakes of P1 to P4 are 0.26 wt%, 1.62 wt%, 1.08 wt% and 1.18 wt% at 273 K/1.0 bar, respectively. One can observe that methane-uptake capacities increases corresponding to the BET surface areas and total pore volume, as the order of the methane uptake ability is P2 > P4 > P3 > P1.The dominant interaction of methane with the surface is the Van der Waals attraction. Goddard and coworkers have found that CH4 adsorption in polymers can occur with the coexistence of layer formation and pore filling.41 Porous materials show potential applications in the purification of clean-burning natural gas,36 as the existence of CO2 in natural gas can reduce the energy density of fuel. A promising material for practical application requires not only a high CO2 uptake but also excellent CO2 selectivity. The CO2/CH4 selectivities of P1 to P4 are calculated using the initial slopes at low pressure in the Henry's law region. The calculations show that the CO2/CH4 selectivities of P1 to P4 are 4.34, 6.9, 5.7 and 5.71, respectively. As the four polymers are polymerized from the same monomers, the differences in selectivity may be caused mainly by the pore structure.
Conclusions
We report two facile strategies with alternative polymerization sequences to prepare (N-vinyl carbazole)-based porous polymers for gas storage. The two strategies can synthesize polymers with high-yielding, cost-effective and full utilization of the two different active sites of the N-vinyl carbazole unit by free radical polymerization and FeCl3 oxidative polymerization. The BET surface areas of the obtained materials P1 to P4 show a large range from 68.65 m2 g−1 to 878.46 m2 g−1. In path 1, in contrast to the linear PIMs analogue P1 with a dominant pore size of 3.94 nm, P2 is an interpenetrating ultramicropore conjugated polymer (dominant pore size 0.54 nm). In path 2, P4 shows slightly higher BET surface areas than, and similar pore width distribution to, P3, indicating that little crosslinking reaction occurs at the next polymerization stage. The CO2, H2, and CH4 adsorption isothermals show that P2 has the highest gas-uptake capacity among the four polymers (for CO2 it is 12.95 wt% at 273 K/1.0 bar, for H2 it is 1.36 wt% at 77 K/1.0 bar and for CH4 it is 1.62 wt% at 273 K/1.0 bar), which is comparable to the highest reported for CMPs under similar conditions. The stabilities of P2 and P4 were investigated. It turns out they are quite stable after 5 cycles. The gas uptake results indicate that path 1, which is cost effective, high-yielding and pollution free, can be regarded as the optimum way to prepare poly(N-vinyl carbazole) CMPs. This work clearly reveals that selection of the appropriate monomer and the right polymerization strategies are crucial considerations for materials with excellent gas-uptake capacities.Acknowledgements
This work was supported by National Natural Science Foundation of China (21274161, 51173199, 51303197), the Ministry of Science and Technology of China (2014CB643501, 2010DFA52310), Shandong Provincial Natural Science Foundation (ZR2011BZ007), and Qingdao Municipal Science and Technology Program (11-2-4-22-hz).Notes and references
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS PubMed.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163–5176 CrossRef CAS.
- W. Lu, D. Yuan, J. Sculley, D. Zhao, R. Krishna and H.-C. Zhou, J. Am. Chem. Soc., 2011, 133, 18126–18129 CrossRef CAS PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng and J. M. Simmons, Angew. Chem., Int. Ed., 2009, 121, 9457–9460 CrossRef PubMed.
- P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS.
- L. Chen, Y. Yang and D. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- J. Xia, S. Yuan, Z. Wang, S. Kirklin, B. Dorney, D.-J. Liu and L. Yu, Macromolecules, 2010, 43, 3325–3330 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- M. Tsyurupa and V. Davankov, React. Funct. Polym., 2006, 66, 768–779 CrossRef CAS PubMed.
- J. H. Choi, K. M. Choi, H. J. Jeon, Y. J. Choi, Y. Lee and J. K. Kang, Macromolecules, 2010, 43, 5508–5511 CrossRef CAS.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky and Y. Z. Khimyak, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., 2011, 123, 8912–8916 CrossRef.
- S. Qiao, Z. Du and R. Yang, J. Mater. Chem. A, 2014, 2, 1877–1885 CAS.
- J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS PubMed.
- Y. Xu, L. Chen, Z. Guo, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2011, 133, 17622–17625 CrossRef CAS PubMed.
- Q. Chen, M. Luo, P. Hammershøj, D. Zhou, Y. Han, B. W. Laursen, C.-G. Yan and B.-H. Han, J. Am. Chem. Soc., 2012, 134, 6084–6087 CrossRef CAS PubMed.
- S. Qiao, Z. Du, C. Yang, Y. Zhou, D. Zhu, J. Wang, X. Chen and R. Yang, Polymer, 2014, 55, 1177–1182 CrossRef CAS PubMed.
- C. Gu, N. Huang, J. Gao, F. Xu, Y. Xu and D. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4850–4855 CrossRef CAS PubMed.
- N. Tsutsumi, K. Kinashi, A. Nonomura and W. Sakai, Materials, 2012, 5, 1477–1486 CrossRef CAS PubMed.
- T. H. Kuo, C. J. Weng, C. L. Chen, Y. L. Chen, C. H. Chang and J. M. Yeh, Polym. Compos., 2012, 33, 275–281 CrossRef CAS.
- K. I. Kim, C. Basavaraja and D. S. Huh, Bull. Korean Chem. Soc., 2013, 34, 1391–1396 CrossRef CAS.
- D. S. Das, D. P. Chatterjee and A. K. Nandi, J. Mater. Chem. A, 2014, 2, 12031–12042 Search PubMed.
- Y. Zhu, H. Long and W. Zhang, Chem. Mater., 2013, 25, 1630–1635 CrossRef CAS.
- A. T. Gokceoren and C. Erbil, Polym. Compos., 2013, 34, 1986–1998 CrossRef.
- Q. Zhang, Y. Yang and S. Zhang, Chem.–Eur. J., 2013, 19, 10024–10029 CrossRef CAS PubMed.
- Z. Wang, B. Zhang, H. Yu, G. Li and Y. Bao, Soft Matter, 2011, 7, 5723–5730 RSC.
- A. Samanta, A. Zhao, G. K. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2011, 51, 1438–1463 CrossRef.
- X. Zhu, C. Tian, S. M. Mahurin, S.-H. Chai, C. Wang, S. Brown, G. M. Veith, H. Luo, H. Liu and S. Dai, J. Am. Chem. Soc., 2012, 134, 10478–10484 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- Q. Chen, J.-X. Wang, Q. Wang, N. Bian, Z.-H. Li, C.-G. Yan and B.-H. Han, Macromolecules, 2011, 44, 7987–7993 CrossRef CAS.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS PubMed.
- R. Dawson, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1173–1177 RSC.
- Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 4, 1–7 RSC.
- W.-C. Song, X.-K. Xu, Q. Chen, Z.-Z. Zhuang and X.-H. Bu, Polym. Chem., 2013, 4, 4690–4696 RSC.
- M. G. Rabbani and H. M. El-Kaderi, Chem. Mater., 2011, 23, 1650–1653 CrossRef CAS.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel and R. Krishna, Chem. Mater., 2010, 22, 5964–5972 CrossRef CAS.
- T. B. Lee, D. Kim, D. H. Jung, S. B. Choi, J. H. Yoon, J. Kim, K. Choi and S.-H. Choi, Catal. Today, 2007, 120, 330–335 CrossRef CAS PubMed.
- X. Zhu and Y.-P. Zhao, J. Phys. Chem. C, 2014, 118, 17737–17744 CAS.
- J. L. Mendoza-Cortes, S. S. Han, H. Furukawa, O. M. Yaghi and W. A. Goddard III, J. Phys. Chem. A, 2010, 114, 10824–10833 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed NMR (1H NMR and 13C NMR) of materials Fig. S1 and S2; XRD for P1 to P4 (Fig. S3), additional gas adsorption properties Fig. S4 and S5, selectivity of CO2/CH4 (Fig. S6). See DOI: 10.1039/c4ra11611a |
| This journal is © The Royal Society of Chemistry 2014 |