
Biogas upgrading through kinetic separation of carbon dioxide and methane over Rb- and Cs-ZK-5 zeolites†
Abstract
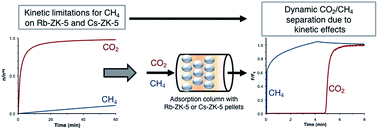
Eight-membered ring (8 MR) zeolites hold large potential for industrial CO2 separations such as biogas separation. They offer large selectivity due to the constrained environment for adsorption, especially when large cations are present in the interconnecting windows. The Rb- and Cs-exchanged ZK-5 zeolites (8 MR KFI type zeolites) were studied for kinetic CO2/CH4 separation. First, Rb-ZK-5 and Cs-ZK-5 were thoroughly characterized via chemical analysis, argon porosimetry, X-ray diffraction and Rietveld refinements. Afterwards, the CO2/CH4 separation potential of both adsorbents was assessed via the measurement of kinetic and equilibrium data (T = 261.15 - 323 K), breakthrough measurements at 303 K (P = 1 - 8 bar), and simulations of their performance. The high occupation of the central 8 MR sites with large cations causes strong diffusional limitations for CH4 on Rb-ZK-5 and Cs-ZK-5. As a result, both zeolites effectively separate CH4 from CO2 with very high selectivities (α = 17 at 1 bar and 303 K). Despite their very large CO2 selectivities, the performance of Rb-ZK-5 and Cs-ZK-5 was still lower than for the benchmark 13X zeolite on a larger scale. Future research needs to further unravel the adsorption mechanism on low-silica 8 MR zeolites and their corresponding potential in separation processes such as biogas purification.
 Please wait while we load your content...
Please wait while we load your content...

