
Direct visualization of microphase separation in block copoly(3-alkylthiophene)s†
Pieter Willota,
Joan Teyssandierb,
Wouter Dujardina,
Jinne Adisoejosob,
Steven De Feyterb,
David Moermanc,
Philippe Leclèrec,
Roberto Lazzaronic and
Guy Koeckelberghs*a
aLaboratory for Polymer Synthesis, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: guy.koeckelberghs@chem.kuleuven.be
bLaboratory of Photochemistry and Spectroscopy, Division of Molecular Imaging and Photonics, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
cLaboratory for Chemistry of Novel Materials, Center of Innovation and Research in Materials & Polymers (CIRMAP), University of Mons – UMONS/ Materia Nova, Place du Parc 20, B7000 Mons, Belgium
First published on 22nd December 2014
Abstract
A poly(3-octylthiophene)-block-poly(3-butylthiophene) block copolymer was synthesized in a one-pot block copolymerization reaction, starting from a functional o-tolyl initiator in order to maximize A–B diblock copolymer formation. First, the composition of this block copolymer is extensively studied using gel permeation chromatography (GPC) and 1H NMR measurements. A complete block copolymer formation is obtained with almost equal block length; B–A–B block copolymer contamination is shown to be very limited. In a second part, the self-assembly was analysed through differential scanning calorimetry (DSC), atomic force microscopy (AFM) and scanning tunnelling microscopy (STM) measurements, focusing on the microphase separation. A direct visualization of the different microphases can be obtained with STM.
Introduction
All-conjugated block copolymers are promising materials for application in organic electronics as they can combine and improve the properties of the different constituting blocks in one molecule.1,2 Moreover, they distinguish themselves from polymer blends through unique aspects such as intrinsic stability, self-assembly, microphase separation, etc. The production of new all-conjugated block copolymers has been growing dramatically in the last few years, because of the broadening insight into the controlled polymerization mechanisms which are being applied to an ever increasing number of conjugated monomer systems.3–23 The increased control over the polymerization allows for a much better control over the molecular and, also, supramolecular structure.13,24–28 Applied to block copolymers with sufficiently differing blocks, this can result in a control over the microscopic morphology which is created by the microphases of the constituting blocks. In turn, a thermodynamically stable morphology can significantly enhance the performance and lifetime of e.g. bulk heterojunction solar cells.29 Of course, as the synthetic possibilities broaden, the implementation of accurate and reliable analytical techniques needs to keep pace.The microphase separation behavior of block copolymers is most often studied with X-ray diffraction techniques, differential scanning calorimetry (DSC), atomic force microscopy (AFM) and transmitting electron microscopy. These techniques give insight into different aspects of the morphology of the polymers. The X-ray diffraction techniques such as (Grazing Incidence) Wide Angle X-ray Scattering ((GI)WAXS),24,26,30,31 Wide Angle X-ray Diffraction (WAXD),27,31 Small Angle X-ray Scattering (SAXS)31 and X-ray diffraction (XRD)28,31,32 can identify the presence of different crystalline microphases by correlating the different observed refraction signals with the different microphases and these refraction signals also give insight in the lattices and stacking distances. The obtained stacking distances can this way directly be correlated with the different microphases that are present. Also Differential Scanning Calorimetry (DSC) measurements are frequently used to confirm the presence of separate microphases.24,26–28,31,32 This is often reflected in the presence of different melting peaks, that are normally ascribed to different microphases in the block copolymer, with their own semi-crystallinity.24,31,32 Nevertheless, these techniques do not provide an actual visualization of the phase separation. Atomic Force Microscopy (AFM)24,27–29,31–34 and Scanning or Transmitting Electron Microscopy (SEM29 or TEM25,31 respectively) images are recorded to reveal the structure down to the nanometer scale. These techniques are capable of visualizing the supramolecular morphology of the block copolymers. Different domains in the polymer film can often be identified and are generally tentatively ascribed to the two microphases which are present in the block copolymer.24,28 These techniques, however, do not provide information on the chemical nature of the different domains, therefore these domains cannot unambiguously be assigned to one of the constituting blocks.
In order to extend the structural characterization to the molecular scale, Scanning Tunneling Microscopy (STM) is of prime interest as it is capable of imaging materials with sub-angström resolution, also at the liquid–solid interface.35 STM measurements have already been performed on several conjugated oligomers36–41 and polymers,42–44 including poly(3-alkylthiophene)s (P3AT).45–53 In those studies it has been demonstrated that molecularly resolved individual strands of regioregular P3AT can clearly be visualized and also that the length of the alkyl side-chains has a clearly marked influence on the chain-to-chain distance of the lamellae. Hence, STM could possibly be used to directly visualize the microphase separation. This type of information is of major importance in the analysis of microphase separation in block copolymers.
Microphase separation has been observed in an array of P3AT block copolymers with different combinations of side-chains. It has been found that microphase separation occurs if the difference in length of the alkyl chains between the blocks is more than 2 carbon atoms.32 In this work, we therefore consider a poly(3-octylthiophene)-b-poly(3-butylthiophene) (P3OT-b-P3BT) block copolymer, i.e., with an alkyl chain length difference of 4 carbon atoms, to ensure microphase separation and to enable an optimal study thereof.
Results and discussion
Polymer synthesis
For poly(3-octylthiophene)-b-poly(3-butylthiophene), Wu et al. have already confirmed by DSC and X-ray studies that microphase separation occurs in this polymer, making it an ideal candidate for this study.31 The most straightforward synthetic approach towards this block copolymer is by using a Ni(dppp)-catalyzed Kumada Catalyst Transfer Protocol (KCTP) and forming the block copolymer by successive monomer addition in a one-pot polymerization.54,55 However, recent advances in this field have shown that the chain-walking of the Ni-catalyst over the polymer chain has a clear effect on the block copolymerization, leading to the formation of not only A–B diblock copolymers, but also a significant amount of B–A–B triblock copolymers.56,57 This results in ill-defined materials which are not ideal for further investigation of the microphase formation. To avoid these B–A–B contaminants as much as possible, a functional o-tolyl initiator is used, in order to maximize unidirectional growth and a preferential formation of the A–B block copolymer (Fig. 1A).57,58 | ||
| Fig. 1 (A) Polymerization pathway in the synthesis of P3OT and P3OT-b-P3BT. (B) GPC elution curves of P3OT and P3OT-b-P3BT. | ||
The initiating entity (2) is formed from 1 through a ligand exchange with 2 eq. of diphenylphosphinopropane (dppp) after which a solution of the 3-octylthiophene monomer (4) is added. After a polymerization time of 1 hour, a part of the polymerization mixture is quenched with acidified THF to yield the homopolymer P3OT, while to the remainder, the 3-butylthiophene monomer (6) is added. After an additional block copolymerization time of 2 h, the block copolymer P3OT-b-P3BT is quenched with acidified THF, precipitated in MeOH and filtrated. Note that no longer reaction times were chosen to avoid disproportionation, which would result in undesired B–A–B triblock chains. The block copolymer is then further extracted with MeOH and CHCl3 in a Soxhlet extraction to remove salts and catalyst residues. The CHCl3-fraction is again precipitated in MeOH, filtered off and used for analysis. GPC analysis of the homopolymer P3OT and the block copolymer P3OT-b-P3BT shows a clear and complete shift from a ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n-value of 9.3 kg mol−1 for P3OT to 14.5 kg mol−1 for P3OT-b-P3BT while the dispersity remains very low (Ð = 1.1 for both polymers) (Fig. 1B).
n-value of 9.3 kg mol−1 for P3OT to 14.5 kg mol−1 for P3OT-b-P3BT while the dispersity remains very low (Ð = 1.1 for both polymers) (Fig. 1B).
These results indicate the successful formation of the block copolymer. The structure of P3OT-b-P3BT is further analyzed using 1H NMR spectroscopy to identify the composition of the block copolymer. The degree of polymerization can be calculated from the 1H NMR spectrum, by comparing the relative integration values of signals corresponding to the different end groups with those of the repeating units. In Fig. 2, all relevant signals have been assigned to the corresponding protons. These values are used in the chain length determination as described by Hardeman et al.:59
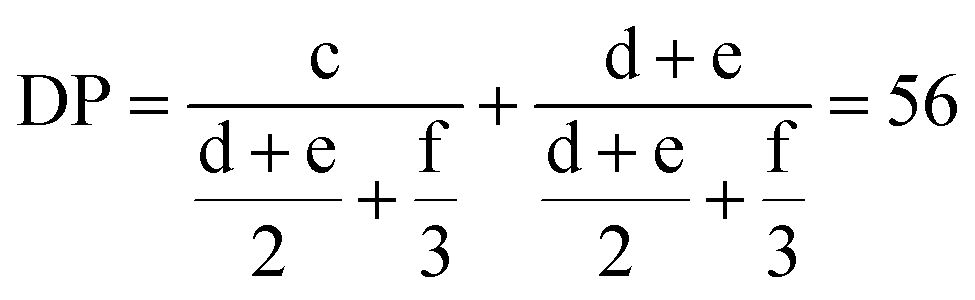
 | ||
| Fig. 2 Interpretation of the 1H NMR spectrum of P3OT-b-P3BT. The signals that are needed for the analysis of the chain-length and composition are assigned to the corresponding protons. | ||
A degree of polymerization of 56 units is found using this equation. Also note that the integration value of the aromatic signals at 6.98 ppm confirms this DP if they are scaled using the same method, corrected for the fact that the aromatic signal only corresponds to one proton per unit. This is also shown in the integration value, which equals half the integration value of signal c. If we then consider the 1H NMR signals of the CH3-groups in the sidechains of both 3-butylthiophene (signal g) and 3-octylthiophene (signal h), the relative integration of 3-butylthiophene is 1.07 times higher than that of 3-octylthiophene. Considering also the total DP of 56 units, it can be concluded that the 3-octylthiophene block has a length of about 27 units and the 3-butylthiophene block consists of 29 units. Thorough analysis of the α-methylene region of the 1H NMR spectrum (signals c–f) further indicates that ±96% of the polymer chains is indeed an A–B block copolymer, and that the amount of contaminants is limited (see ESI†).
Microphase separation study
With this understanding of the composition, the structural behavior of the block copolymer can be investigated. Differential scanning calorimetry (DSC) results of P3OT-b-P3BT are in line with previous results.31 Two melting transitions are observed: one broad signal at 163 °C and one sharp peak at 252 °C. The signal at 163 °C can be ascribed to the P3OT block and the signal at 252 °C to the P3BT block. During the cooling cycle, two corresponding crystallization peaks are also observed (Fig. S3 and S4, ESI†). The presence of two different melting transitions indicates that there is indeed microphase separation in the P3OT-b-P3BT block copolymer. The morphological properties of the synthesized block copolymer are analyzed by AFM measurements to study the behavior in thin film. For this purpose, the block copolymer is dissolved in chlorobenzene and dropcasted onto an ITO-coated glass substrate. The solvent is evaporated overnight in a solvent-saturated atmosphere to favour self-assembly of the polymer chains. A representative image is depicted in Fig. 3. A zoom is present in ESI† (Fig. S5) The AFM measurements clearly show the formation of a fibre-like morphology for the block copolymer. The width of the fibres is 26.1 ± 2.8 nm and they are several microns long. These results are very similar as in P3AT homopolymers.60–62 Such a morphology is the result of the π–π stacking of polymer chains oriented perpendicular to the fiber axis. Note that the white spots probably correspond to aggregates due to the incomplete dissolution of the polymer, even after prolonged heating of the solutions at 60 °C. There are no apparent features visible (e.g., the presence of an internal structure within the fibers) that could indicate microphase separation. This however does not imply the absence of phase separation, but may be simply due to the limited resolution of the AFM measurements at that scale. To obtain more insight into the microphase separation, we need to image this copolymer at a smaller scale. For that purpose, STM measurements of the P3OT-b-P3BT block copolymer were performed. | ||
| Fig. 3 Height (left) and phase (right) tapping-mode AFM image of a thin film of P3OT-b-P3BT. | ||
In order to study the morphology on highly ordered pyrolytic graphite (HOPG), a solution of P3OT-b-P3BT in 1,2,4-trichlorobenzene (TCB) with a concentration of 7.31 × 10−3 mg mL−1 is used to record in situ images at the HOPG–TCB interface. In these images, a sub-molecular resolution can be obtained, enabling the identification of individual thiophene units (Fig. S6, ESI†). A typical image at sub-monolayer coverage is shown in Fig. 4a. In this image, individual strands of the block copolymer are visible. These strands are laterally stacked, as has been observed in previous measurements of P3ATs.47 However, in case of the P3OT-b-P3BT block copolymer, the formation of two types of domains can clearly be observed, in which the lateral distance between the strands differs. These lateral distances have been evaluated by analysis of several series of images. The average value of the most dense domains (x) is 1.2 ± 0.1 nm, while the less dense domains (y) show a periodicity of 1.7 ± 0.1 nm. This difference in lateral distance can be correlated with the side-chain length in the two blocks. Indeed, previous reports showed an increasing lateral distance for longer side-chains.46,47,49,51,52 Taking this into account, the denser domains can be ascribed to the P3BT-block and the less dense domains are ascribed to the P3OT-block. These values correlate very well with the previously obtained values for hexyl and dodecyl side-chains (Fig. 4b). From these values, we can tentatively establish a linear correlation between the side-chain length and the lateral stacking distance. These STM results clearly demonstrate that the two different blocks assemble only with blocks of the same nature and do not mix. This leads to the formation of nanometer-scale domains of each block, alternatively present over the surface, i.e. microphase separation within the P3OT-b-P3BT block copolymer.
 | ||
| Fig. 4 (a) STM-image recorded at a concentration of 7.31 × 10−3 mg mL−1 on HOPG (60 × 60 nm2). The denser areas (x) are ascribed to the P3BT block and the less dense areas (y) to the P3OT block. Parameters of imaging: It = 25 pA; Vt = −1200 mV. (b) Linear correlation between lateral spacing of polymer strands and the number of carbon atoms in the side-chain. Values for hexyl, decyl and dodecyl side-chains are adapted from literature.46,47,49,51,52 (c) STM-image recorded at a concentration of 1.30 × 10−2 mg mL−1 on HOPG (70 × 70 nm2). The brighter areas are polymer strands of the second layer. Parameters of imaging: It = 300 pA; Vt = −800 mV. | ||
In Fig. 4a, a sub-monolayer coverage is present. Further study of the formation of microphases can be performed at a higher surface coverage. This can be achieved by increasing the concentration of the solution. A representative image, recorded 105 min after dropcasting the original solution and equivalent with a concentration of 1.3 × 10−2 mg mL−1 due to evaporation, is shown in Fig. 4c. This evidences that at full monolayer coverage, the same properties are maintained and different blocks exclusively assemble with themselves, resulting in microphase separation within the monolayer. Furthermore, chain-folding (hairpin bends within a polymer chain) can be observed in the monolayer. This chain-folding is frequently present in the P3OT-domains, but is seldom seen in the P3BT-domains even though the block lengths are very similar. This could be explained by the smaller folding radius expected in P3BT, which would cause more strain on the chain compared to P3OT, in which this folding radius is larger because of the larger lateral distance between the chains. The brighter areas in Fig. 4c are ascribed to the presence of a second polymer layer. The assembly is, however, dynamic and slightly unstable, which can be observed with the ‘disappearance’ of some strands of the second layer in consecutive images, as well as reorganization in the first layer. However, from the images it is clear that the second layer consistently conserves the same orientation as the chains underneath. Furthermore, the high difference in contrast between the different layers shows that STM measurements can also be used for studying multilayers of polymers and the homogeneity of the films, and therefore the film-forming properties if higher concentrations are used (Fig. S7, ESI†). It is observed that also in the case of multilayers (>2), the upper layers conserve the orientation that is present in the first layer and that thicker films seem to be relatively homogenous since the difference between the highest and the lowest topographic levels corresponds to the height of two layers.
Conclusions
In conclusion, a P3OT-b-P3BT block copolymer was synthesized with very little non-diblock copolymer impurities by successive monomer addition in a one-pot reaction using the Kumada Catalyst Transfer Protocol. 1H NMR study showed a P3OT block length of 27 units and a P3BT block length of 29 with complete further growth and low Ð-values according to GPC. The self-assembling properties of P3OT-b-P3BT were studied using DSC and AFM measurements. Furthermore, the microphase separation in this block copolymer caused by the difference in the side-chain length in the two blocks was directly visualized by Scanning Tunneling Microscopy (STM). The different microphases could be assigned to the corresponding block by relating the lateral stacking distances between adjacent chains to the side-chain length of the polymer block. This allows to study the impact of molecular changes on the supramolecular structure, making STM a valuable tool towards further understanding and controlling microphase separation in conjugated block copolymers.Acknowledgements
We are grateful to the Onderzoeksfonds KU Leuven/Research Fund KU Leuven and the Fund for Scientific Research (FWO-Vlaanderen). PW is grateful to the agency for Innovation by Science and Technology (IWT) for a doctoral fellowship. JA thanks Fund for Scientific Research (FWO-Vlaanderen) for a postdoctoral fellowship. Part of the research leading to these results has also received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013)/ERC grant agreement no. 340324.The Leuven-Mons collaboration is supported by the Science Policy Office of the Belgian Federal Government (BELSPO – IAP 7/05). Research in Mons is also supported by the European Commission and Région Wallonne (FEDER program; SMARTFILM project) and FNRS/FRFC. DM is grateful to FRIA for a doctoral fellowship. PhL is senior research associate of FNRS.Notes and references
- U. Scherf, A. Gutacker and N. Koenen, Acc. Chem. Res., 2008, 41, 1086–1097 CrossRef CAS PubMed.
- C. Guo, Y.-H. Lin, M. D. Witman, K. A. Smith, C. Wang, A. Hexemer, J. Strzalka, E. D. Gomez and R. Verduzco, Nano Lett., 2013, 13, 2957–2963 CrossRef CAS PubMed.
- M. C. Iovu, E. E. Sheina, R. R. Gil and R. D. McCullough, Macromolecules, 2005, 38, 8649–8656 CrossRef CAS.
- T. Yokozawa, I. Adachi, R. Miyakoshi and A. Yokoyama, High Perform. Polym., 2007, 19, 684–699 CrossRef CAS PubMed.
- Y. Zhang, K. Tajima, K. Hirota and K. Hashimoto, J. Am. Chem. Soc., 2008, 130, 7812–7813 CrossRef CAS PubMed.
- K. Van den Bergh, J. Huybrechts, T. Verbiest and G. Koeckelberghs, Chem.–Eur. J., 2008, 14, 9122–9125 CrossRef CAS PubMed.
- K. Van den Bergh, I. Cosemans, T. Verbiest and G. Koeckelberghs, Macromolecules, 2010, 43, 3794–3800 CrossRef CAS.
- M. Verswyvel, K. Goossens and G. Koeckelberghs, Polym. Chem., 2013, 4, 5310–5320 RSC.
- F. Boon, N. Hergué, G. Deshayes, D. Moerman, S. Desbief, J. De Winter, P. Gerbaux, Y. H. Geerts, R. Lazzaroni and P. Dubois, Polym. Chem., 2013, 4, 4303–4307 RSC.
- Z. J. Bryan, M. L. Smith and A. J. McNeil, Macromol. Rapid Commun., 2012, 33, 842–847 CrossRef CAS PubMed.
- P. Willot, S. Govaerts and G. Koeckelberghs, Macromolecules, 2013, 46, 8888–8895 CrossRef CAS.
- A. E. Javier, S. R. Varshney and R. D. McCullough, Macromolecules, 2010, 43, 3233–3237 CrossRef CAS.
- J. Hollinger, A. A. Jahnke, N. Coombs and D. S. Seferos, J. Am. Chem. Soc., 2010, 132, 8546–8547 CrossRef CAS PubMed.
- E. Goto, S. Nakamura, S. Kawauchi, H. Mori, M. Ueda and T. Higashihara, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2287–2296 CrossRef CAS.
- V. Senkovskyy, R. Tkachov, H. Komber, M. Sommer, M. Heuken, B. Voit, W. T. S. Huck, V. Kataev, A. Petr and A. Kiriy, J. Am. Chem. Soc., 2011, 133, 19966–19970 CrossRef CAS PubMed.
- Y. Nanashima, A. Yokoyama and T. Yokozawa, Macromolecules, 2012, 45, 2609–2613 CrossRef CAS.
- A. Yokoyama, A. Kato, R. Miyakoshi and T. Yokozawa, Macromolecules, 2008, 41, 7271–7273 CrossRef CAS.
- S. Wu, L. Bu, L. Huang, X. Yu, Y. Han, Y. Geng and F. Wang, Polymer, 2009, 50, 6245–6251 CrossRef CAS PubMed.
- A. Amarjargal, L. D. Tijing, C.-H. Park, I.-T. Im and C. S. Kim, Eur. Polym. J., 2013, 49, 3796–3805 CrossRef CAS PubMed.
- R. Miyakoshi, K. Shimono, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2006, 128, 16012–16013 CrossRef CAS PubMed.
- C. R. Bridges, H. Yan, A. A. Pollit and D. S. Seferos, ACS Macro Lett., 2014, 3, 671–674 CrossRef CAS.
- S. Wu, Y. Sun, L. Huang, J. Wang, Y. Zhou, Y. Geng and F. Wang, Macromolecules, 2010, 43, 4438–4440 CrossRef CAS.
- A. Sui, X. Shi, S. Wu, H. Tian, Y. Geng and F. Wang, Macromolecules, 2012, 45, 5436–5443 CrossRef CAS.
- R. Dattani, J. H. Bannock, Z. Fei, R. C. I. MacKenzie, A. A. Y. Guilbert, M. S. Vezie, J. Nelson, J. C. de Mello, M. Heeney, J. T. Cabral and A. J. Nedoma, J. Mater. Chem. A, 2014, 2, 14711 CAS.
- F. Li, K. G. Yager, N. M. Dawson, Y.-B. Jiang, K. J. Malloy and Y. Qin, Chem. Mater., 2014, 26, 3747–3756 CrossRef CAS.
- K. A. Smith, B. Stewart, K. G. Yager, J. Strzalka and R. Verduzco, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 900–906 CrossRef CAS.
- J. Hollinger, P. M. DiCarmine, D. Karl and D. S. Seferos, Macromolecules, 2012, 45, 3772–3778 CrossRef CAS.
- Y. Chen, H. Cui, L. Li, Z. Tian and Z. Tang, Polym. Chem., 2014, 5, 4441–4445 Search PubMed.
- H. Hoppe, M. Niggemann, C. Winder, J. Kraut, R. Hiesgen, A. Hinsch, D. Meissner and N. S. Sariciftci, Adv. Funct. Mater., 2004, 14, 1005–1011 CrossRef CAS.
- Y.-H. Lin, K. A. Smith, C. N. Kempf and R. Verduzco, Polym. Chem., 2013, 4, 229–232 RSC.
- P.-T. Wu, G. Ren, C. Li, R. Mezzenga and S. A. Jenekhe, Macromolecules, 2009, 42, 2317–2320 CrossRef CAS.
- J. Ge, M. He, F. Qiu and Y. Yang, Macromolecules, 2010, 43, 6422–6428 CrossRef CAS.
- P. Samorì, M. Surin, V. Palermo, R. Lazzaroni and P. Leclère, Phys. Chem. Chem. Phys., 2006, 8, 3927–3938 RSC.
- L. Sardone, C. Sabatini, G. Latini, F. Barigelletti, G. Marletta, F. Cacialli and P. Samor, J. Mater. Chem., 2007, 17, 1387–1391 RSC.
- K. S. Mali, J. Adisoejoso, E. Ghijsens, I. De Cat and S. De Feyter, Acc. Chem. Res., 2012, 45, 1309–1320 CrossRef CAS PubMed.
- P. Samorí, V. Francke, K. Müllen and J. P. Rabe, Chem.–Eur. J., 1999, 5, 2312–2317 CrossRef.
- J.-R. Gong, H.-J. Yan, Q.-H. Yuan, L.-P. Xu, Z.-S. Bo and L.-J. Wan, J. Am. Chem. Soc., 2006, 128, 12384–12385 CrossRef CAS PubMed.
- Q. Chen, T. Chen, X. Zhang, L.-J. Wan, H.-B. Liu, Y.-L. Li and P. Stang, Chem. Commun., 2009, 3765–3767 RSC.
- S.-B. Lei, K. Deng, Y.-L. Yang, Q.-D. Zeng, C. Wang, Z. Ma, P. Wang, Y. Zhou, Q.-L. Fan and W. Huang, Macromolecules, 2007, 40, 4552–4560 CrossRef CAS.
- S. Lei, K. Deng, Z. Ma, W. Huang and C. Wang, Chem. Commun., 2011, 47, 8829–8831 RSC.
- A. Saywell, J. K. Sprafke, L. J. Esdaile, A. J. Britton, A. Rienzo, H. L. Anderson, J. N. O'Shea and P. H. Beton, Angew. Chem., Int. Ed., 2010, 49, 9136–9139 CrossRef CAS PubMed.
- Y. Okawa and M. Aono, Nature, 2001, 409, 683–684 CrossRef CAS PubMed.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687–691 CrossRef CAS PubMed.
- D. F. Perepichka and F. Rosei, Science, 2009, 323, 216–217 CrossRef CAS PubMed.
- L. Xu, L. Yang and S. Lei, Nanoscale, 2012, 4, 4399–4415 RSC.
- T. Kirschbaum, R. Azumi, E. Mena-Osteritz and P. Bäuerle, New J. Chem., 1999, 23, 241–250 RSC.
- E. Mena-Osteritz, A. Meyer, B. M. W. Langeveld-Voss, R. A. J. Janssen, E. W. Meijer and P. Bäuerle, Angew. Chem., Int. Ed., 2000, 39, 2679–2684 CrossRef.
- R. Azumi, G. Götz, T. Debaerdemaeker and P. Bäuerle, Chem.–Eur. J., 2000, 6, 735–744 CrossRef CAS.
- X. Ma, Y. Guo, T. Wang and Z. Su, J. Chem. Phys., 2013, 139, 014701 CrossRef PubMed.
- P. Keg, A. Lohani, D. Fichou, Y. M. Lam, Y. Wu, B. S. Ong and S. G. Mhaisalkar, Macromol. Rapid Commun., 2008, 29, 1197–1202 CrossRef CAS.
- B. Greévin, P. Rannou, R. Payerne, A. Pron and J. P. Travers, J. Chem. Phys., 2003, 118, 7097–7102 CrossRef PubMed.
- A. Bocheux, I. Tahar-Djebbar, C. Fiorini-Debuisschert, L. Douillard, F. Mathevet, A.-J. Attias and F. Charra, Langmuir, 2011, 27, 10251–10255 CrossRef CAS PubMed.
- H. Peeters, P. Couturon, S. Vandeleene, D. Moerman, P. Leclère, R. Lazzaroni, I. De Cat, S. De Feyter and G. Koeckelberghs, RSC Adv., 2013, 3, 3342 RSC.
- E. E. Sheina, J. Liu, M. C. Iovu, D. W. Laird and R. D. McCullough, Macromolecules, 2004, 37, 3526–3528 CrossRef CAS.
- A. Yokoyama, R. Miyakoshi and T. Yokozawa, Macromolecules, 2004, 37, 1169–1171 CrossRef CAS.
- R. Tkachov, V. Senkovskyy, H. Komber, J.-U. Sommer and A. Kiriy, J. Am. Chem. Soc., 2010, 132, 7803–7810 CrossRef CAS PubMed.
- M. Verswyvel, F. Monnaie and G. Koeckelberghs, Macromolecules, 2011, 44, 9489–9498 CrossRef CAS.
- A. Smeets, K. Van den Bergh, J. De Winter, P. Gerbaux, T. Verbiest and G. Koeckelberghs, Macromolecules, 2009, 42, 7638–7641 CrossRef CAS.
- T. Hardeman, P. Willot, J. De Winter, T. Josse, P. Gerbaux, P. Shestakova, E. Nies and G. Koeckelberghs, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 804–809 CrossRef CAS.
- G. Derue, S. Coppee, S. Gabriele, M. Surin, V. Geskin, F. Monteverde, P. Leclere, R. Lazzaroni and P. Damman, J. Am. Chem. Soc., 2005, 127, 8018–8019 CrossRef CAS PubMed.
- M. Surin, P. Leclere, R. Lazzaroni, J. D. Yuen, G. Wang, D. Moses, A. J. Heeger, S. Cho and K. Lee, J. Appl. Phys., 2006, 100, 033712/1–033712/6 CrossRef CAS PubMed.
- P. Willot, J. Steverlynck, D. Moerman, P. Leclère, R. Lazzaroni and G. Koeckelberghs, Polym. Chem., 2013, 4, 2662–2671 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H NMR spectra & calculations, DSC, AFM and STM results. See DOI: 10.1039/c4ra11461b |
| This journal is © The Royal Society of Chemistry 2015 |